Acceptor Specificity of β-N-Acetylhexosaminidase from Talaromyces flavus: A Rational Explanation

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. Screening of the Glycosylation of Various Acceptors
2.1.1. Glycosylation Reaction Catalyzed by TfHex WT: Screening of Natural Monosaccharides
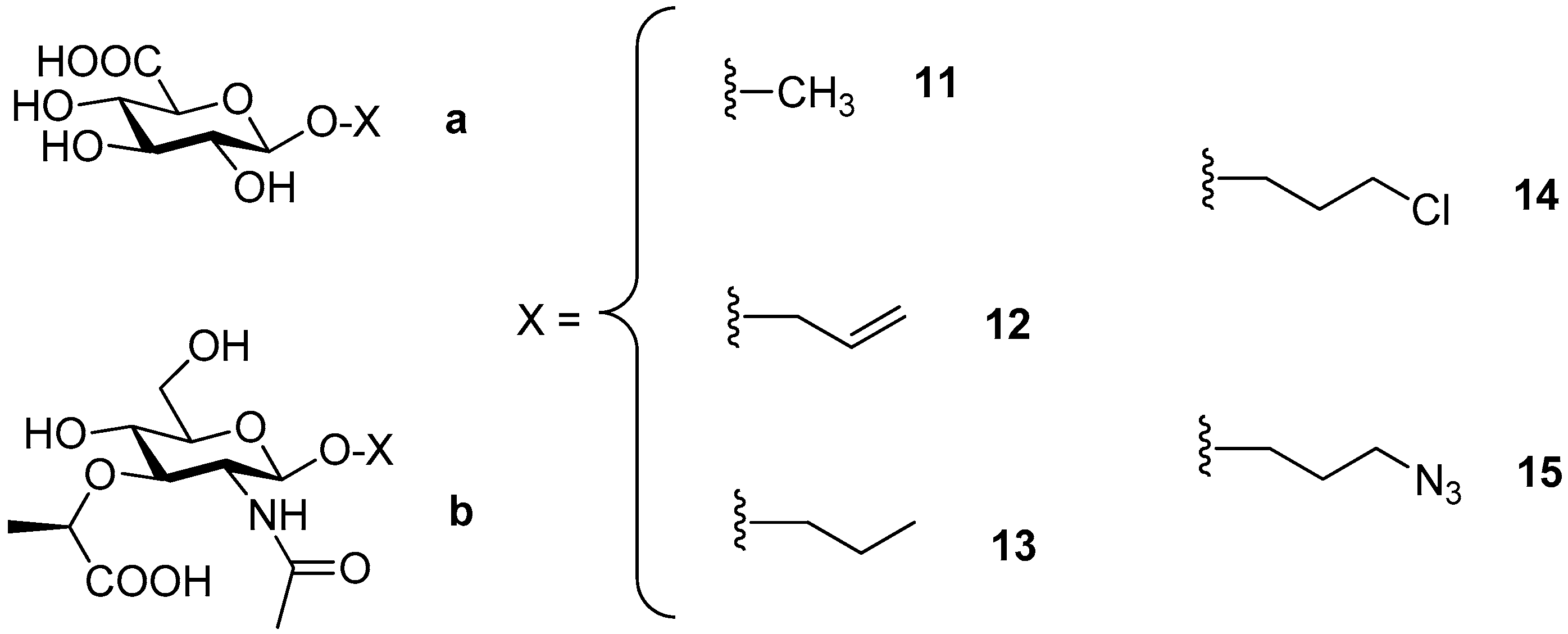
2.1.2. Glycosylation Reaction Catalyzed by TfHex Y470H: Screening of GlcA and MurNAc Glycosides
2.2. Docking and Molecular Dynamics
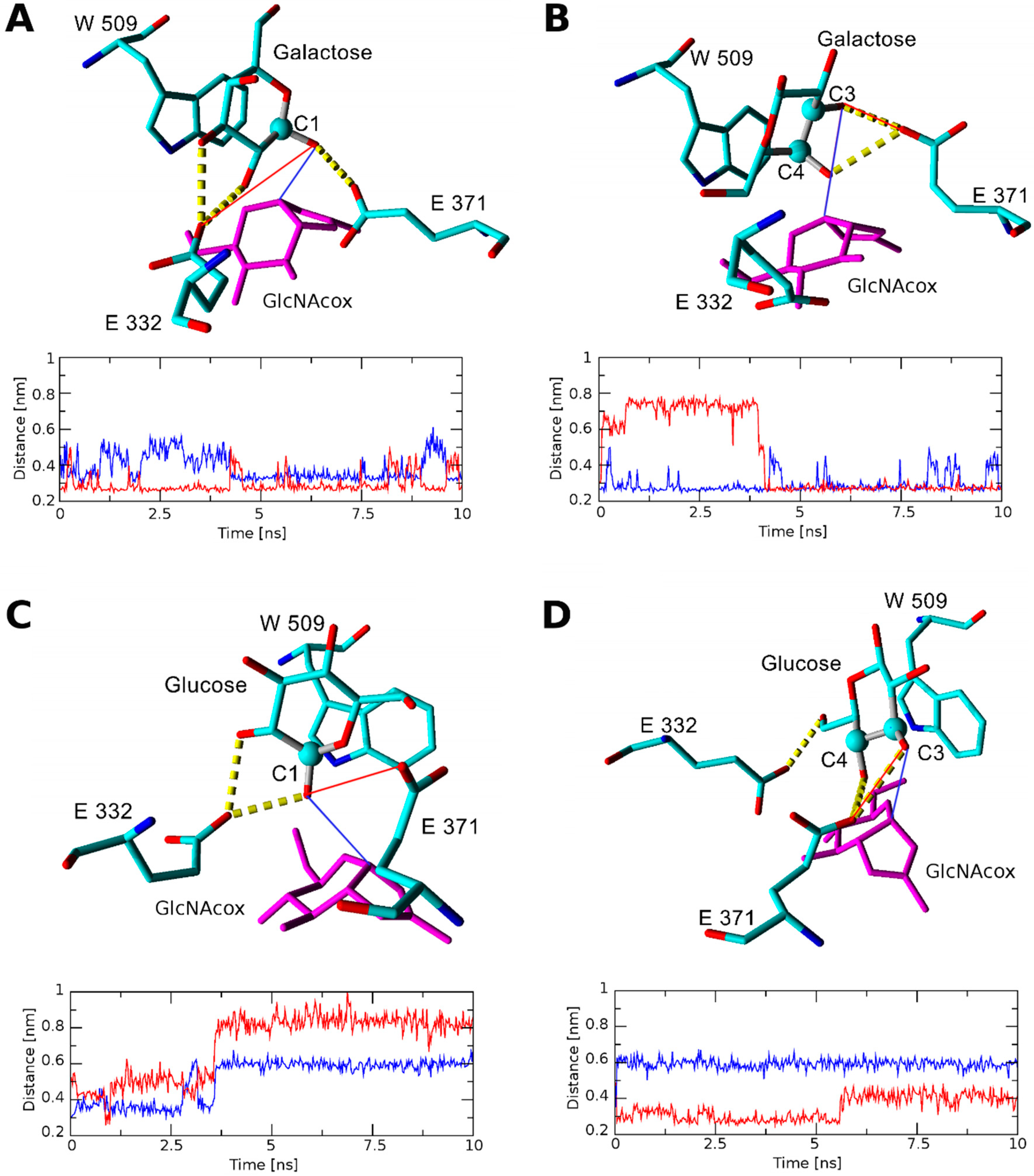
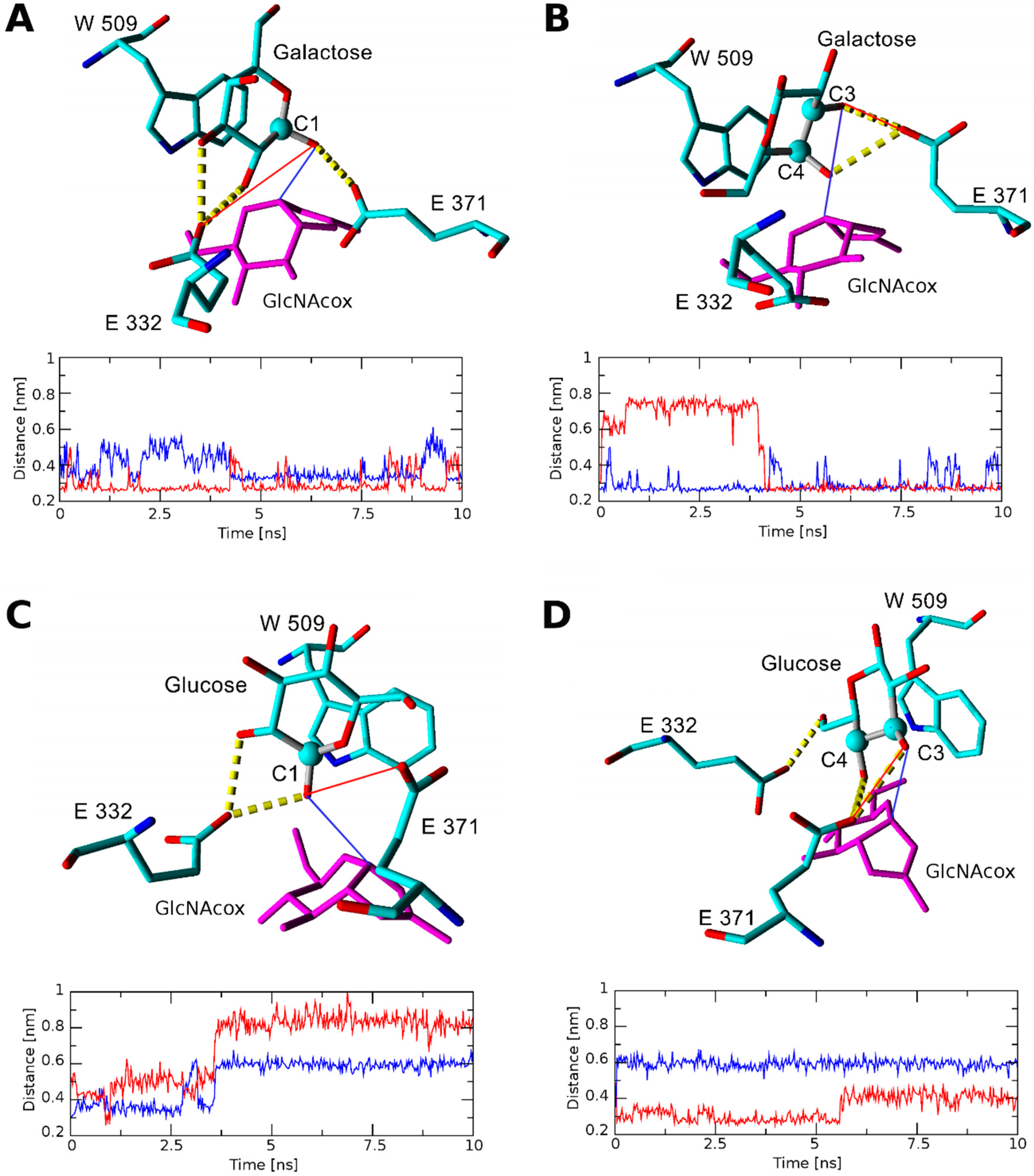
2.2.1. Docking and Molecular Dynamics Simulation of Selected Transglycosylation Acceptors in TfHex WT
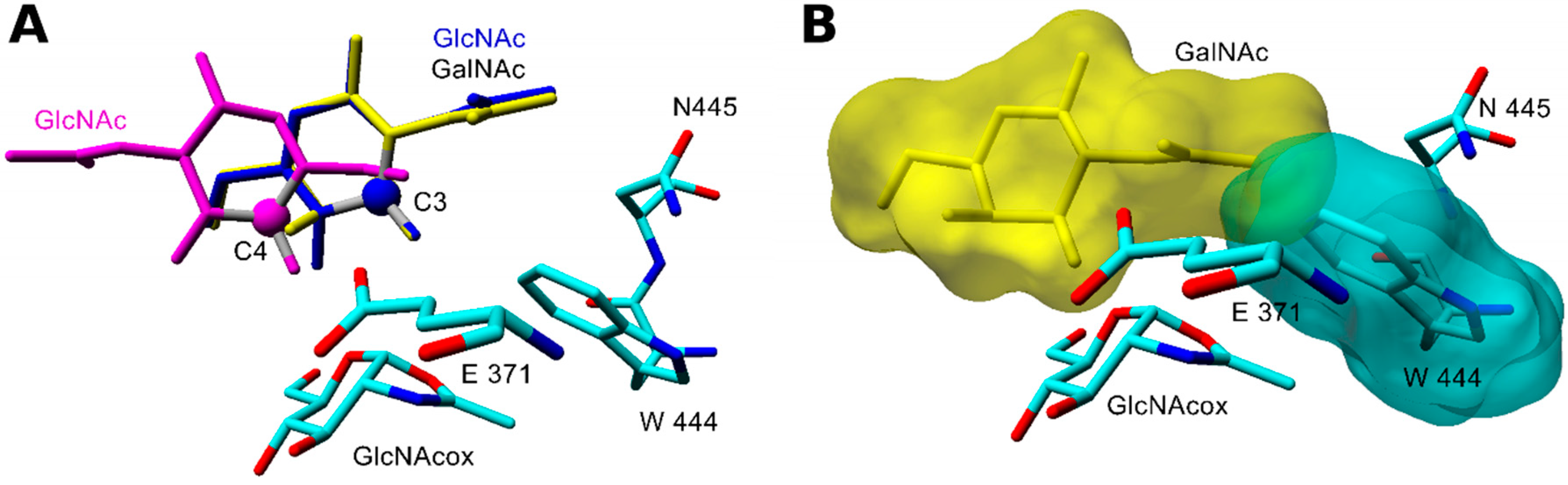
2.2.2. Docking and Molecular Dynamics Simulation of Selected Transglycosylation Acceptors in TfHex Y470H
3. Discussion
4. Materials and Methods
4.1. General Procedures
4.2. Structural Analysis of Compounds
4.2.1. ESI-MS Analysis
4.2.2. NMR Analysis
4.3. Enzymatic Activity Assay
4.4. Analytical Transglycosylation Reactions
4.5. Preparative Transglycosylation Reactions
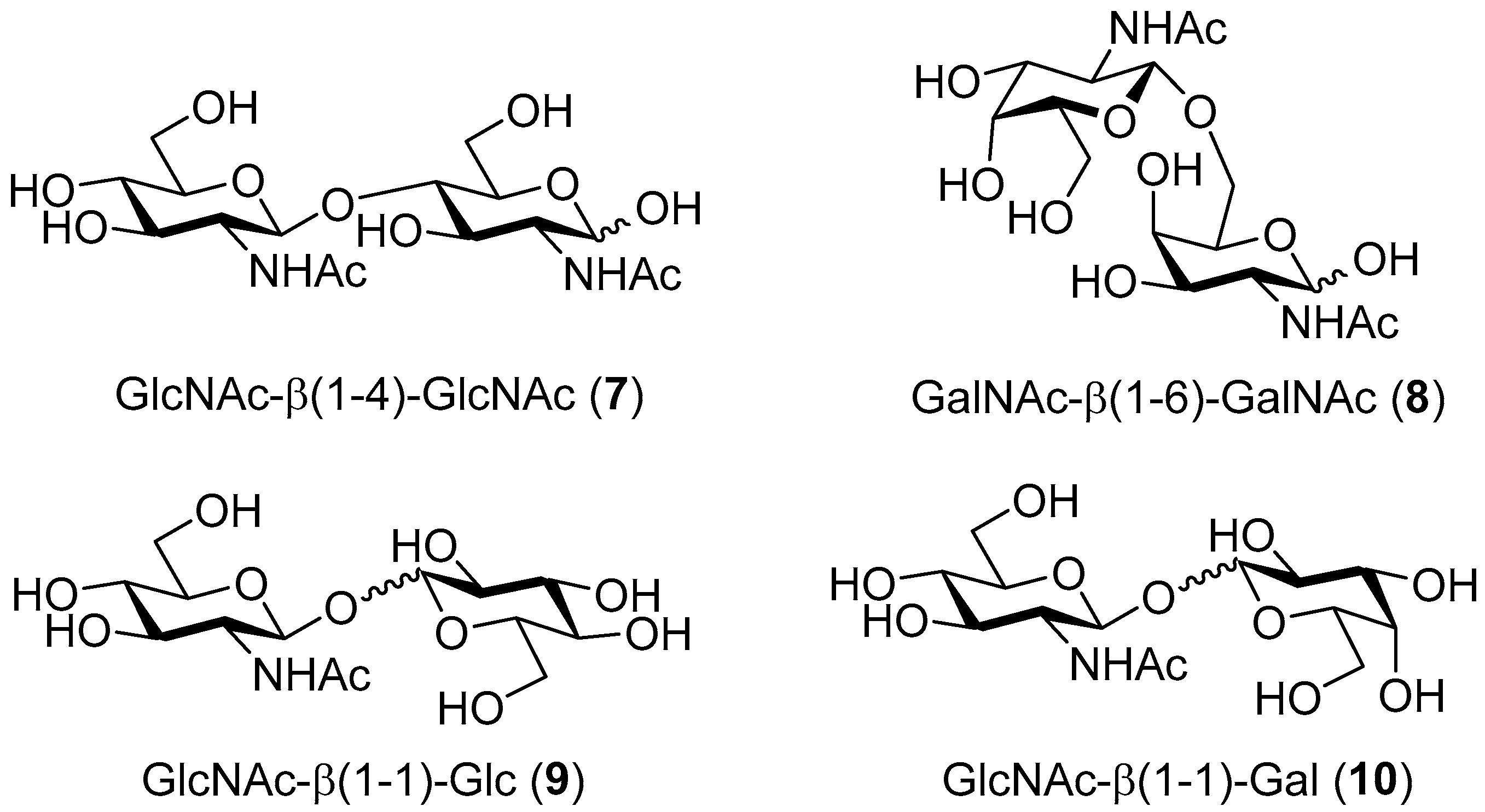
4.5.1. 2-Acetamido-2-Deoxy-β-d-Glucopyranosyl-(1-4)-2-Acetamido-2-Deoxy-d-Glucopyranose (GlcNAc-β(1-4)-GlcNAc; 7)
4.5.2. 2-Acetamido-2-Deoxy-β-d-Galactopyranosyl-(1-6)-2-Acetamido-2-Deoxy-d-Galactopyranose (GalNAc-β(1-6)-GalNAc; 8)
4.5.3. 2-Acetamido-2-Deoxy-β-d-Glucopyranosyl-(1-1)-β-d-Glucopyranoside (GlcNAc-β(1-1)-Glc; 9)
4.5.4. 2-Acetamido-2-Deoxy-β-d-Glucopyranosyl-(1-1)-β-d-Galactopyranoside (GlcNAc-β(1-1)-Gal; 10)
4.5.5. 2-Acetamido-2-Deoxy-β-d-Glucopyranosyl N-Acetylmuramic acid (GlcNAc-β(1-X)-MurNAc)
4.5.6. Propyl 2-Acetamido-2-Deoxy-β-d-Glucopyranosyl-(1-6)-N-Acetylmuramic acid (GalNAc-β(1-6)-MurNAc-OPr; 16)
4.6. Docking and Molecular Dynamics of the TfHex Active Site in the Complex with Tested Acceptors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ESI-MS | Electrospray ionization mass spectrometry |
| Gal | Galactose |
| GalNAc | N-Acetylgalactosamine |
| GalNAcox | N-Acetylgalactosamine oxazoline (catalytic intermediate during enzymatic hydrolysis) |
| Glc | Glucose |
| GlcA | Glucuronic acid |
| GlcNAc | N-Acetylglucosamine |
| GlcNAcox | N-Acetylglucosamine oxazoline (catalytic intermediate during enzymatic hydrolysis) |
| HB | Hydrogen bond |
| MurNAc | N-acetylmuramic acid |
| MurNAc-OPr | Propyl glycoside of N-acetylmuramic acid |
| pNP-GalNAc | 4-Nitrophenyl N-acetyl-β-d-galactosaminide |
| pNP-GlcNAc | 4-Nitrophenyl N-acetyl-β-d-glucosaminide |
| TfHex WT | Wild type β-N-acetylhexosaminidase from Talaromyces flavus |
| TfHex Y470H | Mutant β-N-acetylhexosaminidase from Talaromyces flavus, Tyr470 is exchanged for His |
References
- Hakomori, S. Carbohydrate-to-carbohydrate interaction, through glycosynapse, as a basis of cell recognition and membrane organization. Glycoconj. J. 2004, 21, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Hevey, R. Strategies for the development of glycomimetic drug candidates. Pharmaceuticals 2019, 12, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourin, M.C.; Lindahl, U. Glycosaminoglycans and the regulation of blood coagulation. Biochem. J. 1993, 289, 313–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Middleton, D.R.; Wantuch, P.L.; Ozdilek, A.; Avci, F.Y. Carbohydrates as T-cell antigens with implications in health and disease. Glycobiology 2016, 26, 1029–1040. [Google Scholar] [CrossRef] [PubMed]
- Slamová, K.; Bojarová, P. Engineered N-acetylhexosamine-active enzymes in glycoscience. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2070–2087. [Google Scholar] [CrossRef] [PubMed]
- Slamová, K.; Bojarová, P.; Petrásková, L.; Křen, V. β-N-Acetylhexosaminidase: What’s in a name...? Biotechnol. Adv. 2010, 28, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Bojarová, P.; Bruthans, J.; Křen, V. β-N-Acetylhexosaminidases-the wizards of glycosylation. Appl. Microbiol. Biotechnol. 2019, 103, 7869–7881. [Google Scholar] [CrossRef]
- Bojarová, P.; Slámová, K.; Křenek, K.; Gažák, R.; Kulik, N.; Ettrich, R.; Pelantová, H.; Kuzma, M.; Riva, S.; Adámek, D.; et al. Charged hexosaminides as new substrates for β-N-acetylhexosaminidase-catalyzed synthesis of immunomodulatory disaccharides. Adv. Synth. Catal. 2011, 353, 2409–2420. [Google Scholar] [CrossRef]
- Loft, K.J.; Bojarová, P.; Slámová, K.; Křen, V.; Williams, S.J. Synthesis of sulfated glucosaminides for profiling substrate specificities of sulfatases and fungal β-N-acetylhexosaminidases. ChemBioChem 2009, 10, 565–576. [Google Scholar] [CrossRef]
- Bojarová, P.; Křenek, K.; Kuzma, M.; Petrásková, L.; Bezouška, K.; Namdjou, D.-J.; Elling, L.; Křen, V. N-Acetylhexosamine triad in one molecule: Chemoenzymatic introduction of 2-acetamido-2-deoxy-β-D-galactopyranosyluronic acid residue into a complex oligosaccharide. J. Mol. Catal. B Enzym. 2008, 50, 69–73. [Google Scholar] [CrossRef]
- Slámová, K.; Krejzová, J.; Marhol, P.; Kalachova, L.; Kulik, N.; Pelantová, H.; Cvačka, J.; Křen, V. Synthesis of derivatized chitooligomers using transglycosidases engineered from the fungal GH20 β-N-acetylhexosaminidase. Adv. Synth. Catal. 2015, 357, 1941–1950. [Google Scholar] [CrossRef]
- Lodhi, G.; Kim, Y.-S.; Hwang, J.-W.; Kim, S.-K.; Jeon, Y.-J.; Je, J.-Y.; Ahn, C.-B.; Moon, S.-H.; Jeon, B.-T.; Park, P.-J. Chitooligosaccharide and its derivatives: Preparation and biological applications. Biomed. Res. Int. 2014, 2014, 654913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojarová, P.; Kulik, N.; Hovorková, M.; Slámová, K.; Pelantová, H.; Křen, V. The β-N-acetylhexosaminidase in the synthesis of bioactive glycans: Protein and reaction engineering. Molecules 2019, 24, 599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojarová, P.; Tavares, M.R.; Laaf, D.; Bumba, L.; Petrásková, L.; Konefal, R.; Bláhová, M.; Pelantová, H.; Elling, L.; Etrych, T.; et al. Biocompatible glyconanomaterials based on HPMA-copolymer for specific targeting of galectin-3. J. Nanobiotechnol. 2018, 16, 73. [Google Scholar] [CrossRef] [PubMed]
- Slamová, K.; Gažák, R.; Bojarová, P.; Kulik, N.; Ettrich, R.; Pelantová, H.; Sedmera, P.; Křen, V. 4-Deoxy-substrates for β-N-acetylhexosaminidases: How to make use of their loose specificity. Glycobiology 2010, 20, 1002–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Oliva, C.; Cabanillas, A.H.; Perona, A.; Hoyos, P.; Rumbero, Á.; Hernáiz, M.J. Efficient synthesis of muramic and glucuronic acid glycodendrimers as dengue virus antagonist. Chem. Eur. J. 2019. [Google Scholar] [CrossRef]
- Škerlová, J.; Bláha, J.; Pachl, P.; Hofbauerová, K.; Kukačka, Z.; Man, P.; Pompach, P.; Novák, P.; Otwinowski, Z.; Brynda, J.; et al. Crystal structure of native β-N-acetylhexosaminidase isolated from Aspergillus oryzae sheds light onto its substrate specificity, high stability, and regulation by propeptide. FEBS J. 2018, 285, 580–598. [Google Scholar] [CrossRef] [Green Version]
- Křen, V.; Rajnochová, E.; Huňková, Z.; Dvořáková, J.; Sedmera, P. Unusual nonreducing sugar GlcNAcβ(1-1)Manβ formation by β-N-acetylhexosaminidase from Aspergillus oryzae. Tetrahedron Lett. 1998, 39, 9777–9780. [Google Scholar] [CrossRef]
- Bojarová, P.; Chytil, P.; Mikulová, B.; Bumba, L.; Konefał, R.; Pelantová, H.; Krejzová, J.; Slámová, K.; Petrásková, L.; Kotrchová, L.; et al. Glycan-decorated HPMA copolymers as high-affinity lectin ligands. Polym. Chem. 2017, 8, 2647–2658. [Google Scholar] [CrossRef] [Green Version]
- Kulik, N.; Slámová, K.; Ettrich, R.; Křen, V. Computational study of β-N-acetylhexosaminidase from Talaromyces flavus, a glycosidase with high substrate flexibility. BMC Bioinform. 2015, 16, 28. [Google Scholar] [CrossRef] [Green Version]
- Brás, N.F.; Fernandes, P.A.; Ramos, M.J. Docking and molecular dynamics studies on the stereoselectivity in the enzymatic synthesis of carbohydrates. Theor. Chem. Acc. 2009, 122, 283. [Google Scholar] [CrossRef]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins 2004, 57, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Mark, B.L.; Vocadlo, D.J.; Knapp, S.; Triggs-Raine, B.L.; Withers, S.G.; James, M.N. Crystallographic evidence for substrate-assisted catalysis in a bacterial β-hexosaminidase. J. Biol. Chem. 2001, 276, 10330–10337. [Google Scholar] [CrossRef] [Green Version]




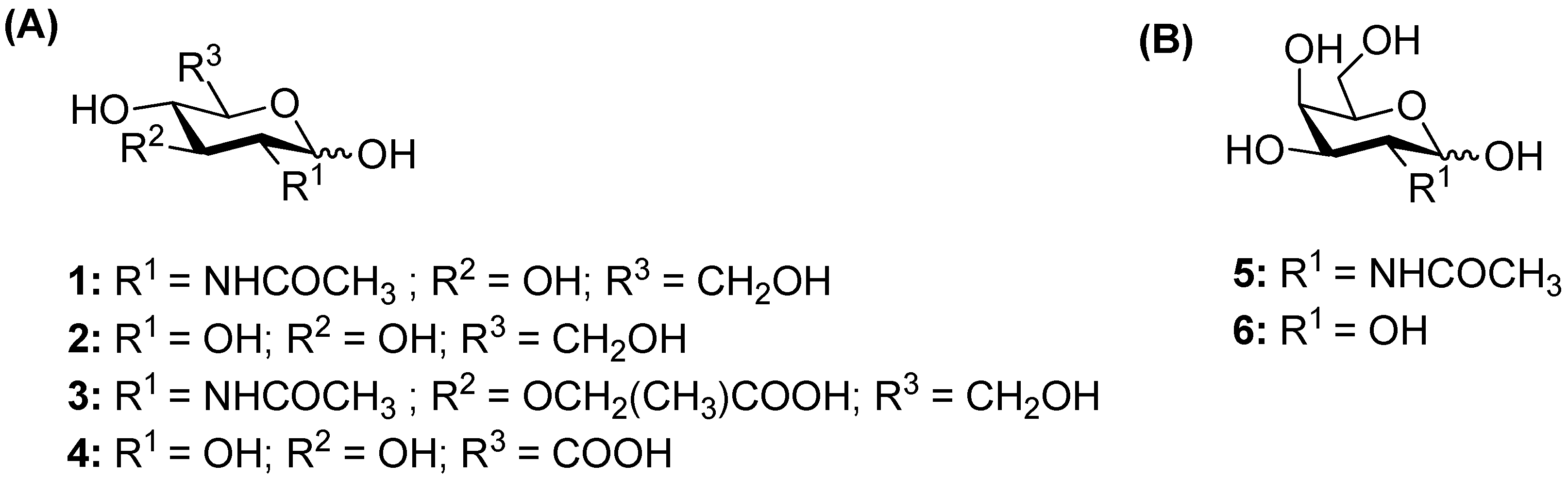{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Enzyme | Donor | Acceptor | Donor/Acceptor [mM] 1 | Product | Yield |
|---|---|---|---|---|---|---|
| I | WT | pNP-GlcNAc | GlcNAc | 50/300 | GlcNAc-β(1-4)-GlcNAc | 24% |
| II | WT | pNP-GalNAc | GalNAc | 50/300 | GalNAc- β (1-6)-GalNAc | 31% |
| III | WT | pNP-GlcNAc | Glc | 50/300 | GlcNAc- β (1-1)-Glc | 14% |
| IV | WT | pNP-GlcNAc | Gal | 50/300 | GlcNAc- β (1-1)-Gal | 10% |
| V | Y470H | pNP-GlcNAc | MurNAc | 50 (x2)/100 | GlcNAc- β (1-X)-MurNAc | n.q. 1 |
| VI | Y470H | pNP-GalNAc | MurNAc-OPr | 50/100 | GalNAc- β (1-6)-MurNAc-OPr | ≈1% |
| Acceptor | Position of Respective Acceptor Hydroxyl close to Catalytic Glu371 | ||||||
|---|---|---|---|---|---|---|---|
| None (Site 1) | None (Site 2) | C-1 | C-2 | C-3 | C-4 | C-6 | |
| TfHex WT in complex with GlcNAcox/GalNAcox | |||||||
| GlcNAc (1) | n.b.1/−3.99 | −2.28/n.b. | n.b./n.b. | n.b./n.b. | −2.37/n.b. | −4.97/−3.36 | −2.29/−2.09 |
| Glc (2) | −5.24/−2.68 | n.b./n.b. | -2.35/n.b. | -2.14/n.b. | −4.44/n.b. | −4.70/n.b. | −2.27/n.b. |
| MurNAc (3) | n.b./ −4.46 | −4.75/−2.93 | n.b./n.b. | n.b./n.b. | n.b./n.b. | n.b./n.b. | −3.66/n.b. |
| GlcA (4) | −5.25/−5.47 | −2.31/−2.60 | n.b./n.b. | -2.03/n.b. | n.b./n.b. | −2.221/n.b. | n.b./n.b. |
| GalNAc (5) | −4.98/−2.62 | −2.65/n.b. | n.b./n.b. | n.b./n.b. | −4.03/−2.28 | n.b./n.b. | n.b./−2.23 |
| Gal (6) | −2.57/−3.47 | −2.36/n.b. | -2.29/n.b. | n.b./n.b. | −2.81/n.b. | n.b./n.b. | n.b./n.b. |
| MurNAc-OPr (XX) | −2.42/−4.34 | −4.41/−2.66 | n.b./n.b. | n.b./n.b. | n.b./n.b. | n.b./n.b. | −4.12/−2.93 |
| TfHex Y470H in complex with GlcNAcox/GalNAcox | |||||||
| GlcA (4) | −5.45/3.80 | n.b./−5.36 | n.b./n.b. | n.b./n.b. | n.b./n.b. | n.b./n.b. | n.b./n.b. |
| MurNAc (3) | n.b./n.b. | −4.68/−4.61 | n.b./n.b. | n.b./n.b. | n.b./n.b. | n.b./n.b. | n.b./−2.69 |
| MurNAc-OPr (XX) | −2.96/−3.19 | −5.05/−4.50 | n.b./n.b. | n.b./n.b. | n.b./n.b. | n.b./n.b. | n.b./−3.69 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Oliva, C.; Hoyos, P.; Petrásková, L.; Kulik, N.; Pelantová, H.; Cabanillas, A.H.; Rumbero, Á.; Křen, V.; Hernáiz, M.J.; Bojarová, P. Acceptor Specificity of β-N-Acetylhexosaminidase from Talaromyces flavus: A Rational Explanation. Int. J. Mol. Sci. 2019, 20, 6181. https://doi.org/10.3390/ijms20246181
Garcia-Oliva C, Hoyos P, Petrásková L, Kulik N, Pelantová H, Cabanillas AH, Rumbero Á, Křen V, Hernáiz MJ, Bojarová P. Acceptor Specificity of β-N-Acetylhexosaminidase from Talaromyces flavus: A Rational Explanation. International Journal of Molecular Sciences. 2019; 20(24):6181. https://doi.org/10.3390/ijms20246181
Chicago/Turabian StyleGarcia-Oliva, Cecilia, Pilar Hoyos, Lucie Petrásková, Natalia Kulik, Helena Pelantová, Alfredo H. Cabanillas, Ángel Rumbero, Vladimír Křen, María J. Hernáiz, and Pavla Bojarová. 2019. "Acceptor Specificity of β-N-Acetylhexosaminidase from Talaromyces flavus: A Rational Explanation" International Journal of Molecular Sciences 20, no. 24: 6181. https://doi.org/10.3390/ijms20246181
APA StyleGarcia-Oliva, C., Hoyos, P., Petrásková, L., Kulik, N., Pelantová, H., Cabanillas, A. H., Rumbero, Á., Křen, V., Hernáiz, M. J., & Bojarová, P. (2019). Acceptor Specificity of β-N-Acetylhexosaminidase from Talaromyces flavus: A Rational Explanation. International Journal of Molecular Sciences, 20(24), 6181. https://doi.org/10.3390/ijms20246181