Structural Insights into the Intracellular Region of the Human Magnesium Transport Mediator CNNM4


 ,
,  ,
,  ,
,  and
and
Abstract

1. Introduction
2. Results
2.1. Crystal Structure of CNNM4BAT
2.2. CNNM4BAT Interaction with ATP Depends on Mg2+
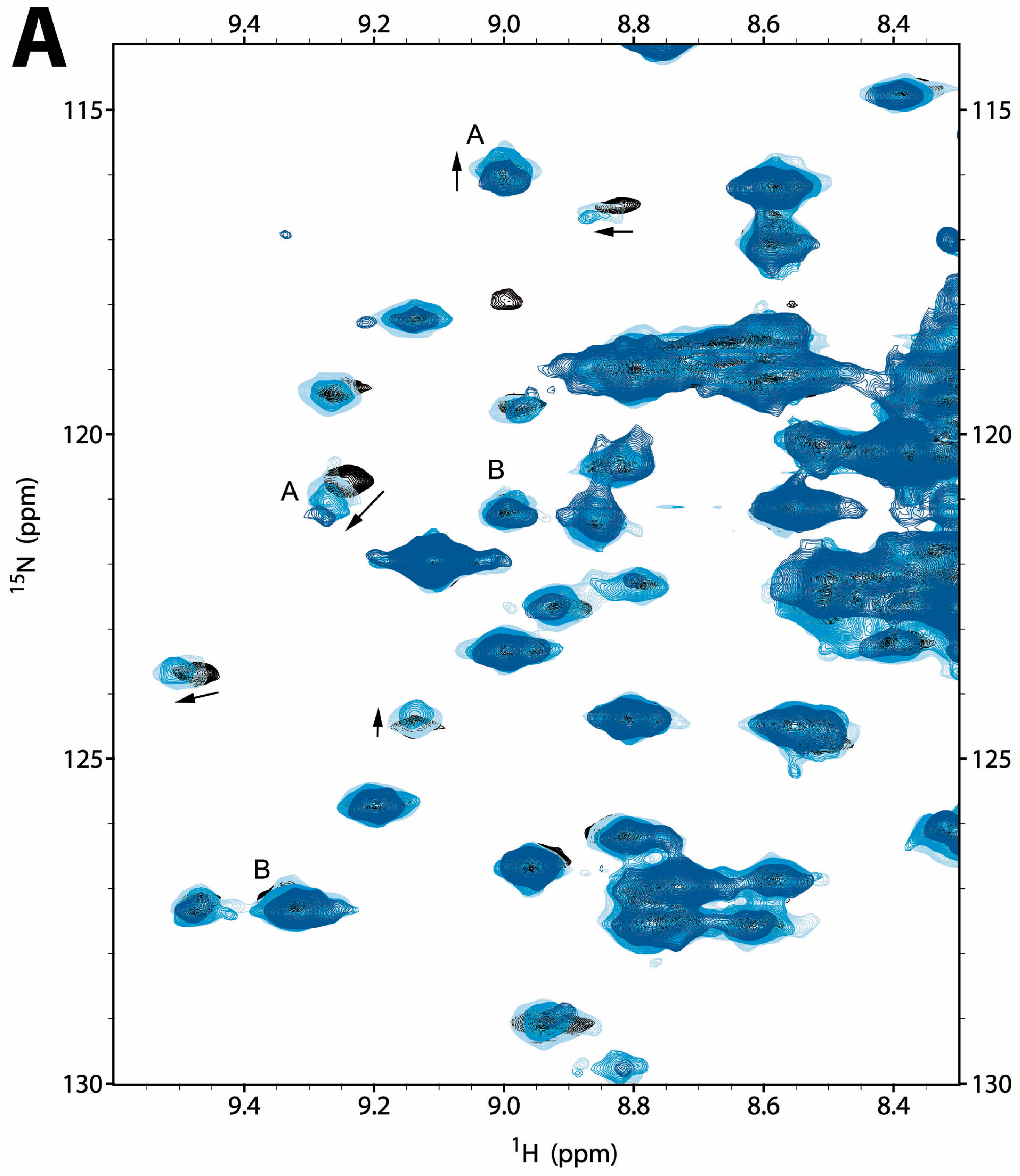
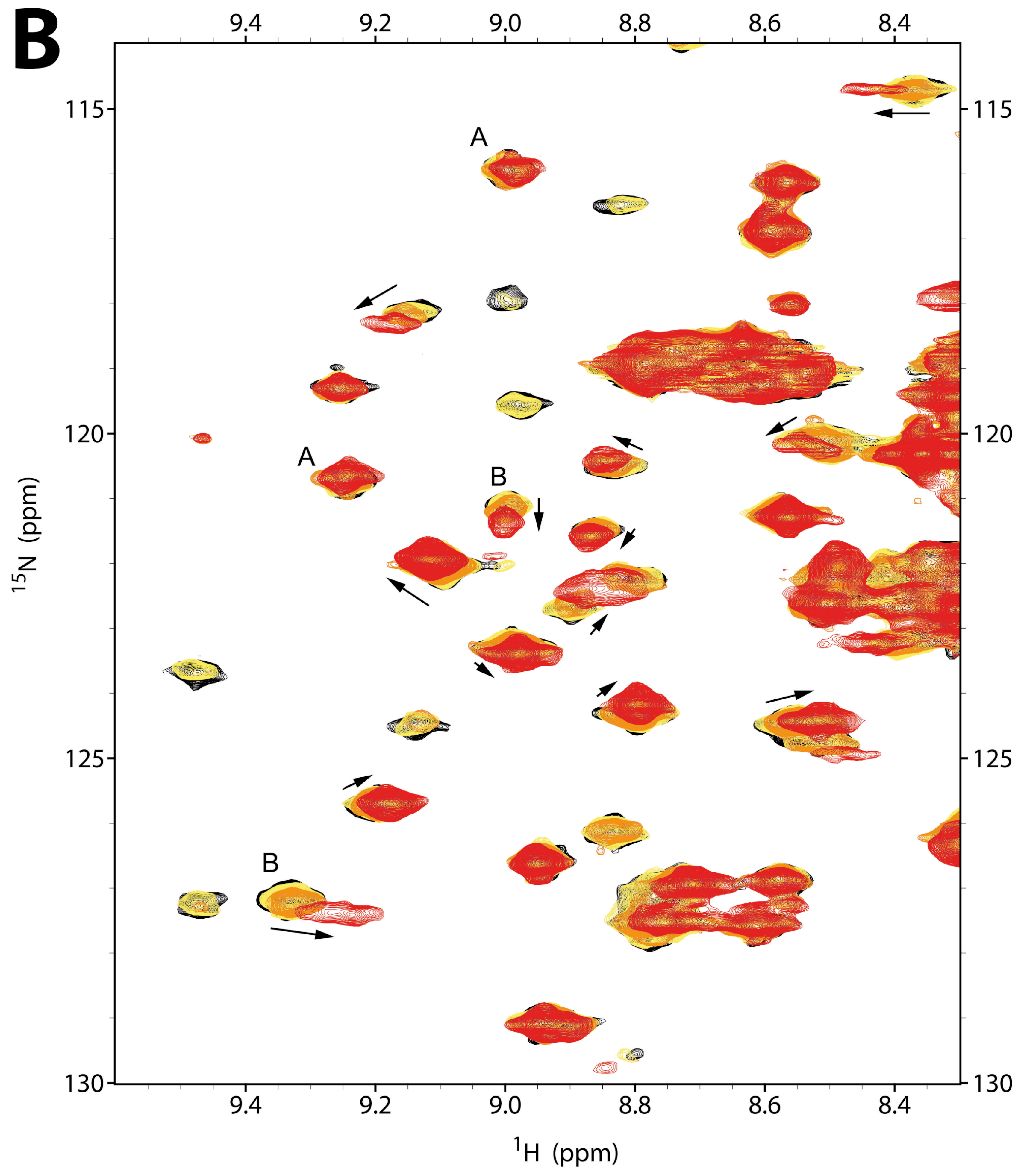
2.2.1. NMR Titration Studies with Mg2+
2.2.2. NMR Titration Studies with ADPNP
2.2.3. NMR Co-Titration Studies with Mg2+ and ADPNP
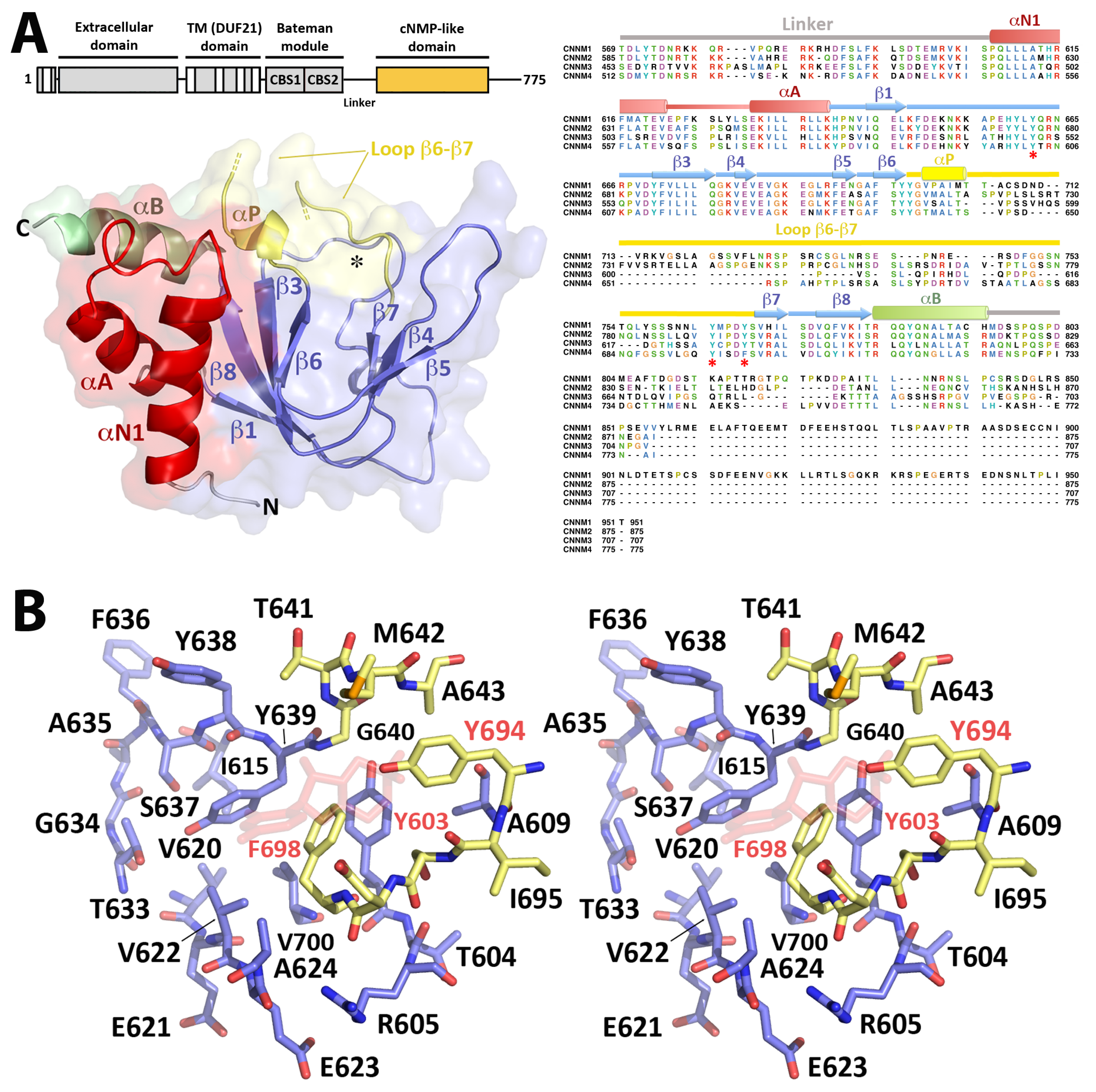
2.3. Crystal Structure of CNNM4cNMP
2.3.1. The CNNM4cNMP Module Is Unable to Bind Cyclic Nucleotides
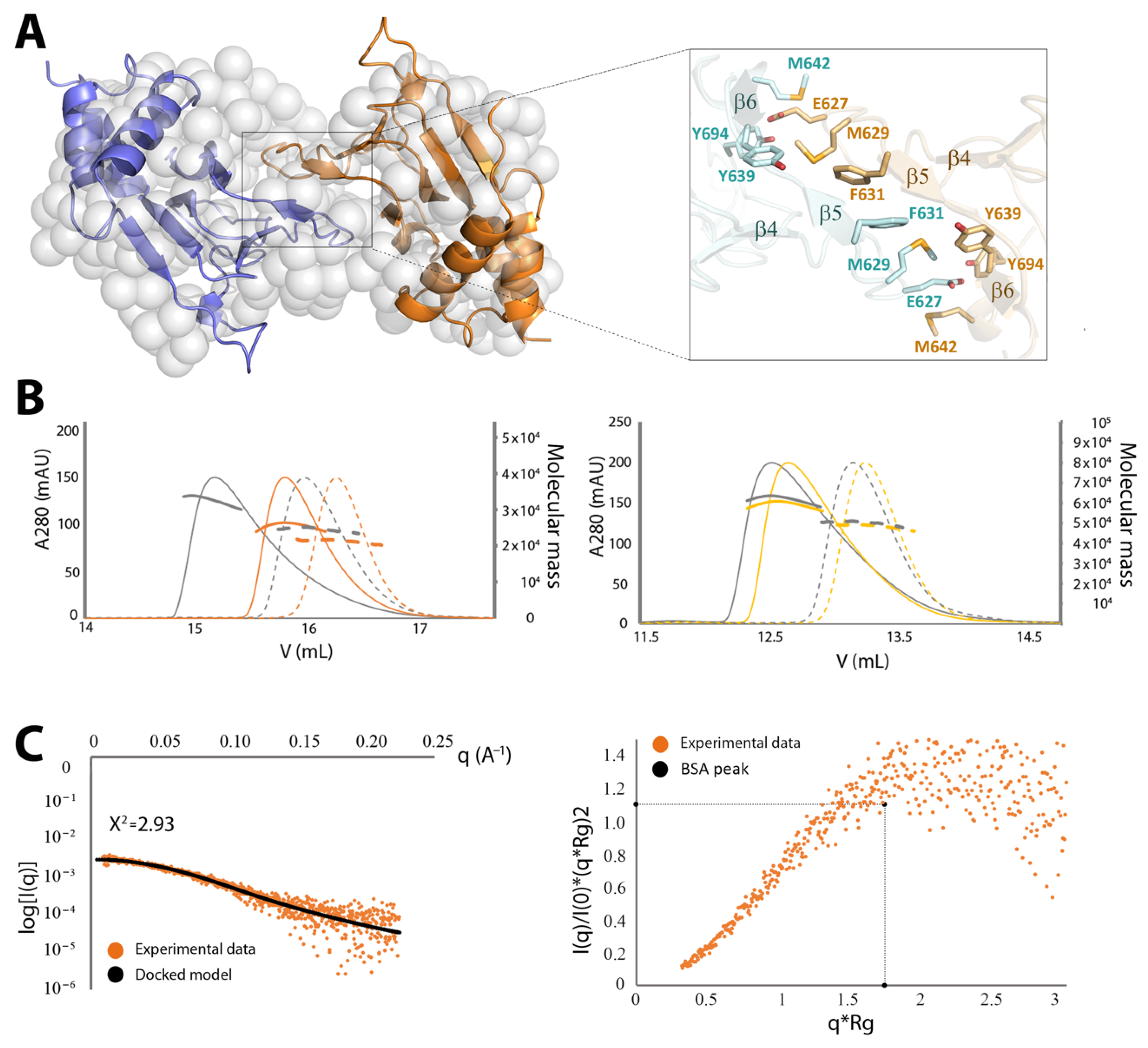
2.3.2. CNNM4cNMP Forms Symmetric Homodimers
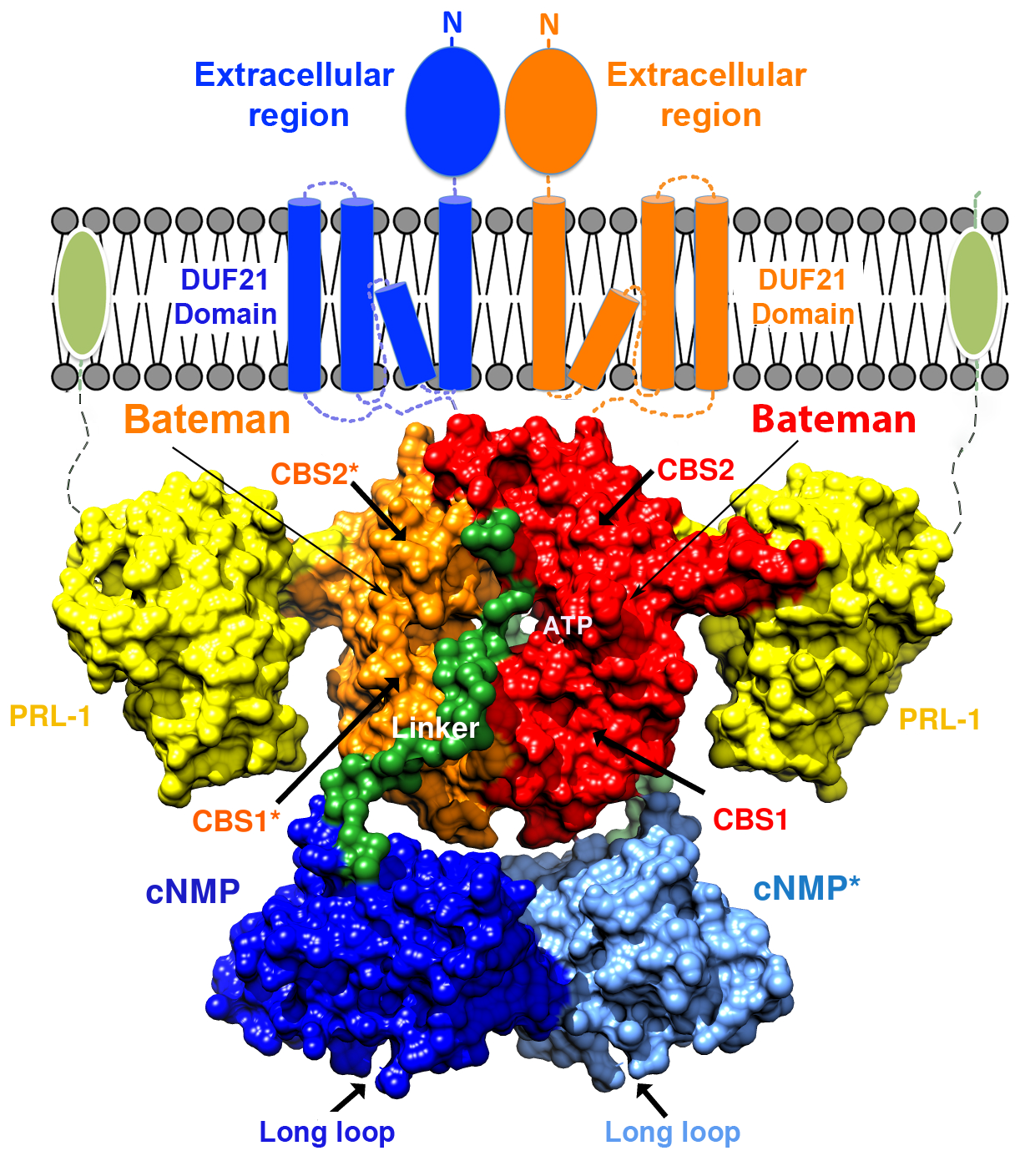
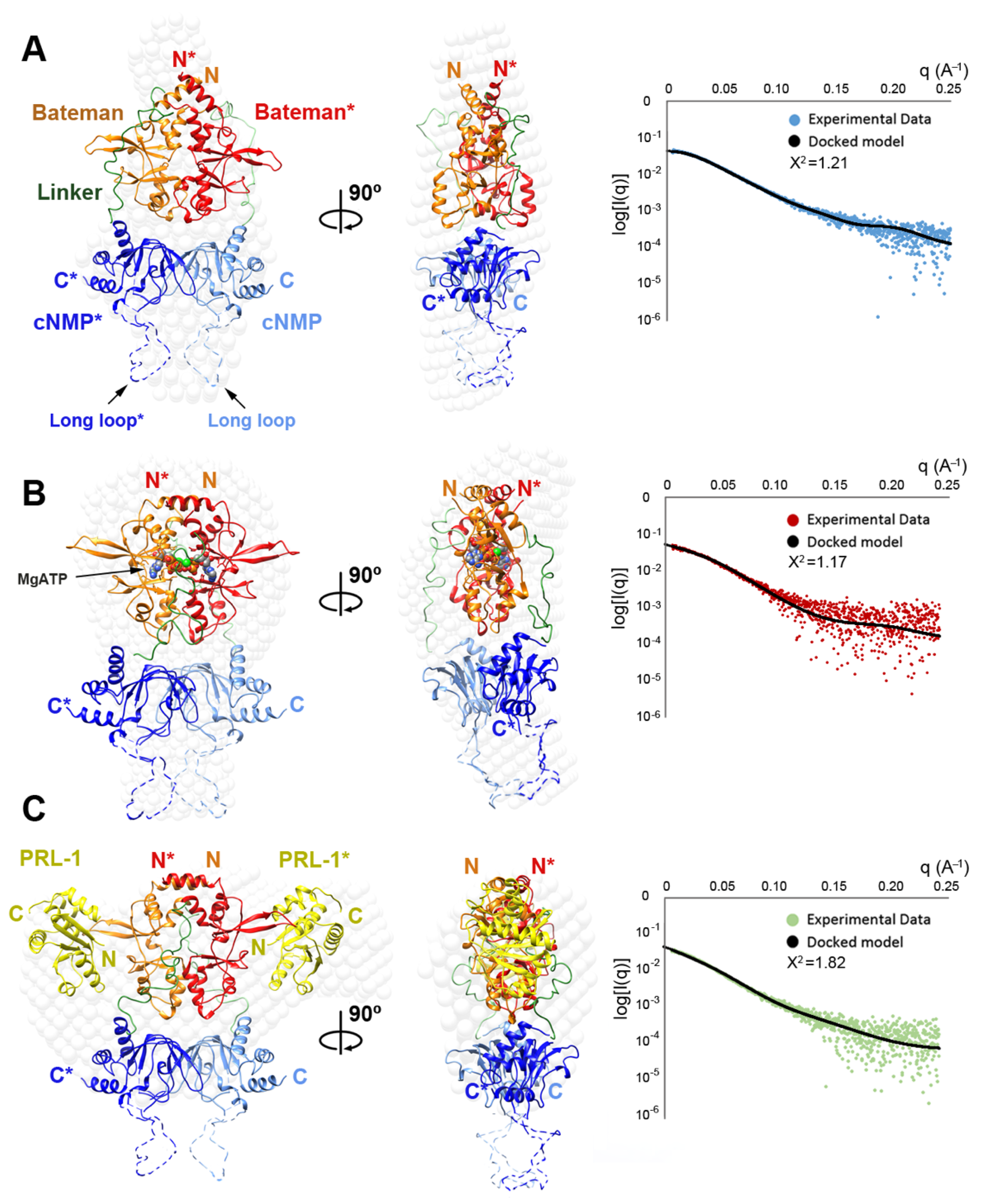
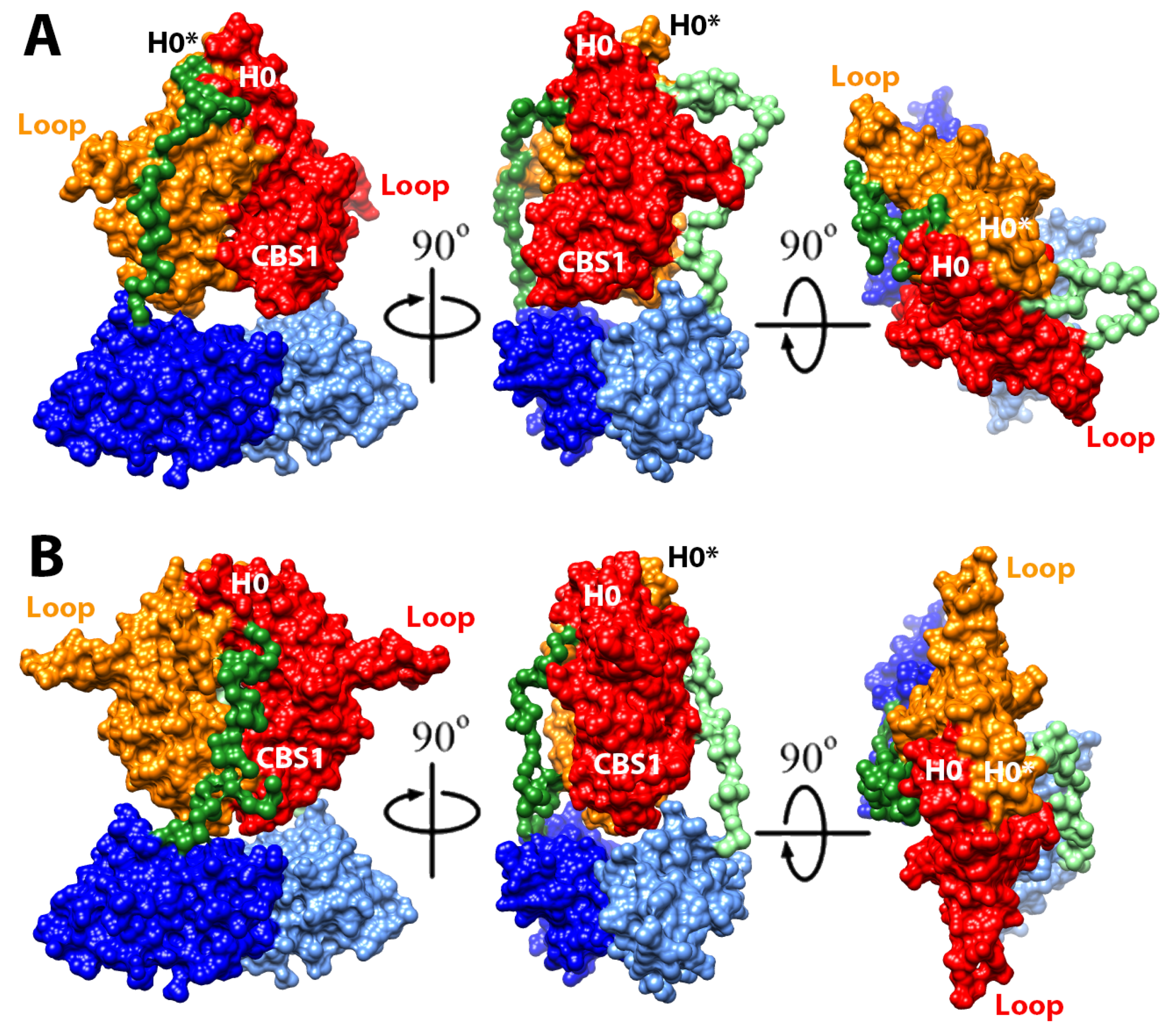
2.4. Structural Model of the Complete Intracellular Region of CNNM4
2.5. Structural Model of the Full Intracellular Region of CNNM4 in Complex with PRL-1
3. Discussion
4. Materials and Methods
4.1. Cloning and Protein Purification
4.2. NMR Analysis
4.3. Size Exclusion Chromatography with Multiangle Light Scattering (SEC-MALS)
4.4. Isothermal Titration Calorimetry (ITC)
4.5. Small Angle X-Ray Scattering (SAXS)
4.6. Protein Crystallization
4.7. X-Ray Structure Determination and Refinement
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Quamme, G.A. Molecular identification of ancient and modern mammalian magnesium transporters. Am. J. Physiol. Cell Physiol. 2010, 298, C407–C429. [Google Scholar] [CrossRef] [PubMed]
- Stuiver, M.; Lainez, S.; Will, C.; Terryn, S.; Gu, D.; Sommer, K.; Kopplin, K.; Thumfart, J.; Kampik, N.B.; Querfeld, U.; et al. CNNM2 encoding a basolateral protein required for renal Mg2+ handling, is mutated in Dominant Hypomagnesemia. Am. J. Hum. Genet. 2011, 88, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, D.; Funato, Y.; Miura, J.; Sato, S.; Toyosawa, S.; Furutani, K.; Kurachi, Y.; Omori, Y.; Furukawa, T.; Tsuda, T.; et al. Basolateral Mg2+ extrusion via CNNM4 mediates transcellular Mg2+ transport across epithelia: A mouse model. PLoS Genet. 2013, 9, e1003983. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Da Shi, J.; Yang, P.; Kumar, P.G.; Li, Q.Z.; Run, Q.G.; Su, Y.C.; Scott, H.S.; Kao, K.J.; She, J.X. Molecular cloning and characterization of a novel gene family of four ancient conserved domain proteins (ACDP). Gene 2003, 306, 37–44. [Google Scholar] [CrossRef]
- Corral-Rodríguez, M.Á.; Stuiver, M.; Abascal-Palacios, G.; Diercks, T.; Oyenarte, I.; Ereño-Orbea, J.; de Opakua, A.I.; Blanco, F.J.; Encinar, J.A.; Spiwok, V.; et al. Nucleotide binding triggers a conformational change of the CBS module of the magnesium transporter CNNM2 from a twisted towards a flat structure. Biochem. J. 2014, 464, 23–34. [Google Scholar] [CrossRef]
- Giménez-Mascarell, P.; Oyenarte, I.; Hardy, S.; Breiderhoff, T.; Stuiver, M.; Kostantin, E.; Diercks, T.; Pey, A.L.; Ereño-Orbea, J.; Martínez-Chantar, M.L.; et al. Structural basis of the oncogenic interaction of phosphatase PRL-1 with the magnesium transporter CNNM2. J. Biol. Chem. 2017, 292, 786–801. [Google Scholar] [CrossRef]
- De Baaij, J.H.F.; Stuiver, M.; Meij, I.C.; Lainez, S.; Kopplin, K.; Venselaar, H.; Müller, D.; Bindels, R.J.M.; Hoenderop, J.G.J. Membrane topology and intracellular processing of cyclin M2 (CNNM2). J. Biol. Chem. 2012, 287, 13644–13655. [Google Scholar] [CrossRef]
- Goytain, A.; Quamme, G.A. Functional characterization of ACDP2 (ancient conserved domain protein), a divalent metal transporter. Physiol. Genomics. 2005, 22, 382–389. [Google Scholar] [CrossRef]
- Kolisek, M.; Sponder, G.; Pilchova, I.; Cibulka, M.; Tatarkova, Z.; Werner, T.; Racay, P. Magnesium extravaganza: A critical compendium of current research into cellular Mg2+ transporters other than TRPM6/7. Rev. Physiol. Biochem. Pharmacol. 2018, 176, 65–105. [Google Scholar]
- Arjona, F.J.; de Baaij, J.H.F. CrossTalk opposing view: CNNM proteins are not Na+/Mg2+ exchangers but Mg2+ transport regulators playing a central role in transepithelial Mg2+(re)absorption. J. Physiol. 2018, 596, 747–750. [Google Scholar] [CrossRef]
- Funato, Y.; Furutani, K.; Kurachi, Y.; Miki, H. CrossTalk proposal: CNNM proteins are Na+/Mg2+ exchangers playing a central role in transepithelial Mg2+ (re)absorption. J. Physiol. 2018, 596, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Schäffers, O.J.M.; Hoenderop, J.G.J.; Bindels, R.J.M.; de Baaij, J.H.F. The rise and fall of novel renal magnesium transporters. Am. J. Physiol. Physiol. 2018, 314, F1027–F1033. [Google Scholar] [CrossRef] [PubMed]
- Funato, Y.; Furutani, K.; Kurachi, Y.; Miki, H. Rebuttal from Yosuke Funato, Kazuharu Furutani, Yoshihisa Kurachi and Hiroaki Miki. J. Physiol. 2018, 596, 751. [Google Scholar] [CrossRef] [PubMed]
- Alderton, A.; Davies, P.; Illman, K.; Brown, D.R. Ancient conserved domain protein-1 binds copper and modifies its retention in cells. J. Neurochem. 2007, 103, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Funato, Y.; Yamazaki, D.; Mizukami, S.; Du, L.; Kikuchi, K.; Miki, H. Membrane protein CNNM4–dependent Mg2+ efflux suppresses tumor progression. J. Clin. Investig. 2014, 124, 5398–5410. [Google Scholar] [CrossRef] [PubMed]
- Sponder, G.; Mastrototaro, L.; Kurth, K.; Merolle, L.; Zhang, Z.; Abdulhanan, N.; Smorodchenko, A.; Wolf, K.; Fleig, A.; Penner, R.; et al. Human CNNM2 is not a Mg2+ transporter per se. Pflugers Arch. Eur. J. Physiol. 2016, 468, 1223–1240. [Google Scholar] [CrossRef]
- Parry, D.A.; Mighell, A.J.; El-Sayed, W.; Shore, R.C.; Jalili, I.K.; Dollfus, H.; Bloch-Zupan, A.; Carlos, R.; Carr, I.M.; Downey, L.M.; et al. Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. Am. J. Hum. Genet. 2008, 84, 266–273. [Google Scholar] [CrossRef]
- Polok, B.; Escher, P.; Ambresin, A.; Chouery, E.; Bolay, S.; Meunier, I.; Nan, F.; Hamel, C.; Munier, F.L.; Thilo, B.; et al. Mutations in CNNM4 cause recessive cone-rod dystrophy with amelogenesis imperfecta. Am. J. Hum. Genet. 2008, 84, 259–265. [Google Scholar] [CrossRef]
- Luder, H.U.; Gerth-Kahlert, C.; Ostertag-Benzinger, S.; Schorderet, D.F. Dental phenotype in Jalili Syndrome due to a c.1312 dupC homozygous mutation in the CNNM4 gene. PLoS ONE 2013, 8, 6–12. [Google Scholar] [CrossRef]
- Cherkaoui Jaouad, I.; Lyahyai, J.; Guaoua, S.; El Alloussi, M.; Zrhidri, A.; Doubaj, Y.; Boulanouar, A.; Sefiani, A. Novel splice site mutation in CNNM4 gene in a family with Jalili syndrome. Eur. J. Med. Genet. 2017, 60, 239–244. [Google Scholar] [CrossRef]
- Topçu, V.; Alp, M.Y.; Alp, C.K.; Bakir, A.; Geylan, D.; Yilmazoğlu, M.Ö. A new familial case of Jalili syndrome caused by a novel mutation in CNNM4. Ophthalmic Genet. 2016, 38, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Hirji, N.; Bradley, P.D.; Li, S.; Vincent, A.; Pennesi, M.E.; Thomas, A.S.; Heon, E.; Bhan, A.; Mahroo, O.A.; Robson, A.; et al. Jalili Syndrome: Cross-sectional and longitudinal features of seven patients with Cone-Rod Dystrophy and Amelogenesis Imperfecta. Am. J. Ophthalmol. 2018, 188, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Maia, C.M.F.; Machado, R.A.; Gil-da-Silva-Lopes, V.L.; Lustosa-Mendes, E.; Rim, P.H.H.; Dias, V.O.; Martelli, D.R.B.; Nasser, L.S.; Coletta, R.D.; Martelli-Júnior, H. Report of two unrelated families with Jalili syndrome and a novel nonsense heterozygous mutation in CNNM4 gene. Eur. J. Med. Genet. 2018, 61, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, D.; Miyata, H.; Funato, Y.; Fujihara, Y.; Ikawa, M.; Miki, H. The Mg2+ transporter CNNM4 regulates sperm Ca2+ homeostasis and is essential for reproduction. J. Cell Sci. 2016, 129, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- Rezende, F.M.; Dietsch, G.O.; Peñagaricano, F. Genetic dissection of bull fertility in US Jersey dairy cattle. Anim. Genet. 2018, 49, 393–402. [Google Scholar] [CrossRef]
- Accogli, A.; Scala, M.; Calcagno, A.; Napoli, F.; Di Iorgi, N.; Arrigo, S.; Mancardi, M.M.; Prato, G.; Pisciotta, L.; Nagel, M.; et al. CNNM2 homozygous mutations cause severe refractory hypomagnesemia, epileptic encephalopathy and brain malformations. Eur. J. Med. Genet. 2018, 62, 198–203. [Google Scholar] [CrossRef]
- Rose, E.J.; Hargreaves, A.; Morris, D.; Fahey, C.; Tropea, D.; Cummings, E.; Caltagirone, C.; Bossù, P.; Chiapponi, C.; Piras, F.; et al. Effects of a novel schizophrenia risk variant rs7914558 at CNNM2 on brain structure and attributional style. Br. J. Psychiatry 2014, 204, 115–121. [Google Scholar] [CrossRef]
- Ohi, K. Influences of schizophrenia risk variant rs7914558 at CNNM2 on brain structure. Br. J. Psychiatry 2015, 206, 343. [Google Scholar] [CrossRef][Green Version]
- Guan, F.; Zhang, T.; Li, L.; Fu, D.; Lin, H.; Chen, G.; Chen, T. Two-stage replication of previous genome-wide association studies of AS3MT-CNNM2-NT5C2 gene cluster region in a large schizophrenia case–control sample from Han Chinese population. Schizophr. Res. 2016, 176, 125–130. [Google Scholar] [CrossRef]
- Paparelli, A.; Iwata, K.; Wakuda, T.; Iyegbe, C.; Murray, R.M.; Takei, N. Perinatal asphyxia in rat alters expression of novel Schizophrenia risk genes. Front. Mol. Neurosci. 2017, 10, 1–10. [Google Scholar] [CrossRef]
- Funato, Y.; Yamazaki, D.; Miki, H. Renal function of cyclin M2 Mg2+ transporter maintains blood pressure. J. Hypertens. 2017, 35, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.; Uetani, N.; Wong, N.; Kostantin, E.; Labbé, D.P.; Bégin, L.R.; Mes-Masson, A.; Miranda-Saavedra, D.; Tremblay, M.L. The protein tyrosine phosphatase PRL-2 interacts with the magnesium transporter CNNM3 to promote oncogenesis. Oncogene 2015, 34, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Gulerez, I.; Funato, Y.; Wu, H.; Yang, M.; Kozlov, G.; Miki, H.; Gehring, K. Phosphocysteine in the PRL-CNNM pathway mediates magnesium homeostasis. EMBO Rep. 2016, 17, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Kostantin, E.; Hardy, S.; Valinsky, W.C.; Kompatscher, A.; De Baaij, J.H.F.; Zolotarov, Y.; Landry, M.; Uetani, N.; Martínez-Cruz, L.A.; Hoenderop, J.G.J.; et al. Inhibition of PRL-2·CNNM3 protein complex formation decreases breast cancer proliferation and tumor growth. J. Biol. Chem. 2016, 291, 10716–10725. [Google Scholar] [CrossRef]
- Hirata, Y.; Funato, Y.; Takano, Y.; Miki, H. Mg2+-dependent interactions of ATP with the cystathionine- β-synthase (CBS) domains of a magnesium transporter. J. Biol. Chem. 2014, 289, 14731–14739. [Google Scholar] [CrossRef]
- Zhang, H.; Kozlov, G.; Li, X.; Wu, H.; Gulerez, I.; Gehring, K. PRL3 phosphatase active site is required for binding the putative magnesium transporter CNNM3. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Shabb, J.B.; Corbin, J.D. Cyclic nucleotide-binding domains in proteins having diverse functions. J. Biol. Chem. 1992, 267, 5723–5726. [Google Scholar]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Gómez García, I.; Oyenarte, I.; Martínez-Cruz, L.A. Purification, crystallization and preliminary crystallographic analysis of the CBS pair of the human metal transporter CNNM4. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 349–353. [Google Scholar] [CrossRef]
- Baykov, A.A.; Tuominen, H.K.; Lahti, R. The CBS Domain: A protein module with an emerging prominent role in regulation. ACS Chem. Biol. 2011, 6, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Ereño-Orbea, J.; Oyenarte, I.; Martínez-Cruz, L.A. CBS domains: Ligand binding sites and conformational variability. Arch. Biochem. Biophys. 2013, 540, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Armitano, J.; Redder, P.; Guimarães, V.A.; Linder, P. An essential factor for high Mg2+ tolerance of Staphylococcus aureus. Front. Microbiol. 2016, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.M.; Bagga, D.A.; Miller, C.G.; Maguire, M.E. Magnesium transport in Salmonella typhimurium: the influence of new mutations conferring Co2+ resistance on the CorA Mg2+ transport system. Mol. Microbiol. 1991, 5, 2753–2762. [Google Scholar] [CrossRef]
- Hattori, M.; Tanaka, Y.; Fukai, S.; Ishitani, R.; Nureki, O. Crystal structure of the MgtE Mg2+ transporter. Nature 2007, 448, 1072–1075. [Google Scholar] [CrossRef]
- Tomita, A.; Zhang, M.; Jin, F.; Zhuang, W.; Takeda, H.; Maruyama, T.; Osawa, M.; Hashimoto, K.I.; Kawasaki, H.; Ito, K.; et al. ATP-dependent modulation of MgtE in Mg2+ homeostasis. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Meyer, S.; Dutzler, R. Crystal structure of the cytoplasmic domain of the chloride channel ClC-0. Structure 2006, 14, 299–307. [Google Scholar] [CrossRef]
- Sigel, H. Isomeric equilibria in complexes of adenosine 5′-triphosphate with divalent metal ions. Solution structures of M(ATP)2- complexes. Eur. J. Biochem. 1987, 165, 65–72. [Google Scholar] [CrossRef]
- Berman, H.M.; Ten Eyck, L.F.; Goodsell, D.S.; Haste, N.M.; Kornev, A.; Taylor, S.S. The cAMP binding domain: An ancient signaling module. Proc. Natl. Acad. Sci. USA 2005, 102, 45–50. [Google Scholar] [CrossRef]
- Rehmann, H.; Wittinghofer, A.; Bos, J.L. Capturing cyclic nucleotides in action: Snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 2007, 8, 63–73. [Google Scholar] [CrossRef]
- Haitin, Y.; Carlson, A.E.; Zagotta, W.N. The structural mechanism of KCNH-channel regulation by the eag domain. Nature 2013, 501, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Kozlov, G.; Fakih, R.; Funato, Y.; Miki, H.; Gehring, K. The cyclic nucleotide-binding homology domain of the integral membrane protein CNNM mediates dimerization and is required for Mg2+ efflux activity. J. Biol. Chem. 2018, 293, 19998–20007. [Google Scholar] [CrossRef] [PubMed]
- Rambo, R.P.; Tainer, J.A. Super-Resolution in Solution X-Ray Scattering and its applications to tructural Systems Biology. Annu. Rev. Biophys. 2013, 42, 415–441. [Google Scholar] [CrossRef] [PubMed]
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.T.; Konarev, P.V.; Svergun, D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012, 45, 342–350. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Sheffield, P.; Garrard, S. and Derewenda, Z. Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif. 1999, 15, 34–39. [Google Scholar] [CrossRef]
- Marley, J.; Lu, M.; Bracken, C. A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 2001, 20, 71–75. [Google Scholar] [CrossRef]
- Tyler, R.C.; Sreenath, H.K.; Singh, S.; Aceti, D.J.; Bingman, C.A.; Markley, J.L.; Fox, B.G. Auto-induction medium for the production of [U-15N]- and [U-13C, U-15N]-labeled proteins for NMR screening and structure determination. Protein Expr. Purif. 2005, 40, 268–278. [Google Scholar] [CrossRef]
- Wishart, D.S.; Bigam, C.G.; Yao, J.; Abildgaard, F.; Dyson, H.J.; Oldfield, E.; Markley, J.L.; Sykes, B.D. H-1, C-13 and N-15 chemical-shift referencing in Biomolecular NMR. J. Biomol. NMR 1995, 6, 135–140. [Google Scholar] [CrossRef]
- Glatter, O. A new method for the evaluation of small-angle scattering data. J. Appl. Crystallogr. 1977, 10, 415–421. [Google Scholar] [CrossRef]
- Svergun, D.I.; Petoukhov, M.V.; Koch, M.H.J. Determination of domain structure of proteins from X-Ray Solution Scattering. Biophys. J. 2001, 80, 2946–2953. [Google Scholar] [CrossRef]
- Volkov, V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003, 36, 860–864. [Google Scholar] [CrossRef]
- Valentini, E.; Kikhney, A.G.; Previtali, G.; Jeffries, C.M.; Svergun, D.I. SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 2015, 43, D357–D363. [Google Scholar] [CrossRef]
- Slabinski, L.; Jaroszewski, L.; Rychlewski, L.; Wilson, I.A.; Lesley, S.A.; Godzik, A. XtalPred: A web server for prediction of protein crystallizability. Bioinformatics 2007, 23, 3403–3405. [Google Scholar] [CrossRef]
- Strong, M.; Sawaya, M.R.; Wang, S.; Phillips, M.; Cascio, D.; Eisenberg, D. Toward the structural genomics of complexes: Crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2006, 103, 8060–8065. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 479–485. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Adams, P.D.; Read, R.J.; McCoy, A.J.; Moriarty, N.W.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Zwart, P.H.; Hung, L.W. Decision-making in structure solution using Bayesian estimates of map quality: The PHENIX AutoSol wizard. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 582–601. [Google Scholar] [CrossRef]
- Cowtan, K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. Sect. D Biol. Crystallogr. 2006, 62, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | CNNM4BAT | CNNM4cNMP |
|---|---|---|
| Data Collection and Process | ||
| Beamline | ESRF, ID14-1 | DIAMOND, I03 |
| Radiation wavelength (Å) | 0.934 | 0.9794 |
| Space group/PDB ID | C2/6RS2 | P3221/6G52 |
| a (Å) b (Å) c (Å) | 91.39 141.36 87.89 | 116.74 116.74 243.46 |
| Molecules per a.u. | 4 | 9 |
| Resolution (Å) | 43.94–3.69 (3.76–3.69) | 243.46–3.69 (3.99–3.69) |
| Rsym a | 0.047 (0.437) | 0.179 (1.661) |
| Rmeas b | 0.052 (0.485) | 0.184 (1.707) |
| Rpim c | 0.023 (0.209) | 0.042 (0.387) |
| No. of observations | 62,620 | 413,463 |
| No. of unique reflections | 11,864 | 21,343 |
| Mean I/I | 21 (3.4) | 13.9 (2.8) |
| CC1/2 | 0.99 (0.91) | 0.99 (0.87) |
| Completeness (%) | 98.7 (90.7) | 99.7 (98.5) |
| Redundancy | 5.3 (5.2) | 19.4 (19.3) |
| Mosaicity (°) | 0.2 | 0.1 |
| Refinement Statistics | ||
| No. of working/test reflections | 44,826/1178 | 20,676/1989 |
| Rwork d/Rfree e | 0.23/0.28 | 0.2844/0.3026 |
| No. of atoms | ||
| Protein | 4567 | 9252 |
| Ligand | - | - |
| Water | - | - |
| Average B factors (Å2) | ||
| Protein | 105,94 | 100 |
| Ligand | - | - |
| Water | - | - |
| RMSDs | ||
| Bond lengths (Å)/angles (°) | 0.004/0.683 | 0.003/0.748 |
| Ramachandran plot statistics (%) | ||
| Residues in most favored regions | 97.5 | 99 |
| Residues in additionally allowed regions | 2.5 | 1 |
| Residues in disallowed regions | 0 | 0 |
| Data Collection Parameters | |||||
| Beamline | B21, Diamond Light Source, Harwell (UK) | ||||
| Detector | Pilatus 2M | ||||
| Beam size | 0.2 × 0.2 mm | ||||
| Energy | 12.4 keV | ||||
| Sample-to-detector distance (mm) | 4014 | ||||
| q range (A−1) | 0.0038–0.42 | ||||
| Exposure time (s) | 3 | ||||
| Number of frames | 620 | ||||
| Temperature (K) | 293 | ||||
| Mode | SEC online | ||||
| Structural Parameters | |||||
| Protein construct | CNNM4cNMP | CNNM4BAT-cNMP-Ctail | CNNM4BAT-cNMP-Ctail + MgATP | CNNM4BAT-cNMP-Ctail + PRL-1 | CNNM4BAT-cNMP-Ctail + MgATP + PRL-1 |
| SASBDB access code | SASDER8 | SASDEQ8 | SASDES8 | SASDEP8 | SASDEN8 |
| Concentration range (mg/mL) | 7.0 | 5.0 | 4.7 | 8.5 | 5.0 |
| q Interval for Fourier inversion (Å−1) | 0.014–0.2018 | 0.007–0.164 | 0.010–0.109 | 0.09–0.173 | 0.009–0.125 |
| Rg [from P(r)] (Å) | 25.67 ± 1.32 | 41.62 ± 2.12 | 39.15 ± 1.38 | 47.90 ± 5.24 | 47.20 ± 2.55 |
| Rg [from Guiner approximation] (Å) | 25.52 ± 1.65 | 40.22 ± 0.54 | 39.23 ± 1.98 | 46.55 ± 0.81 | 44.48 ± 1.42 |
| sRg limits [from Guiner approx.] | 0.34–1.30 | 0.31–1.30 | 0.41–1.30 | 0.36–1.29 | 0.38–1.29 |
| Dmax (Å) | 96 | 166 | 143 | 185 | 183 |
| Porod volume estimate (nm3) | 102 | 156 | 149 | 226 | 216 |
| Porod exponent | 2.3 | 3.2 | 3.3 | 3.1 | 3.1 |
| Molecular Mass (kDa) | |||||
| from Porod volume (×0.58) | 59 | 90 | 86 | 131 | 125 |
| from amino acid sequence | 21 | 48 | 48 | 68 | 68 |
| Software Employed | |||||
| Primary data reduction | DAWN pipeline (Diamond Light Source, UK) | ||||
| Data processing | ScÅtter v3.1v | ||||
| Ab initio modelling | GASBOR | ||||
| Validation and averaging | DAMAVER/DAMCLUST | ||||
| Computation of model intensities | CRYSOL/CORAL | ||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giménez-Mascarell, P.; Oyenarte, I.; González-Recio, I.; Fernández-Rodríguez, C.; Corral-Rodríguez, M.Á.; Campos-Zarraga, I.; Simón, J.; Kostantin, E.; Hardy, S.; Díaz Quintana, A.; et al. Structural Insights into the Intracellular Region of the Human Magnesium Transport Mediator CNNM4. Int. J. Mol. Sci. 2019, 20, 6279. https://doi.org/10.3390/ijms20246279
Giménez-Mascarell P, Oyenarte I, González-Recio I, Fernández-Rodríguez C, Corral-Rodríguez MÁ, Campos-Zarraga I, Simón J, Kostantin E, Hardy S, Díaz Quintana A, et al. Structural Insights into the Intracellular Region of the Human Magnesium Transport Mediator CNNM4. International Journal of Molecular Sciences. 2019; 20(24):6279. https://doi.org/10.3390/ijms20246279
Chicago/Turabian StyleGiménez-Mascarell, Paula, Iker Oyenarte, Irene González-Recio, Carmen Fernández-Rodríguez, María Ángeles Corral-Rodríguez, Igone Campos-Zarraga, Jorge Simón, Elie Kostantin, Serge Hardy, Antonio Díaz Quintana, and et al. 2019. "Structural Insights into the Intracellular Region of the Human Magnesium Transport Mediator CNNM4" International Journal of Molecular Sciences 20, no. 24: 6279. https://doi.org/10.3390/ijms20246279
APA StyleGiménez-Mascarell, P., Oyenarte, I., González-Recio, I., Fernández-Rodríguez, C., Corral-Rodríguez, M. Á., Campos-Zarraga, I., Simón, J., Kostantin, E., Hardy, S., Díaz Quintana, A., Zubillaga Lizeaga, M., Merino, N., Diercks, T., Blanco, F. J., Díaz Moreno, I., Martínez-Chantar, M. L., Tremblay, M. L., Müller, D., Siliqi, D., & Martínez-Cruz, L. A. (2019). Structural Insights into the Intracellular Region of the Human Magnesium Transport Mediator CNNM4. International Journal of Molecular Sciences, 20(24), 6279. https://doi.org/10.3390/ijms20246279