Mechanisms of Metastatic Tumor Dormancy and Implications for Cancer Therapy




Abstract
:1. Introduction
2. Mechanisms Regulating Metastatic Dormancy
2.1. Mechanisms That Sustain Metastatic Dormancy
2.2. Mechanisms That Promote Escape from Dormancy
2.3. Epigenetic Alterations That Regulate Metastatic Dormancy
3. Microenvironment
3.1. The Role of the Extracellular Matrix Components in Regulating Dormancy
3.2. Lung Microenvironment Controls the Metastatic Cell Dormant State
3.3. Brain Metastasis: Microenvironmental Factors Regulating Dormancy
3.4. Metastasis to the Bone: The Role of the Bone Marrow and the Bone Microenvironment in Dormancy
3.4.1. Bone Marrow and the Regulation of Dormancy
3.4.2. The Role of the Bone Microenvironment in Dormancy
4. Therapeutic Approaches against Dormancy
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 5-Aza-C | 5-azadeoxycytidine |
| ATG7 | Autophagy-related 7 |
| BM | Bone marrow |
| BMP | Bone morphogenetic protein |
| CDK4 | Cyclin-dependent kinase 4 |
| Col-I | Type I collagen |
| CSC | Cancer Stem Cells |
| CXCL12 | CXC-chemokine ligand 12 |
| CXCR4 | C-X-C motif chemokine receptor 4 |
| CYR61 | Cysteine-rich protein 61 |
| DTCs | Disseminating tumor cells |
| EMT | Epithelial to mesenchymal transition |
| ERs | Estrogen Receptors |
| FAK | Focal adhesion kinase |
| FBXW7 | F-box/WD repeat-containing protein 7 |
| FN | Fibronectin |
| GAS6 | Growth arrest-specific 6 |
| GFAP | Glial fibrillary acidic protein |
| GJIC | Gap junctional intercellular communication |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| HNSCC | Head and neck squamous cell carcinoma |
| HSCs | Hematopoietic stem cells |
| ID | Inhibitor of differentiation (Id) family of proteins |
| IDC | Ductal carcinoma |
| IL-8 | Interleukin-8 |
| KISS1 | Kisspeptin 1 |
| LIFR | Leukemia inhibitory factor receptor |
| LOX | Oxidase Lysyl oxidase |
| LPA1 | Lysophosphatidic acid receptor 1 |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MDSCs | Myeloid-derived suppressor cells |
| MLCK | Activating its downstream target myosin light chain by MLC kinase |
| MMP-9 | Metalloproteinase-9 |
| MSCs | Mesenchymal stem cells |
| MSK1 | Mitogen- and stress-activated protein kinase-1 |
| NDRG1 | N-myc downstream regulated gene 1 |
| NK | Natural killer |
| POSTN | Periostin |
| PRAME | Preferentially expressed antigen in melanoma |
| PRRX1 | Paired-related homeobox transcription factor |
| PTHRP | Parathyroid hormone-related protein |
| RANKL | Receptor activator of nuclear factor-κB ligand |
| Rb | Retinoblastoma |
| ROR2 | Receptor Tyrosine Kinase Like Orphan Receptor 2 |
| SCF-type | Skp1-Cul1-F box-type |
| SIAH2 | Siah E3 Ubiquitin Protein Ligase 2 |
| SDF-1 | Stromal cell-derived factor 1 |
| SOCS | Suppressor of cytokine signaling |
| TBK1 | Tank-binding kinase-1 |
| TBVA | Tumor-associated blood vessel Ags |
| TRAIL | TNF-Related Apoptosis Inducing Ligand |
| TGF-β1 | Transforming growth factor-β1 |
| TGF-β2 | Transforming growth factor-β2 |
| TGF-β3 | Transforming growth factor-β3 |
| TGF-βR | Transforming growth factor-β receptor |
| TNFα | Tumor necrosis factor-α |
| TSP-1 | Thrombospondin-1 |
| TGF-β | Transforming growth factor-β |
| uPAR | Urokinase plasminogen activator receptor |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VEGF-A | Vascular endothelial growth factor A |
| ZEB1 | Zinc finger E-box-binding homeobox 1 |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Pantel, K.; Brakenhoff, R.H. Dissecting the metastatic cascade. Nat. Rev. Cancer 2004, 4, 448–456. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef]
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [Green Version]
- Goddard, E.T.; Bozic, I.; Riddell, S.R.; Ghajar, C.M. Dormant tumour cells, their niches and the influence of immunity. Nat. Cell Biol. 2018, 20, 1240–1249. [Google Scholar] [CrossRef]
- Karrison, T.G.; Ferguson, D.J.; Meier, P. Dormancy of mammary carcinoma after mastectomy. J. Natl. Cancer Inst. 1999, 91, 80–85. [Google Scholar] [CrossRef]
- Freedland, S.J.; Moul, J.W. Prostate specific antigen recurrence after definitive therapy. J. Urol. 2007, 177, 1985–1991. [Google Scholar] [CrossRef]
- McNichols, D.W.; Segura, J.W.; DeWeerd, J.H. Renal cell carcinoma: Long-term survival and late recurrence. J. Urol. 1981, 126, 17–23. [Google Scholar] [CrossRef]
- Tsao, H.; Cosimi, A.B.; Sober, A.J. Ultra-late recurrence (15 years or longer) of cutaneous melanoma. Cancer 1997, 79, 2361–2370. [Google Scholar] [CrossRef]
- Klein, C.A. Framework models of tumor dormancy from patient-derived observations. Curr. Opin. Genet. Dev. 2011, 21, 42–49. [Google Scholar] [CrossRef]
- Schmidt-Kittler, O.; Ragg, T.; Daskalakis, A.; Granzow, M.; Ahr, A.; Blankenstein, T.J.; Kaufmann, M.; Diebold, J.; Arnholdt, H.; Muller, P.; et al. From latent disseminated cells to overt metastasis: Genetic analysis of systemic breast cancer progression. Proc. Natl. Acad. Sci. USA 2003, 100, 7737–7742. [Google Scholar] [CrossRef] [Green Version]
- Harper, K.L.; Sosa, M.S.; Entenberg, D.; Hosseini, H.; Cheung, J.F.; Nobre, R.; Avivar-Valderas, A.; Nagi, C.; Girnius, N.; Davis, R.J.; et al. Mechanism of early dissemination and metastasis in Her2 (+) mammary cancer. Nature 2016, 540, 588–592. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Giancotti, F.G. Mechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750–764. [Google Scholar] [CrossRef] [Green Version]
- Onoyama, I.; Nakayama, K.I. Fbxw7 in cell cycle exit and stem cell maintenance: Insight from gene-targeted mice. Cell Cycle 2008, 7, 3307–3313. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Takeishi, S.; Nakatsumi, H.; Nakayama, K.I. Prevention of cancer dormancy by Fbxw7 ablation eradicates disseminated tumor cells. JCI Insight 2019, 4, 125138. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef] [Green Version]
- Vander Griend, D.J.; Kocherginsky, M.; Hickson, J.A.; Stadler, W.M.; Lin, A.; Rinker-Schaeffer, C.W. Suppression of metastatic colonization by the context-dependent activation of the c-Jun NH2-terminal kinase kinases JNKK1/MKK4 and MKK7. Cancer Res. 2005, 65, 10984–10991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, S.D.; Hickson, J.A.; Hrobowski, Y.; Vander Griend, D.J.; Benson, D.; Montag, A.; Karrison, T.; Huo, D.; Rutgers, J.; Adams, S.; et al. Mitogen-activated protein kinase kinase 4 (MKK4) acts as a metastasis suppressor gene in human ovarian carcinoma. Cancer Res. 2002, 62, 6717–6723. [Google Scholar] [PubMed]
- Hickson, J.A.; Huo, D.; Vander Griend, D.J.; Lin, A.; Rinker-Schaeffer, C.W.; Yamada, S.D. The p38 kinases MKK4 and MKK6 suppress metastatic colonization in human ovarian carcinoma. Cancer Res. 2006, 66, 2264–2270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre-Ghiso, J.A.; Estrada, Y.; Liu, D.; Ossowski, L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK). Cancer Res. 2003, 63, 1684–1695. [Google Scholar] [CrossRef]
- El-Shennawy, L.; Dubrovskyi, O.; Kastrati, I.; Danes, J.M.; Zhang, Y.; Whiteley, H.E.; Creighton, C.J.; Frasor, J. Coactivation of Estrogen Receptor and IKKbeta Induces a Dormant Metastatic Phenotype in ER-Positive Breast Cancer. Cancer Res. 2018, 78, 974–984. [Google Scholar] [CrossRef] [Green Version]
- Nobutani, K.; Shimono, Y.; Mizutani, K.; Ueda, Y.; Suzuki, T.; Kitayama, M.; Minami, A.; Momose, K.; Miyawaki, K.; Akashi, K.; et al. Downregulation of CXCR4 in Metastasized Breast Cancer Cells and Implication in Their Dormancy. PLoS ONE 2015, 10, e0130032. [Google Scholar] [CrossRef]
- Jiang, J.; Zheng, M.; Zhang, M.; Yang, X.; Li, L.; Wang, S.S.; Wu, J.S.; Yu, X.H.; Wu, J.B.; Pang, X.; et al. PRRX1 Regulates Cellular Phenotype Plasticity and Dormancy of Head and Neck Squamous Cell Carcinoma Through miR-642b-3p. Neoplasia 2019, 21, 216–229. [Google Scholar] [CrossRef]
- Jiang, Y.; Berk, M.; Singh, L.S.; Tan, H.; Yin, L.; Powell, C.T.; Xu, Y. KiSS1 suppresses metastasis in human ovarian cancer via inhibition of protein kinase C alpha. Clin. Exp. Metastasis 2005, 22, 369–376. [Google Scholar] [CrossRef]
- Lee, J.H.; Miele, M.E.; Hicks, D.J.; Phillips, K.K.; Trent, J.M.; Weissman, B.E.; Welch, D.R. KiSS-1, a novel human malignant melanoma metastasis-suppressor gene. J. Natl. Cancer Inst. 1996, 88, 1731–1737. [Google Scholar] [CrossRef]
- Lee, J.H.; Welch, D.R. Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res. 1997, 57, 2384–2387. [Google Scholar]
- Lee, J.H.; Welch, D.R. Identification of highly expressed genes in metastasis-suppressed chromosome 6/human malignant melanoma hybrid cells using subtractive hybridization and differential display. Int. J. Cancer 1997, 71, 1035–1044. [Google Scholar] [CrossRef]
- Nash, K.T.; Phadke, P.A.; Navenot, J.M.; Hurst, D.R.; Accavitti-Loper, M.A.; Sztul, E.; Vaidya, K.S.; Frost, A.R.; Kappes, J.C.; Peiper, S.C.; et al. Requirement of KISS1 secretion for multiple organ metastasis suppression and maintenance of tumor dormancy. J. Natl. Cancer Inst. 2007, 99, 309–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khazali, A.S.; Clark, A.M.; Wells, A. Inflammatory cytokine IL-8/CXCL8 promotes tumour escape from hepatocyte-induced dormancy. Br. J. Cancer 2018, 118, 566–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, N.; Millena, A.C.; Walker, L.; Chaudhary, J.; Khan, S.A. Inhibitor of differentiation 1 (Id1) and Id3 proteins play different roles in TGFbeta effects on cell proliferation and migration in prostate cancer cells. Prostate 2013, 73, 624–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.P.; Perk, J.; Acharyya, S.; de Candia, P.; Mittal, V.; Todorova-Manova, K.; Gerald, W.L.; Brogi, E.; Benezra, R.; Massague, J. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc. Natl. Acad. Sci. USA 2007, 104, 19506–19511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swarbrick, A.; Roy, E.; Allen, T.; Bishop, J.M. Id1 cooperates with oncogenic Ras to induce metastatic mammary carcinoma by subversion of the cellular senescence response. Proc. Natl. Acad. Sci. USA 2008, 105, 5402–5407. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012, 150, 764–779. [Google Scholar] [CrossRef] [Green Version]
- Ruppender, N.; Larson, S.; Lakely, B.; Kollath, L.; Brown, L.; Coleman, I.; Coleman, R.; Nguyen, H.; Nelson, P.S.; Corey, E.; et al. Cellular Adhesion Promotes Prostate Cancer Cells Escape from Dormancy. PLoS ONE 2015, 10, e0130565. [Google Scholar] [CrossRef]
- Gawrzak, S.; Rinaldi, L.; Gregorio, S.; Arenas, E.J.; Salvador, F.; Urosevic, J.; Figueras-Puig, C.; Rojo, F.; Del Barco Barrantes, I.; Cejalvo, J.M.; et al. MSK1 regulates luminal cell differentiation and metastatic dormancy in ER (+) breast cancer. Nat. Cell Biol. 2018, 20, 211–221. [Google Scholar] [CrossRef]
- Adam, A.P.; George, A.; Schewe, D.; Bragado, P.; Iglesias, B.V.; Ranganathan, A.C.; Kourtidis, A.; Conklin, D.S.; Aguirre-Ghiso, J.A. Computational identification of a p38SAPK-regulated transcription factor network required for tumor cell quiescence. Cancer Res. 2009, 69, 5664–5672. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguirre Ghiso, J.A.; Kovalski, K.; Ossowski, L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell Biol. 1999, 147, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol. Biol. Cell 2001, 12, 863–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef] [Green Version]
- Rachman-Tzemah, C.; Zaffryar-Eilot, S.; Grossman, M.; Ribero, D.; Timaner, M.; Maki, J.M.; Myllyharju, J.; Bertolini, F.; Hershkovitz, D.; Sagi, I.; et al. Blocking Surgically Induced Lysyl Oxidase Activity Reduces the Risk of Lung Metastases. Cell Rep. 2017, 19, 774–784. [Google Scholar] [CrossRef] [Green Version]
- Cox, T.R.; Bird, D.; Baker, A.M.; Barker, H.E.; Ho, M.W.; Lang, G.; Erler, J.T. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013, 73, 1721–1732. [Google Scholar] [CrossRef] [Green Version]
- Barry-Hamilton, V.; Spangler, R.; Marshall, D.; McCauley, S.; Rodriguez, H.M.; Oyasu, M.; Mikels, A.; Vaysberg, M.; Ghermazien, H.; Wai, C.; et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nat. Med. 2010, 16, 1009–1017. [Google Scholar] [CrossRef]
- Cano, A.; Santamaria, P.G.; Moreno-Bueno, G. LOXL2 in epithelial cell plasticity and tumor progression. Future Oncol. 2012, 8, 1095–1108. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Salvador, F.; Martin, A.; Floristan, A.; Cuevas, E.P.; Santos, V.; Montes, A.; Morales, S.; Castilla, M.A.; Rojo-Sebastian, A.; et al. Lysyl oxidase-like 2 (LOXL2), a new regulator of cell polarity required for metastatic dissemination of basal-like breast carcinomas. EMBO Mol. Med. 2011, 3, 528–544. [Google Scholar] [CrossRef] [Green Version]
- Weidenfeld, K.; Schif-Zuck, S.; Abu-Tayeh, H.; Kang, K.; Kessler, O.; Weissmann, M.; Neufeld, G.; Barkan, D. Dormant tumor cells expressing LOXL2 acquire a stem-like phenotype mediating their transition to proliferative growth. Oncotarget 2016, 7, 71362–71377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Cock, J.M.; Shibue, T.; Dongre, A.; Keckesova, Z.; Reinhardt, F.; Weinberg, R.A. Inflammation Triggers Zeb1-Dependent Escape from Tumor Latency. Cancer Res. 2016, 76, 6778–6784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Huang, Y.J.; Liu, C.; Yang, Y.Y.; Liu, H.; Cui, J.G.; Cheng, Y.; Gao, F.; Cai, J.M.; Li, B.L. Inhibition of TBK1 attenuates radiation-induced epithelial-mesenchymal transition of A549 human lung cancer cells via activation of GSK-3beta and repression of ZEB1. Lab. Investig. J. Tech. Methods Pathol. 2014, 94, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malanchi, I.; Santamaria-Martinez, A.; Susanto, E.; Peng, H.; Lehr, H.A.; Delaloye, J.F.; Huelsken, J. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 2011, 481, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-beta 2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38 alpha/beta signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Zhang, X.H.; Massague, J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell 2011, 20, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Lorger, M.; Felding-Habermann, B. Capturing changes in the brain microenvironment during initial steps of breast cancer brain metastasis. Am. J. Pathol. 2010, 176, 2958–2971. [Google Scholar] [CrossRef]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1alpha induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [Green Version]
- Denkins, Y.; Reiland, J.; Roy, M.; Sinnappah-Kang, N.D.; Galjour, J.; Murry, B.P.; Blust, J.; Aucoin, R.; Marchetti, D. Brain metastases in melanoma: Roles of neurotrophins. Neuro Oncol. 2004, 6, 154–165. [Google Scholar] [CrossRef]
- Izraely, S.; Sagi-Assif, O.; Klein, A.; Meshel, T.; Tsarfaty, G.; Pasmanik-Chor, M.; Nahmias, C.; Couraud, P.O.; Ateh, E.; Bryant, J.L.; et al. The metastatic microenvironment: Brain-residing melanoma metastasis and dormant micrometastasis. Int. J. Cancer 2012, 131, 1071–1082. [Google Scholar] [CrossRef]
- Yeh, A.C.; Ramaswamy, S. Mechanisms of Cancer Cell Dormancy—Another Hallmark of Cancer? Cancer Res. 2015, 75, 5014–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, M.S.; Holmgren, L.; Shing, Y.; Chen, C.; Rosenthal, R.A.; Moses, M.; Lane, W.S.; Cao, Y.; Sage, E.H.; Folkman, J. Angiostatin: A novel angiogenesis inhibitor that mediates the suppression of metastases by a Lewis lung carcinoma. Cell 1994, 79, 315–328. [Google Scholar] [CrossRef]
- Cao, Y.; O’Reilly, M.S.; Marshall, B.; Flynn, E.; Ji, R.W.; Folkman, J. Expression of angiostatin cDNA in a murine fibrosarcoma suppresses primary tumor growth and produces long-term dormancy of metastases. J. Clin. Investig. 1998, 101, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almog, N.; Ma, L.; Raychowdhury, R.; Schwager, C.; Erber, R.; Short, S.; Hlatky, L.; Vajkoczy, P.; Huber, P.E.; Folkman, J.; et al. Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res. 2009, 69, 836–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122. [Google Scholar] [CrossRef]
- Naumov, G.N.; Bender, E.; Zurakowski, D.; Kang, S.Y.; Sampson, D.; Flynn, E.; Watnick, R.S.; Straume, O.; Akslen, L.A.; Folkman, J.; et al. A model of human tumor dormancy: An angiogenic switch from the nonangiogenic phenotype. J. Natl. Cancer Inst. 2006, 98, 316–325. [Google Scholar] [CrossRef]
- Bleau, A.M.; Agliano, A.; Larzabal, L.; de Aberasturi, A.L.; Calvo, A. Metastatic dormancy: A complex network between cancer stem cells and their microenvironment. Histol. Histopathol. 2014, 29, 1499–1510. [Google Scholar] [CrossRef]
- Sanger, N.; Effenberger, K.E.; Riethdorf, S.; Van Haasteren, V.; Gauwerky, J.; Wiegratz, I.; Strebhardt, K.; Kaufmann, M.; Pantel, K. Disseminated tumor cells in the bone marrow of patients with ductal carcinoma in situ. Int. J. Cancer 2011, 129, 2522–2526. [Google Scholar] [CrossRef]
- Hoilund-Carlsen, P.F.; Hess, S.; Werner, T.J.; Alavi, A. Cancer metastasizes to the bone marrow and not to the bone: Time for a paradigm shift! Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 893–897. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Wang, J.; Song, J.; Shiozawa, Y.; Wang, J.; Havens, A.; Wang, Z.; Sun, Y.X.; Emerson, S.G.; Krebsbach, P.H.; et al. Annexin II expressed by osteoblasts and endothelial cells regulates stem cell adhesion, homing, and engraftment following transplantation. Blood 2007, 110, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Axelrod, H.D.; Valkenburg, K.C.; Amend, S.R.; Hicks, J.L.; Parsana, P.; Torga, G.; DeMarzo, A.M.; Pienta, K.J. AXL Is a Putative Tumor Suppressor and Dormancy Regulator in Prostate Cancer. Mol. Cancer Res. 2019, 17, 356–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S. Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef] [PubMed]
- Khoo, W.H.; Ledergor, G.; Weiner, A.; Roden, D.L.; Terry, R.L.; McDonald, M.M.; Chai, R.C.; De Veirman, K.; Owen, K.L.; Opperman, K.S.; et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood 2019, 134, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Taichman, R.S.; Cooper, C.; Keller, E.T.; Pienta, K.J.; Taichman, N.S.; McCauley, L.K. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002, 62, 1832–1837. [Google Scholar] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [Green Version]
- Romero-Moreno, R.; Curtis, K.J.; Coughlin, T.R.; Cristina Miranda-Vergara, M.; Dutta, S.; Natarajan, A.; Facchine, B.A.; Jackson, K.M.; Nystrom, L.; Li, J.; et al. The CXCL5/CXCR2 axis is sufficient to promote breast cancer colonization during bone metastasis. Nat. Commun. 2019, 10, 4404. [Google Scholar] [CrossRef] [Green Version]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Kim Lyerly, H.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra73. [Google Scholar] [CrossRef]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C.; et al. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef] [Green Version]
- Walker, N.D.; Elias, M.; Guiro, K.; Bhatia, R.; Greco, S.J.; Bryan, M.; Gergues, M.; Sandiford, O.A.; Ponzio, N.M.; Leibovich, S.J.; et al. Exosomes from differentially activated macrophages influence dormancy or resurgence of breast cancer cells within bone marrow stroma. Cell Death Dis. 2019, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Casson, J.; Davies, O.G.; Smith, C.A.; Dalby, M.J.; Berry, C.C. Mesenchymal stem cell-derived extracellular vesicles may promote breast cancer cell dormancy. J. Tissue Eng. 2018, 9, 2041731418810093. [Google Scholar] [CrossRef]
- Yin, J.J.; Selander, K.; Chirgwin, J.M.; Dallas, M.; Grubbs, B.G.; Wieser, R.; Massague, J.; Mundy, G.R.; Guise, T.A. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Investig. 1999, 103, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Siegel, P.M.; Shu, W.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Zhang, H.; Zeng, Q.; Dai, J.; Keller, E.T.; Giordano, T.; Gu, K.; Shah, V.; Pei, L.; Zarbo, R.J.; et al. NF-kappaB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat. Med. 2007, 13, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Ren, D.; Dai, Y.; Yang, Q.; Zhang, X.; Guo, W.; Ye, L.; Huang, S.; Chen, X.; Lai, Y.; Du, H.; et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J. Exp. Med. 2019, 216, 428–449. [Google Scholar] [CrossRef]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef] [Green Version]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFbetaRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [Green Version]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.; Seshadri, M.; Komorowski, M.P.; Abrams, S.I.; Kozbor, D. Targeting CXCL12/CXCR4 signaling with oncolytic virotherapy disrupts tumor vasculature and inhibits breast cancer metastases. Proc. Natl. Acad. Sci. USA 2013, 110, E1291–E1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes-Bastos, B.; Jin, L.; Ruge, F.; Owen, S.; Sanders, A.; Cogle, C.; Chester, J.; Jiang, W.G.; Cai, J. Association of breast carcinoma growth with a non-canonical axis of IFNgamma/IDO1/TSP1. Oncotarget 2017, 8, 85024–85039. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, M.S.; Holmgren, L.; Chen, C.; Folkman, J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat. Med. 1996, 2, 689–692. [Google Scholar] [CrossRef]
- El Touny, L.H.; Vieira, A.; Mendoza, A.; Khanna, C.; Hoenerhoff, M.J.; Green, J.E. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J. Clin. Investig. 2014, 124, 156–168. [Google Scholar] [CrossRef] [Green Version]
- Bliss, S.A.; Sinha, G.; Sandiford, O.A.; Williams, L.M.; Engelberth, D.J.; Guiro, K.; Isenalumhe, L.L.; Greco, S.J.; Ayer, S.; Bryan, M.; et al. Mesenchymal Stem Cell-Derived Exosomes Stimulate Cycling Quiescence and Early Breast Cancer Dormancy in Bone Marrow. Cancer Res. 2016, 76, 5832–5844. [Google Scholar] [CrossRef] [Green Version]
- Marshall, J.C.; Collins, J.W.; Nakayama, J.; Horak, C.E.; Liewehr, D.J.; Steinberg, S.M.; Albaugh, M.; Vidal-Vanaclocha, F.; Palmieri, D.; Barbier, M.; et al. Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. J. Natl. Cancer Inst. 2012, 104, 1306–1319. [Google Scholar] [CrossRef]
- Lu, D.; Chen, S.; Tan, X.; Li, N.; Liu, C.; Li, Z.; Liu, Z.; Stupack, D.G.; Reisfeld, R.A.; Xiang, R. Fra-1 promotes breast cancer chemosensitivity by driving cancer stem cells from dormancy. Cancer Res. 2012, 72, 3451–3456. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Eyles, J.; Puaux, A.L.; Wang, X.; Toh, B.; Prakash, C.; Hong, M.; Tan, T.G.; Zheng, L.; Ong, L.C.; Jin, Y.; et al. Tumor cells disseminate early, but immunosurveillance limits metastatic outgrowth, in a mouse model of melanoma. J. Clin. Investig. 2010, 120, 2030–2039. [Google Scholar] [CrossRef]
- Pantel, K.; Schlimok, G.; Kutter, D.; Schaller, G.; Genz, T.; Wiebecke, B.; Backmann, R.; Funke, I.; Riethmuller, G. Frequent down-regulation of major histocompatibility class I antigen expression on individual micrometastatic carcinoma cells. Cancer Res. 1991, 51, 4712–4715. [Google Scholar] [PubMed]
- Hirata, Y.; Furuhashi, K.; Ishii, H.; Li, H.W.; Pinho, S.; Ding, L.; Robson, S.C.; Frenette, P.S.; Fujisaki, J. CD150(high) Bone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege via Adenosine. Cell Stem Cell 2018, 22, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bichsel, C.A.; Wang, L.; Froment, L.; Berezowska, S.; Muller, S.; Dorn, P.; Marti, T.M.; Peng, R.W.; Geiser, T.; Schmid, R.A.; et al. Increased PD-L1 expression and IL-6 secretion characterize human lung tumor-derived perivascular-like cells that promote vascular leakage in a perfusable microvasculature model. Sci. Rep. 2017, 7, 10636. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Bose, A.; Komita, H.; Taylor, J.L.; Chi, N.; Lowe, D.B.; Okada, H.; Cao, Y.; Mukhopadhyay, D.; Cohen, P.A.; et al. Vaccines targeting tumor blood vessel antigens promote CD8(+) T cell-dependent tumor eradication or dormancy in HLA-A2 transgenic mice. J. Immunol. 2012, 188, 1782–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudemont, A.; Jouy, N.; Hetuin, D.; Quesnel, B. NK cells that are activated by CXCL10 can kill dormant tumor cells that resist CTL-mediated lysis and can express B7-H1 that stimulates T cells. Blood 2005, 105, 2428–2435. [Google Scholar] [CrossRef] [PubMed]
- Payne, K.K.; Keim, R.C.; Graham, L.; Idowu, M.O.; Wan, W.; Wang, X.Y.; Toor, A.A.; Bear, H.D.; Manjili, M.H. Tumor-reactive immune cells protect against metastatic tumor and induce immunoediting of indolent but not quiescent tumor cells. J. Leukoc. Biol. 2016, 100, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyengar, N.M.; Hudis, C.A.; Dannenberg, A.J. Obesity and inflammation: New insights into breast cancer development and progression. Am. Soc. Clin. Oncol. Educ. Book 2013, 33, 46–51. [Google Scholar] [CrossRef]
- Quail, D.F.; Olson, O.C.; Bhardwaj, P.; Walsh, L.A.; Akkari, L.; Quick, M.L.; Chen, I.C.; Wendel, N.; Ben-Chetrit, N.; Walker, J.; et al. Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nat. Cell Biol. 2017, 19, 974–987. [Google Scholar] [CrossRef]
- Krall, J.A.; Reinhardt, F.; Mercury, O.A.; Pattabiraman, D.R.; Brooks, M.W.; Dougan, M.; Lambert, A.W.; Bierie, B.; Ploegh, H.L.; Dougan, S.K.; et al. The systemic response to surgery triggers the outgrowth of distant immune-controlled tumors in mouse models of dormancy. Sci. Transl. Med. 2018, 10, eaaan3464. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Fryknas, M.; Hernlund, E.; Fayad, W.; De Milito, A.; Olofsson, M.H.; Gogvadze, V.; Dang, L.; Pahlman, S.; Schughart, L.A.; et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat. Commun. 2014, 5, 3295. [Google Scholar] [CrossRef] [Green Version]
- Saudemont, A.; Hamrouni, A.; Marchetti, P.; Liu, J.; Jouy, N.; Hetuin, D.; Colucci, F.; Quesnel, B. Dormant tumor cells develop cross-resistance to apoptosis induced by CTLs or imatinib mesylate via methylation of suppressor of cytokine signaling 1. Cancer Res. 2007, 67, 4491–4498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Politaki, E.; Agelaki, S.; Apostolaki, S.; Hatzidaki, D.; Strati, A.; Koinis, F.; Perraki, M.; Saloustrou, G.; Stoupis, G.; Kallergi, G.; et al. A Comparison of Three Methods for the Detection of Circulating Tumor Cells in Patients with Early and Metastatic Breast Cancer. Cell. Physiol. Biochem. 2017, 44, 594–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiliotaki, M.; Mavroudis, D.; Kokotsaki, M.; Vetsika, E.K.; Stoupis, I.; Matikas, A.; Kallergi, G.; Georgoulias, V.; Agelaki, S. Expression of insulin-like growth factor-1 receptor in circulating tumor cells of patients with breast cancer is associated with patient outcomes. Mol. Oncol. 2018, 12, 21–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiliotaki, M.; Mavroudis, D.; Kapranou, K.; Markomanolaki, H.; Kallergi, G.; Koinis, F.; Kalbakis, K.; Georgoulias, V.; Agelaki, S. Evaluation of proliferation and apoptosis markers in circulating tumor cells of women with early breast cancer who are candidates for tumor dormancy. Breast Cancer Res. BCR 2014, 16, 485. [Google Scholar] [CrossRef] [Green Version]
- Papadaki, C.; Stratigos, M.; Markakis, G.; Spiliotaki, M.; Mastrostamatis, G.; Nikolaou, C.; Mavroudis, D.; Agelaki, S. Circulating microRNAs in the early prediction of disease recurrence in primary breast cancer. Breast Cancer Res. BCR 2018, 20, 72. [Google Scholar] [CrossRef] [Green Version]
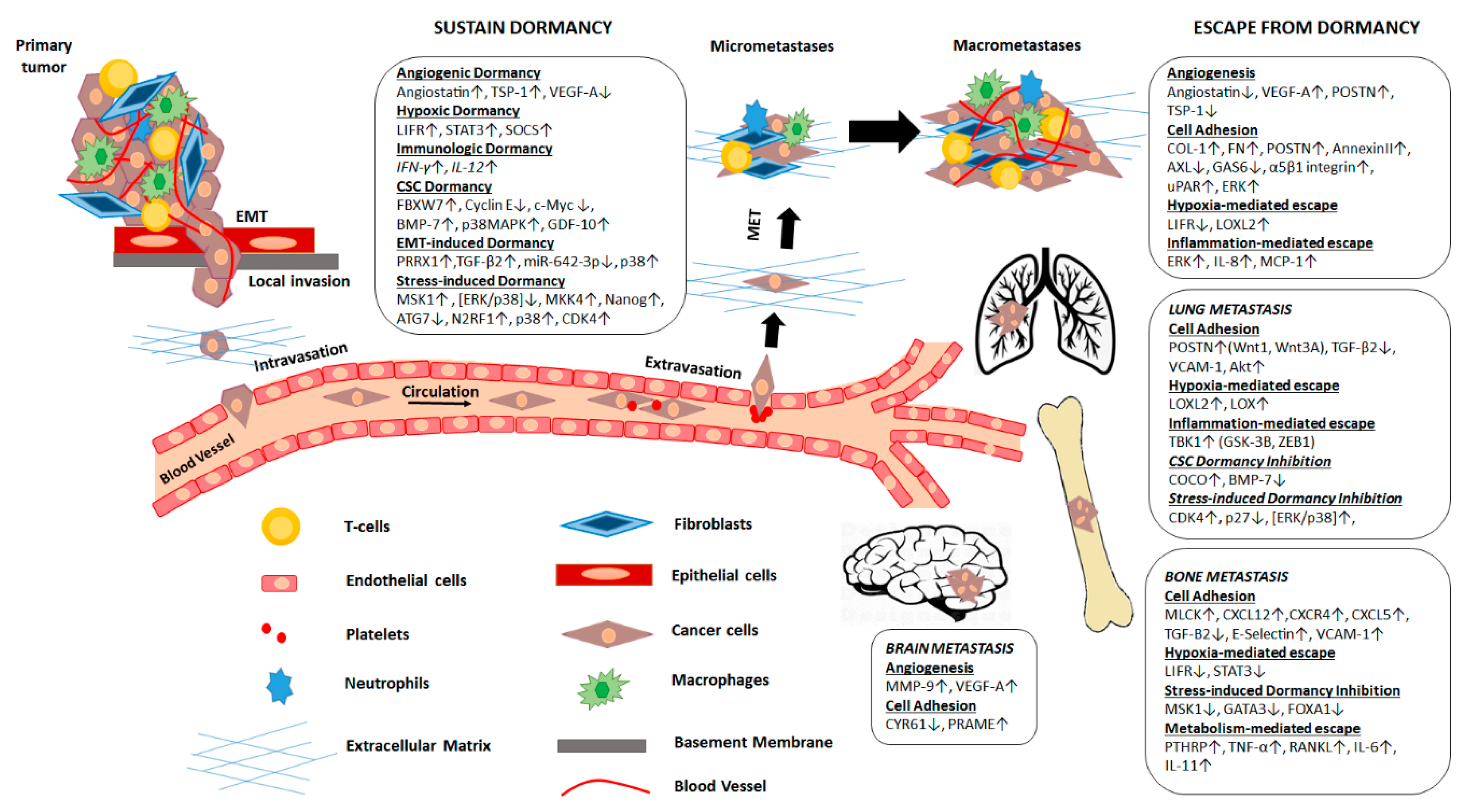

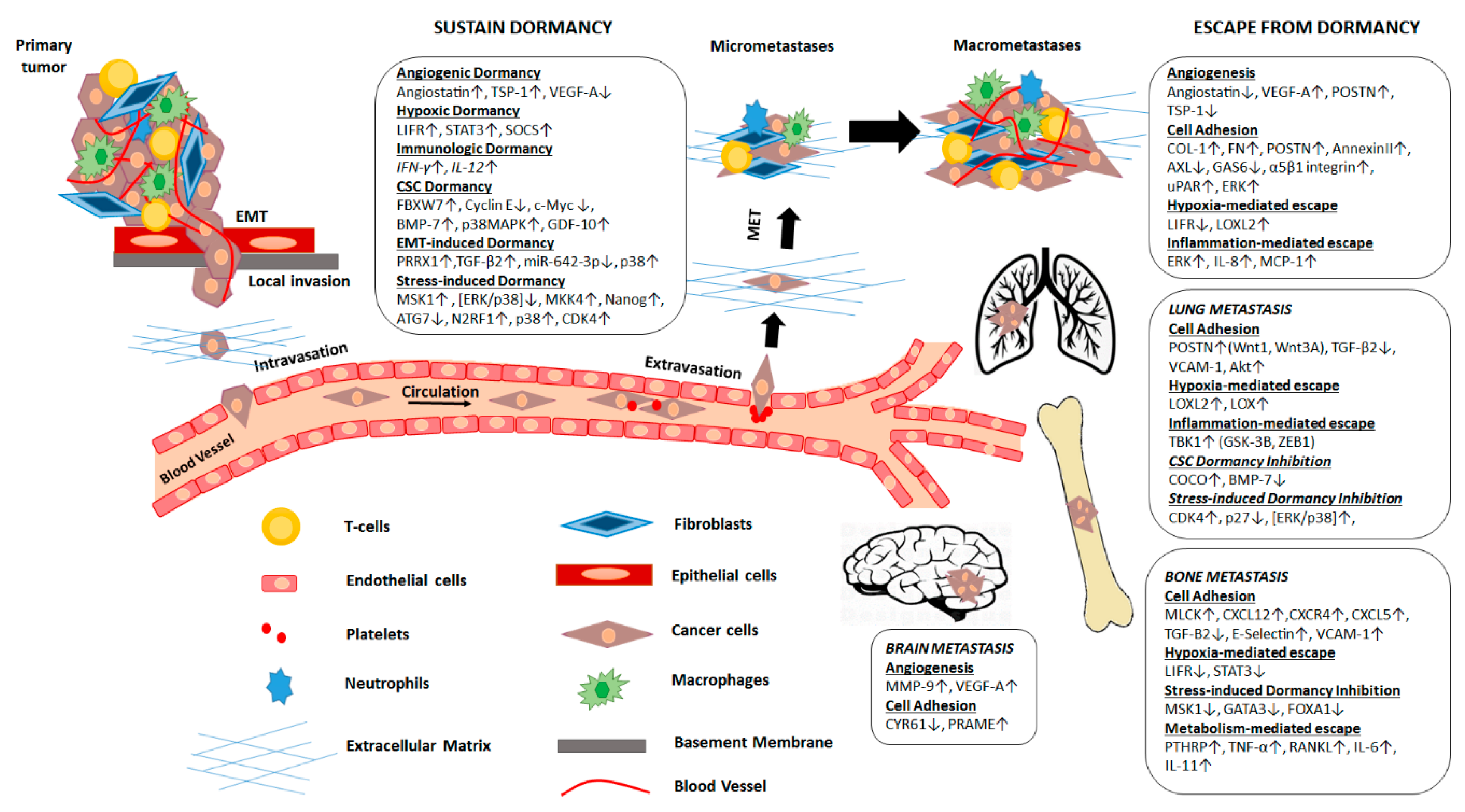{kind=link}
| Factor | Mechanism | Regulation | Cancer Type | Metastatic Site | Model | Ref |
|---|---|---|---|---|---|---|
| Mechanisms that Sustain Dormancy | ||||||
| Fbxw7 | Cell cycle control | Increased levels | Breast | Lung | Mouse | [18] |
| LIFR | Hypoxia | Increased levels | Breast | Bone marrow | Mouse/Human | [19] |
| ATG7 | Autophagy | Increased levels | Breast | Lung | Mouse/Human | [20] |
| MKK4 | Apoptosis, proliferation, differentiation | Increased levels | Ovarian | Intraperitoneal sites | Mouse | [23] |
| IKKβ | Inflammation | Overactivation | Breast | Multiple sites | Mouse | [25] |
| CXCR4 | Cell cycle control, Inflammation, Cell survival | Decreased levels | Breast | Lung | Mouse | [26] |
| PRRX1 | EMT | Increased levels | HNSCC | Lymph nodes | Mouse/Human | [27] |
| KISS 1 | Hormone regulation | Increased levels | Melanoma Breast Ovarian | Lung Intraperitoneal sites | Mouse | [28,29,30] |
| MSK1 | Differentiation | Increased levels | Breast | Bone | Human | [39] |
| N2RF1/NANOG | Development Differentiation | Increased levels | HNSCC Prostate | Bone marrow | Human | [41] |
| TGF-β2 | Development Morphogenesis | Increased levels | HNSCC | Bone marrow | Mouse | [55] |
| GAS6/AXL | Apoptosis Differentiation | Increased levels | Prostate | Liver, Lymph node, Bone | Mouse/Human | [71] |
| BMP-7 | Morphogenesis Differentiation | Increased levels | Prostate | Bone | Mouse/Human | [80] |
| Wnt5a | Development | Increased levels | Prostate | Bone | Mouse/Rat/Human | [87] |
| GDF10/TGF-β2/ TGF-βRIII | Cell cycle regulation | Increased levels | Prostate | Bone | Mouse/Human | [89] |
| IFN-γ, IL-12 | Immune response | Increased levels | Sarcoma | Multiple sites | Mouse | [91] |
| Mechanisms that Promote Escape from Dormancy | ||||||
| IL8/MCP-1 | Inflammation | Increased levels | Breast | Liver | Ex vivo | [33] |
| ID1/ID3 | Proliferation Differentiation | Increased levels | Breast | Lung | Mouse | [35] |
| Coco | Morphogenesis | Increased levels | Breast | Lung | Mouse | [37] |
| MLCK | Proliferation, Actin stress fiber formation | Constitutive activation | Prostate Breast | Bone marrow Lung | Human Mouse | [38,44] |
| Col-I | Induction of fibrosis | Increased levels | Breast | Lung | Mouse | [45] |
| LOX | Development Hypoxia | Increased levels | Breast | Lung | Human/Mouse | [46] |
| LOXL2 | EMT Hypoxia | Increased levels | Breast | Lung | Mouse | [51] |
| Zeb1 | EMT Inflammation | Increased levels | Breast | Lung | Mouse | [52] |
| POSTN | Bone regeneration Cell adhesion | Increased levels | Breast/Cancer stem cells | Lung | Human/Mouse | [54] |
| VCAM-1 | Cell adhesion | Increased levels | Breast | Lung/Bone | Mouse/Human | [56,90] |
| MMP-9 | Metabolic processes | Increased levels | Breast | Brain | Mouse | [57] |
| PRAME | Apoptosis Differentiation | Increased levels | Melanoma | Brain | Human/mouse | [60] |
| VEGF-A | Angiogenesis | Increased levels | Melanoma Lung | Brain | Mouse | [66] |
| TSP-1 | ECM constituent | Elevated levels | Breast | Bone Marrow | Mouse Zebrafish Human | [75] |
| CXCL12/CXCR4 | Embryonic development | Elevated levels | Prostate | Bone marrow | Human/Ex vivo | [76] |
| CXCL5/CXCR2 | Proliferation | Elevated levels | Breast | Bone | Mouse | [78] |
| E-selectin | Cell adhesion | Elevated levels | Breast | Bone marrow | Human/Mouse | [79] |
| PTHRP | Metabolic processes | Elevated levels | Breast | Bone | Mouse | [83] |
| Approach | Mechanism | Therapeutic Method | Effect | Ref |
|---|---|---|---|---|
| Prolonging Dormant State | ||||
| Enhancing tumor-associated microvessel induced dormancy | Regulation of the IFNγ/IDO1/TSP1 axis | Administration of TSP1 | Reduction of proliferation of invasive cells | [93] |
| Inhibiting angiogenesis | Activation of Angiostatin-regulated pathways | Upregulation of Angiostatin | Inhibition of tumor growth, reduction of metastases | [94] |
| Regulation of expression of LPA1 that is inversely correlated with Nm23-H1 expression | Modulating LPA1 levels | Specific LPA1 inhibitor Debio-0719 | Reduced expression of proliferative markers Ki67 and pErk, increase of p-p38 stress kinase | [97] |
| Epigenetic regulation of expression of pluripotent genes | Upregulation of master receptor NR2F1 | 5-AZA demethylating agent | Increase in expression of SOX9, RARβ, and NANOG | [41] |
| Tumor blood vessel antigens | Activation of T-cell dependent immunity | Vaccines against TBVA | Tumor regression | [104] |
| Combination of immune therapy with Adriamycin | Activation of T cells and NK cells against cancer cells | Tumor-sensitized T cells and CD25(+) NKT cells | Sensitization of dormant cells to immunoediting, prolonged animal survival | [106] |
| Elimination of Dormant Cells | ||||
| Inhibition of cell cycle | Prevention of COL1-induced proliferation and upregulation of p27 | Saracatinib with ERK1/2 inhibitor | Apoptotic cell death | [95] |
| Activation of T cells against cancer | Induction of NK cells to express PDL-1 | Vaccination with cells transduced with CXCL10 | Destruction of cancer cells by immune system | [105] |
| Reduce inflammation at metastatic site | Reducing the pro-metastatic effect of GM-CSF and IL-5 | Low-fat diet | Decreased metastatic burden | [108] |
| Reduction of systemic inflammation | Inflammation | Perioperative treatment with NSAIDs | Decreased metastatic burden | [109] |
| Induction of mitochondrial dysfunction | Reduced mitochondrial respiration, leading to bioenergetic catastrophe | Small molecule VLX600 | Tumor cell death | [110] |
| Reducing resistance induced by the JAK/STAT pathways | Inhibition of SOCS1 and IL-3 | Specific inhibitors against SOCS1 and IL-3 | Apoptosis | [111] |
| Sensitization of Dormant Cells to Chemotherapy | ||||
| Blocking communication of cancer cells with microenvironment | Disrupting CXCL12/ CXCR4 binding | CXCR4 antagonists | DTCs are mobilized from the BM, activate cell cycle | [92] |
| Blocking interaction with microenvironment | miRNA contents of exosomes | Administration of MSC loaded with antagomiR222/223 | Breast cancer cells become sensitive to carboplatin | [96] |
| Cancer stem cells | Modulating Fra-1 levels | Enhanced expression of Fra-1 | Decreased tumor incidence, chemosensitivity | [98] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neophytou, C.M.; Kyriakou, T.-C.; Papageorgis, P. Mechanisms of Metastatic Tumor Dormancy and Implications for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 6158. https://doi.org/10.3390/ijms20246158
Neophytou CM, Kyriakou T-C, Papageorgis P. Mechanisms of Metastatic Tumor Dormancy and Implications for Cancer Therapy. International Journal of Molecular Sciences. 2019; 20(24):6158. https://doi.org/10.3390/ijms20246158
Chicago/Turabian StyleNeophytou, Christiana M., Theodora-Christina Kyriakou, and Panagiotis Papageorgis. 2019. "Mechanisms of Metastatic Tumor Dormancy and Implications for Cancer Therapy" International Journal of Molecular Sciences 20, no. 24: 6158. https://doi.org/10.3390/ijms20246158
APA StyleNeophytou, C. M., Kyriakou, T. -C., & Papageorgis, P. (2019). Mechanisms of Metastatic Tumor Dormancy and Implications for Cancer Therapy. International Journal of Molecular Sciences, 20(24), 6158. https://doi.org/10.3390/ijms20246158