NF-κB Signaling Regulates Physiological and Pathological Chondrogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
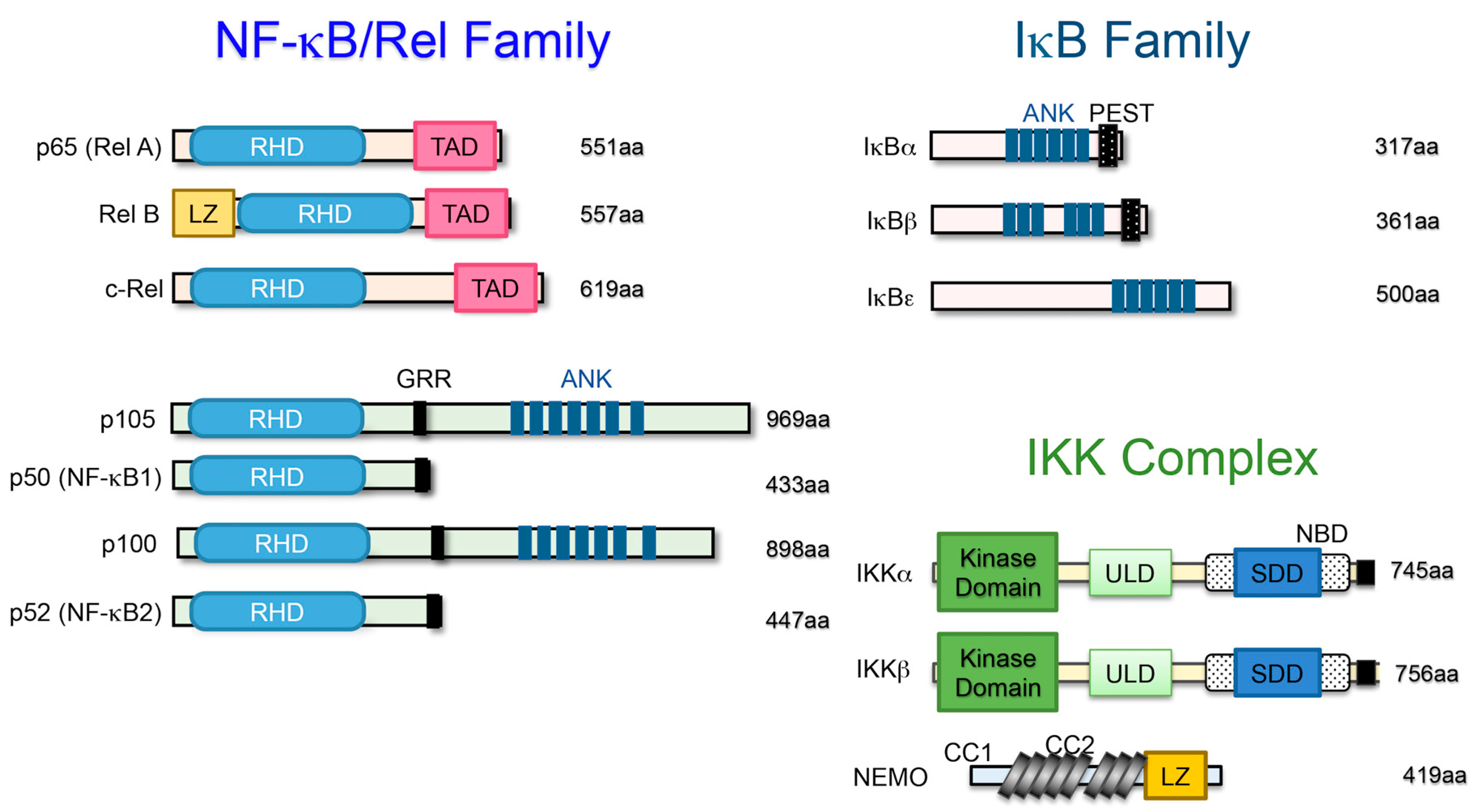
2. The NF-κB Family and Its Signaling
3. The Role of the Classical NF-κB Pathway in Chondrogenic Development
4. The Role of the Alternative NF-κB Pathway in Endochondral Ossification
5. NF-κB Signaling Is a Target for Preventing Rheumatoid Arthritis (RA)
6. The Strength of the Classical NF-κB Signaling Regulates Cartilage Homeostasis and Osteoarthritis (OA) Development
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NF-κB | Nuclear factor-κB |
| IKK | IκB kinase |
| NIK | NF-κB-inducing kinase |
| RA | Rheumatoid arthritis |
| OA | Osteoarthritis |
References
- Long, F.; Ornitz, D.M. Development of the endochondral skeleton. Cold Spring Harb Perspect. Biol. 2013, 5, a008334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, K.; Takahata, Y.; Murakami, T.; Nishimura, R. Transcriptional network controlling endochondral ossification. J. Bone Metab. 2017, 24, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, H.; Chaboissier, M.C.; Martin, J.F.; Schedl, A.; de Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002, 16, 2813–2828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, S.; Bonnamy, J.P.; Owen, M.J.; Ducy, P.; Karsenty, G. Continuous expression of Cbfa1 in nonhypertrophic chondrocytes uncovers its ability to induce hypertrophic chondrocyte differentiation and partially rescues Cbfa1-deficient mice. Genes Dev. 2001, 15, 467–481. [Google Scholar] [CrossRef] [Green Version]
- Jimi, E.; Ghosh, S. Role of nuclear factor-κB in the immune system and bone. Immunol. Rev. 2005, 208, 80–87. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [Green Version]
- De Luca, F. Role of Nuclear Factor-κB (NF-κB) in growth plate chondrogenesis. Pediatr. Endocrinol. Rev. 2016, 13, 720–730. [Google Scholar]
- Novack, D.V. Role of NF-κB in the skeleton. Cell Res. 2011, 21, 169–182. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Flint, J.K.; Rezvani, G.; De Luca, F. Nuclear factor-κB p65 facilitates longitudinal bone growth by inducing growth plate chondrocyteproliferation and differentiation and by preventing apoptosis. J. Biol. Chem. 2007, 282, 33698–33706. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Chang, S.H.; Mori, D.; Itoh, S.; Hirata, M.; Hosaka, Y.; Taniguchi, Y.; Okada, K.; Mori, Y.; Yano, F.; et al. Biphasic regulation of chondrocytes by Rela through induction of anti-apoptotic and catabolic target genes. Nat. Commun. 2016, 7, 13336. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, Z.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target. Ther. 2017, 2, e17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, T. The nuclear factor NF-κB pathway in inflammation. Cold Spring Harb Perspect. Biol. 2009, 6, a001651. [Google Scholar]
- Xu, J.; Wu, H.F.; Ang, E.S.; Yip, K.; Woloszyn, M.; Zheng, M.H.; Tan, R.X. NF-κB modulators in osteolytic bone diseases. Cytokine Growth Factor Rev. 2009, 20, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Britanova, L.; Makeev, V.J.; Kuprash, D.V. In vitro selection of optimal RelB/p52 DNA-binding motifs. Biochem. Biophys. Res. Commun. 2008, 365, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Kanegae, Y.; Tavares, A.T.; Izpisúa Belmonte, J.C.; Verma, I.M. Role of Rel/NF-κB transcription factors during the outgrowth of the vertebrate limb. Nature 1998, 392, 611–614. [Google Scholar] [CrossRef]
- Wu, S.; Fadoju, D.; Rezvani, G.; De Luca, F. Stimulatory effects of insulin-like growth factor-I on growth plate chondrogenesis are mediated by nuclear factor-κB p65. J. Biol. Chem. 2008, 283, 34037–34044. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Morrison, A.; Sun, H.; De Luca, F. Nuclear factor-κB (NF-κB) p65 interacts with Stat5b in growth plate chondrocytes and mediates the effects of growth hormone on chondrogenesis and on the expression of insulin-like growth factor-1 and bone morphogenetic protein-2. J. Biol. Chem. 2011, 286, 24726–24734. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.Q.; Xing, L.; Zhang, J.H.; Zhao, M.; Horn, D.; Chan, J.; Boyce, B.F.; Harris, S.E.; Mundy, G.R.; Chen, D. NF-κB specifically activates BMP-2 gene expression in growth plate chondrocytes in vivo and in a chondrocyte cell line in vitro. J. Biol. Chem. 2003, 278, 29130–29135. [Google Scholar] [CrossRef] [Green Version]
- Iotsova, V.; Caamaño, J.; Loy, J.; Yang, Y.; Lewin, A.; Bravo, R. Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat. Med. 1997, 3, 1285–1289. [Google Scholar] [CrossRef]
- Franzoso, G.; Carlson, L.; Xing, L.; Poljak, L.; Shores, E.W.; Brown, K.D.; Leonardi, A.; Tran, T.; Boyce, B.F.; Siebenlist, U. Requirement for NF-κB in osteoclast and B-cell development. Genes Dev. 1997, 11, 3482–3496. [Google Scholar] [CrossRef] [Green Version]
- Caron, M.M.; Emans, P.J.; Surtel, D.A.; Cremers, A.; Voncken, J.W.; Welting, T.J.; van Rhijn, L.W. Activation of NF-κB/p65 facilitates early chondrogenic differentiation during endochondral ossification. PLoS ONE 2012, 7, e33467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Yang, W.; De Luca, F. Insulin-like growth factor-independent effects of growth hormone on growth plate chondrogenesis and longitudinal bone growth. Endocrinology 2015, 156, 2541–2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeda, K.; Takeuchi, O.; Tsujimura, T.; Itami, S.; Adachi, O.; Kawai, T.; Sanjo, H.; Yoshikawa, K.; Terada, N.; Akira, S. Limb and skin abnormalities in mice lacking IKKα. Science 1999, 284, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lu, Q.; Hwang, J.Y.; Büscher, D.; Lee, K.F.; Izpisua-Belmonte, J.C.; Verma, I.M. IKK1-deficient mice exhibit abnormal development of skin and skeleton. Genes Dev. 1999, 13, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- Nakatomi, C.; Nakatomi, M.; Matsubara, T.; Komori, T.; Doi-Inoue, T.; Ishimaru, N.; Weih, F.; Iwamoto, T.; Matsuda, M.; Kokabu, S.; et al. Constitutive activation of the alternative NF-κB pathway disturbs endochondral ossification. Bone 2019, 121, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Carrasco, D.; Claudio, E.; Ryseck, R.P.; Bravo, R. Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-κB2. J. Exp. Med. 1997, 186, 999–1014. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, R.E.; Foxwell, B.M. Signalling, inflammation and arthritis: NF-κB and its relevance to arthritis and inflammation. Rheumatology 2008, 47, 584–590. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Asahara, H.; Asanuma, M.; Ogawa, N.; Nishibayashi, S.; Inoue, H. High DNA-binding activity of transcription factor NF-κB in synovial membranes of patients with rheumatoid arthritis. Biochem. Mol. Biol. Int. 1995, 37, 827–832. [Google Scholar]
- Gilston, V.; Jones, H.W.; Soo, C.C.; Coumbe, A.; Blades, S.; Kaltschmidt, C.; Baeuerle, P.A.; Morris, C.J.; Blake, D.R.; Winyard, P.G. NF-κB activation in human knee-joint synovial tissue during the early stage of joint inflammation. Biochem. Soc. Trans. 1997, 25, 518S. [Google Scholar] [CrossRef]
- Miyazawa, K.; Mori, A.; Yamamoto, K.; Okudaira, H. Constitutive transcription of the human interleukin-6 gene by rheumatoid synoviocytes: Spontaneous activation of NF-κB and CBF1. Am. J. Pathol. 1998, 152, 793–803. [Google Scholar]
- Han, Z.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. AP-1 and NF-κB regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity 1998, 28, 197–208. [Google Scholar] [CrossRef]
- Miagkov, A.V.; Kovalenko, D.V.; Brown, C.E.; Didsbury, J.R.; Cogswell, J.P.; Stimpson, S.A.; Baldwin, A.S.; Makarov, S.S. NF-κB activation provides the potential link between inflammation and hyperplasia in the arthritic joint. Proc. Natl. Acad. Sci. USA 1998, 95, 13859–13864. [Google Scholar] [CrossRef] [Green Version]
- Palombella, V.J.; Conner, E.M.; Fuseler, J.W.; Destree, A.; Davis, J.M.; Laroux, F.S.; Wolf, R.E.; Huang, J.; Brand, S.; Elliott, P.J.; et al. Role of the proteasome and NF-κB in streptococcal cell wall-induced polyarthritis. Proc. Natl. Acad. Sci. USA 1998, 95, 15671–15676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, P.P.; Gerlag, D.M.; Aupperle, K.R.; van de Geest, D.A.; Overbeek, M.; Bennett, B.L.; Boyle, D.L.; Manning, A.M.; Firestein, G.S. Inhibitor of nuclear factor κB kinase beta is a key regulator of synovial inflammation. Arthritis Rheum. 2001, 44, 1897–1907. [Google Scholar] [CrossRef]
- Ruocco, M.G.; Maeda, S.; Park, J.M.; Lawrence, T.; Hsu, L.C.; Cao, Y.; Schett, G.; Wagner, E.F.; Karin, M. IκB kinase (IKK)β, but not IKKα, is a critical mediator of osteoclast survival and is required for inflammation-induced bone loss. J. Exp. Med. 2005, 201, 1677–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimi, E.; Aoki, K.; Saito, H.; D’Acquisto, F.; May, M.J.; Nakamura, I.; Sudo, T.; Kojima, T.; Okamoto, F.; Fukushima, H.; et al. Selective inhibition of NF-κB blocks osteoclastogenesis and prevents inflammatory bone destruction in vivo. Nat. Med. 2004, 10, 617–624. [Google Scholar] [CrossRef]
- Dai, S.; Hirayama, T.; Abbas, S.; Abu-Amer, Y. The IκB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks osteoclastogenesis and bone erosion in inflammatory arthritis. J. Biol. Chem. 2004, 279, 37219–37222. [Google Scholar] [CrossRef] [Green Version]
- Clohisy, J.C.; Roy, B.C.; Biondo, C.; Frazier, E.; Willis, D.; Teitelbaum, S.L.; Abu-Amer, Y. Direct inhibition of NF-κB blocks bone erosion associated with inflammatory arthritis. J. Immunol. 2003, 171, 5547–5553. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, K.W.; Shuster, D.J.; Gillooly, K.M.; Dambach, D.M.; Pattoli, M.A.; Lu, P.; Zhou, X.D.; Qiu, Y.; Zusi, F.C.; Burke, J.R. A highly selective inhibitor of IκB kinase, BMS-345541, blocks both joint inflammation and destruction in collagen-induced arthritis in mice. Arthritis Rheum. 2003, 48, 2652–2659. [Google Scholar] [CrossRef]
- Ruocco, M.G.; Karin, M. Control of osteoclast activity and bone loss by IKK subunits: New targets for therapy. Adv. Exp. Med. Biol. 2007, 602, 125–134. [Google Scholar] [PubMed]
- Maijer, K.I.; Noort, A.R.; de Hair, M.J.; van der Leij, C.; van Zoest, K.P.; Choi, I.Y.; Gerlag, D.M.; Maas, M.; Tak, P.P.; Tas, S.W. Nuclear Factor-κB-inducing Kinase Is Expressed in Synovial Endothelial Cells in Patients with Early Arthritis and Correlates with Markers of Inflammation: A Prospective Cohort Study. J. Rheumatol. 2015, 42, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Noort, A.R.; van Zoest, K.P.; Weijers, E.M.; Koolwijk, P.; Maracle, C.X.; Novack, D.V.; Siemerink, M.J.; Schlingemann, R.O.; Tak, P.P.; Tas, S.W. NF-κB-inducing kinase is a key regulator of inflammation-induced and tumour-associated angiogenesis. J. Pathol. 2014, 234, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Aya, K.; Alhawagri, M.; Hagen-Stapleton, A.; Kitaura, H.; Kanagawa, O.; Novack, D.V. NF-κB-inducing kinase controls lymphocyte and osteoclast activities in inflammatory arthritis. J. Clin. Investig. 2005, 115, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; McCoy, K.; Davis, J.L.; Schmidt-Supprian, M.; Sasaki, Y.; Faccio, R.; Novack, D.V. NIK stabilization in osteoclasts results in osteoporosis and enhanced inflammatory osteolysis. PLoS ONE 2010, 5, e15383. [Google Scholar] [CrossRef] [PubMed]
- Cheema, G.S.; Roschke, V.; Hilbert, D.M.; Stohl, W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001, 44, 1313–1319. [Google Scholar] [CrossRef]
- Zhang, J.; Roschke, V.; Baker, K.P.; Wang, Z.; Alarcón, G.S.; Fessler, B.J.; Bastian, H.; Kimberly, R.P.; Zhou, T. Cutting edge: A role for B lymphocyte stimulator in systemic lupus erythematosus. J. Immunol. 2001, 166, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Marsters, S.A.; Baker, T.; Chan, B.; Lee, W.P.; Fu, L.; Tumas, D.; Yan, M.; Dixit, V.M.; Ashkenazi, A.; et al. TACI-ligand interactions are required for T cell activation and collagen-induced arthritis in mice. Nat. Immunol. 2001, 2, 632–637. [Google Scholar] [CrossRef]
- Doherty, M. Risk factors for progression of knee osteoarthritis. Lancet 2001, 358, 775–776. [Google Scholar] [CrossRef]
- Ripmeester, E.G.J.; Timur, U.T.; Caron, M.M.J.; Welting, T.J.M. Recent Insights into the contribution of the changing hypertrophic chondrocyte phenotype in the development and progression of osteoarthritis. Front. Bioeng. Biotechnol. 2018, 6, 18. [Google Scholar] [CrossRef]
- Handel, M.L.; McMorrow, L.B.; Gravallese, E.M. Nuclear factor-κB in rheumatoid synovium. Localization of p50 and p65. Arthritis Rheum. 1995, 38, 1762–1770. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, E.; Borzi, R.M.; Vitellozzi, R.; Pagani, S.; Facchini, A.; Battistelli, M.; Penzo, M.; Li, X.; Flamigni, F.; Li, J.; et al. Differential requirements for IKKα and IKKβ in the differentiation of primary human osteoarthritic chondrocytes. Arthritis Rheum. 2008, 58, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Tanaka, S. Molecular mechanisms underlying osteoarthritis development: Notch and NF-κB. Arthritis Res. Ther. 2017, 19, 94. [Google Scholar] [CrossRef] [PubMed]
- Murahashi, Y.; Yano, F.; Kobayashi, H.; Makii, Y.; Iba, K.; Yamashita, T.; Tanaka, S.; Saito, T. Intra-articular administration of IκBα kinase inhibitor suppresses mouse knee osteoarthritis via downregulation of the NF-κB/HIF-2α axis. Sci. Rep. 2018, 8, 16475. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jimi, E.; Huang, F.; Nakatomi, C. NF-κB Signaling Regulates Physiological and Pathological Chondrogenesis. Int. J. Mol. Sci. 2019, 20, 6275. https://doi.org/10.3390/ijms20246275
Jimi E, Huang F, Nakatomi C. NF-κB Signaling Regulates Physiological and Pathological Chondrogenesis. International Journal of Molecular Sciences. 2019; 20(24):6275. https://doi.org/10.3390/ijms20246275
Chicago/Turabian StyleJimi, Eijiro, Fei Huang, and Chihiro Nakatomi. 2019. "NF-κB Signaling Regulates Physiological and Pathological Chondrogenesis" International Journal of Molecular Sciences 20, no. 24: 6275. https://doi.org/10.3390/ijms20246275