Resveratrol Treatment Enhances the Cellular Response to Leptin by Increasing OBRb Content in Palmitate-Induced Steatotic HepG2 Cells

 ,
,  ,
,  , , ,
, , , 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
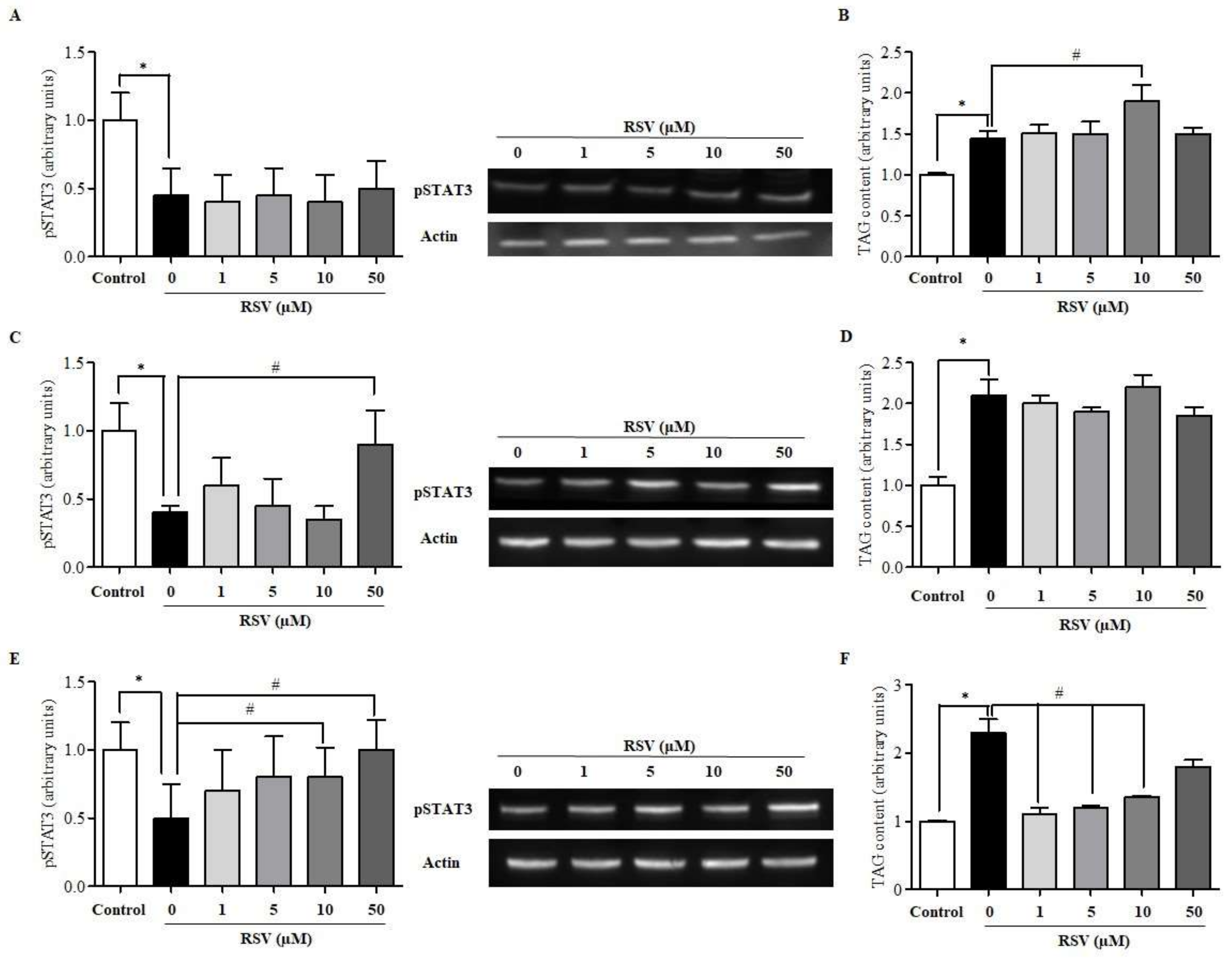
2.1. RSV Improves Leptin Signalling and Lipid Content in Steatotic HepG2 Cells
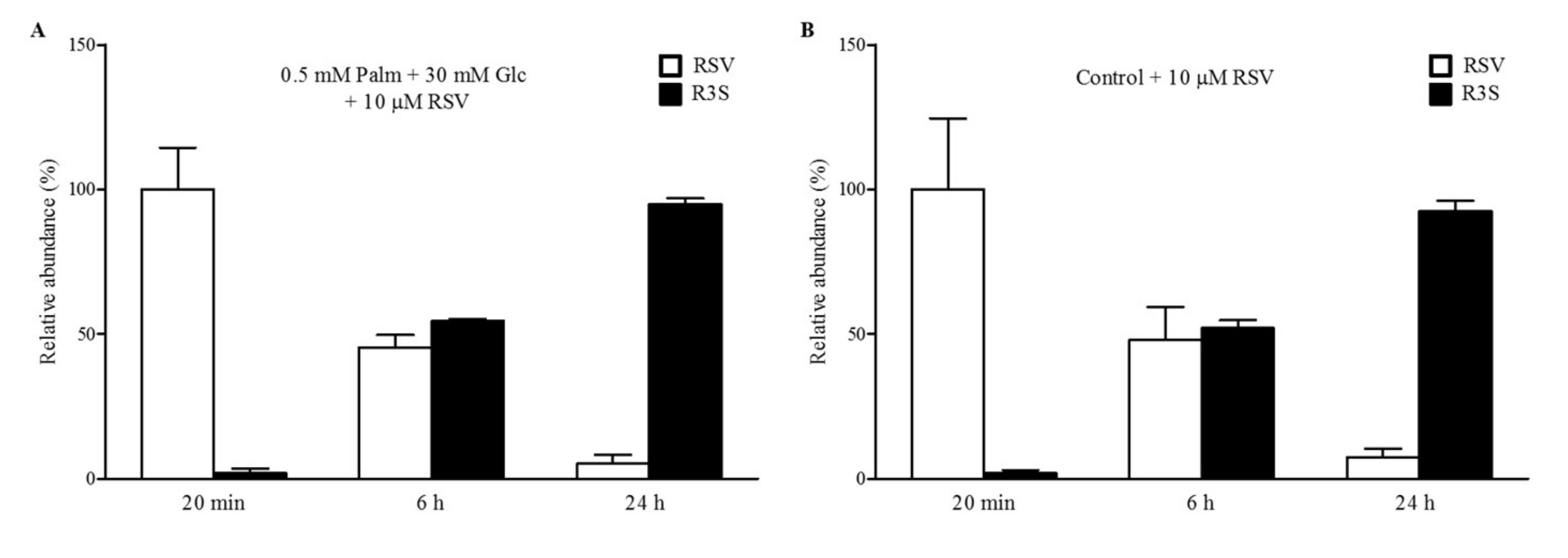
2.2. RSV Is Rapidly Metabolized into RSV-3-Sulfate
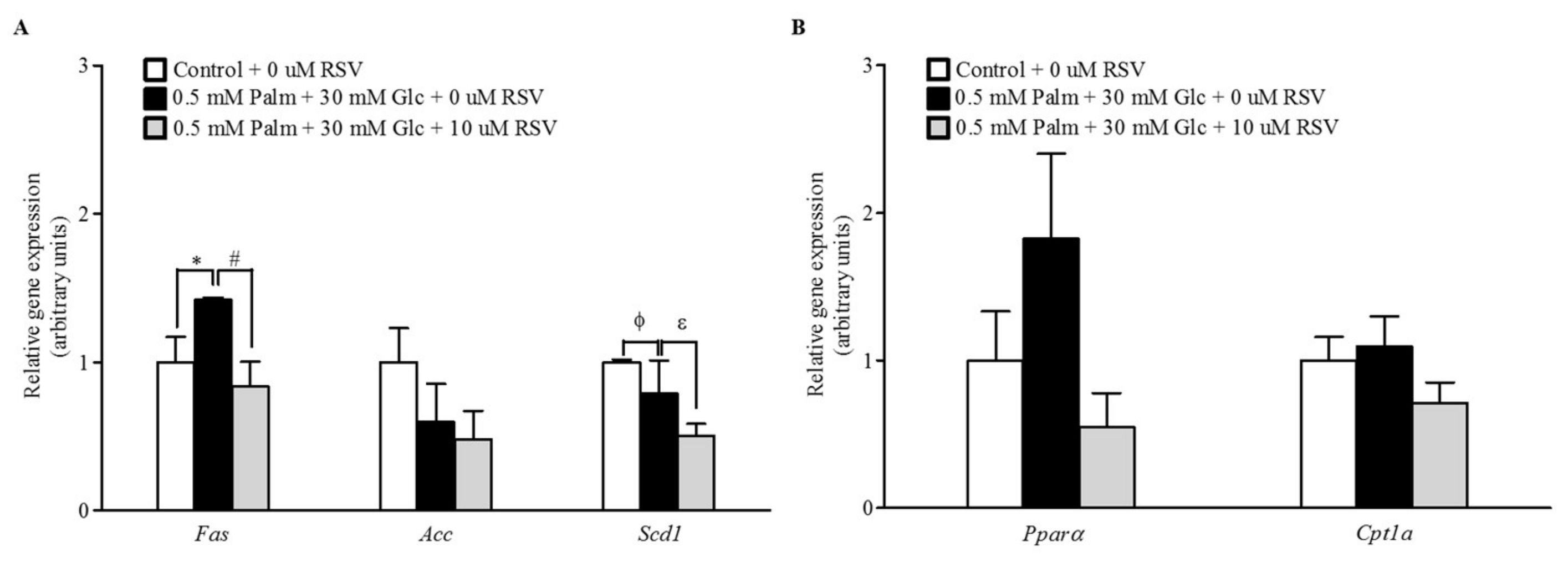
2.3. RSV Modulates Lipogenic Gene Expression but Not Fatty Acid Oxidation
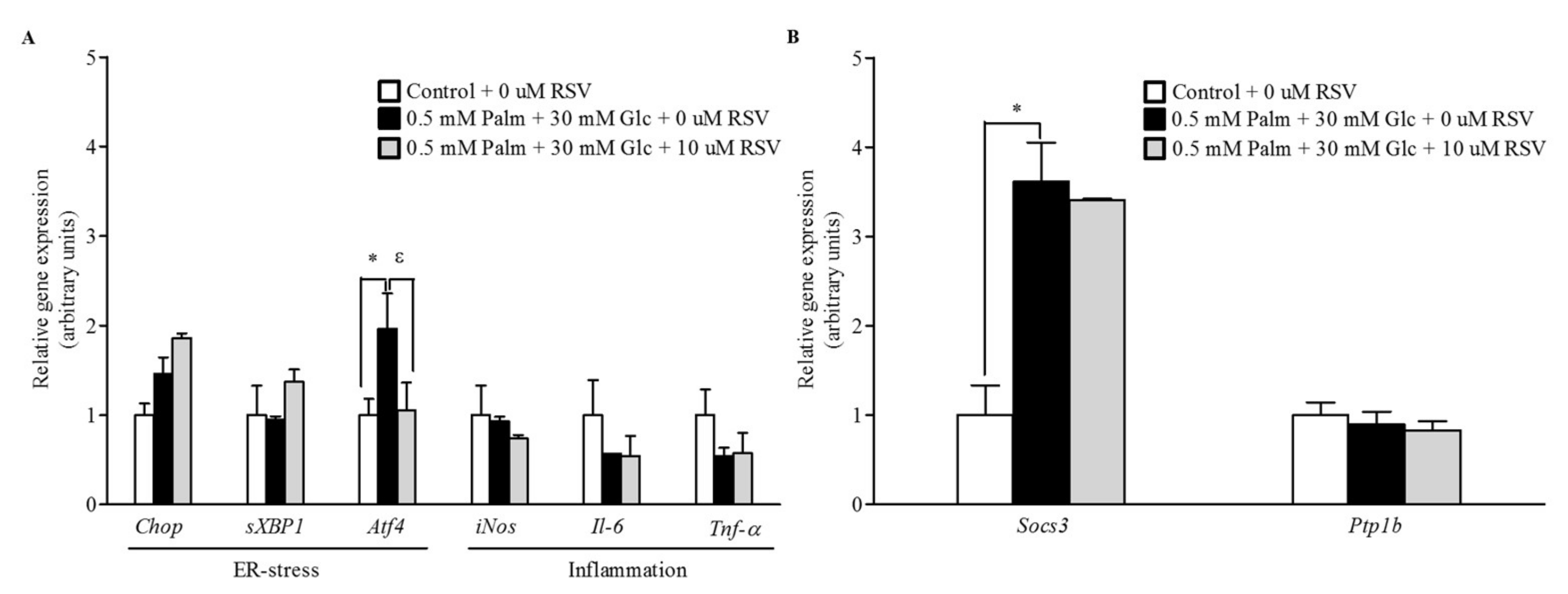
2.4. RSV Does Not Modulate the Expression of Pro-Inflammatory and ER Stress-Related Genes
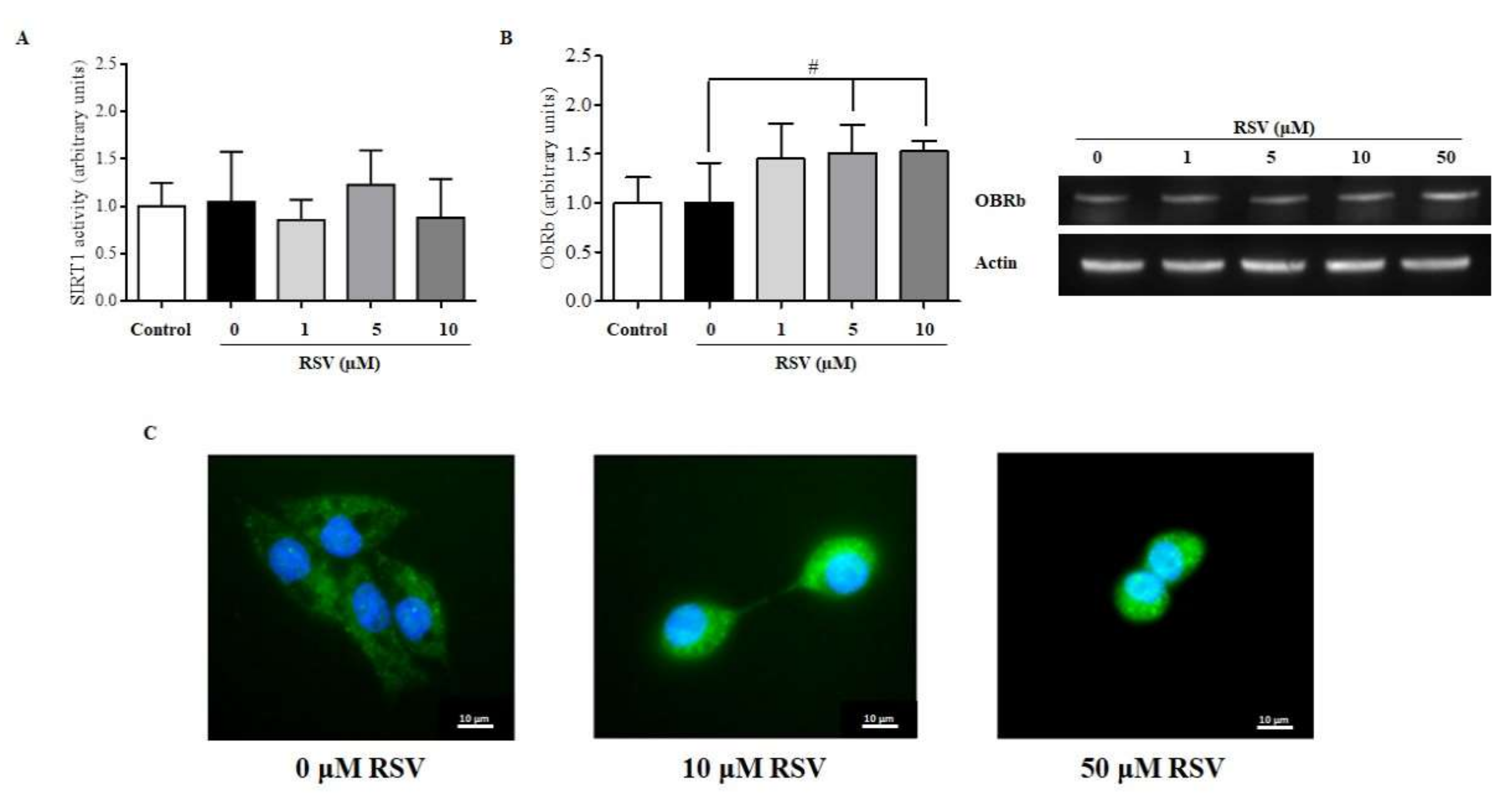
2.5. RSV Increases the mRNA and Protein Levels of Leptin Receptor OBRb but Not SIRT1 Activity
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture and Experimental Design
4.3. Palmitate-BSA Solution Preparation
4.4. Determination of Cell Viability by Neutral Red Assay
4.5. Quantification of Triglyceride Content
4.6. SIRT1 Activity Assay
4.7. Total RNA Isolation and Gene Expression Analysis
4.8. Western Blotting Analysis
4.9. Immunofluorescence Analysis
4.10. UHPLC-MSn Analysis of Cell Media
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BSA | Bovine serum albumin |
| DMEM | Dulbecco’s modified eagle medium |
| FBS | Foetal bovine serum |
| GLC | Glucose |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
| OBRb | Leptin receptor isoform b |
| GLN | Glutamine |
| NAFLD | Non-alcoholic fatty liver disease |
| NEFA | Non-essential fatty acid |
| NEAA | Non-essential amino acids |
| NAD | Nicotinamide adenine dinucleotide |
| PALM | Palmitate |
| P/S | Penicillin/streptomycin |
| PTP1B | Protein-tyrosine phosphatase 1B |
| RSV | Resveratrol |
| R3S | Resveratrol-3-sulfate |
| STAT3 | Signal transducer and activator of transcription-3 |
| SIRT | Sirtuin 1 |
| SOCS3 | Suppressor of cytokine signalling 3 |
References
- Wadden, T.A.; Webb, V.L.; Moran, C.H.; Brooke, A. Lifestyle Modification for Obesity. New Developments in Diet, Physical Activity, and Behavior Therapy. Circulation 2012, 125, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- Baboota, R.K.; Bishnoi, M.; Ambalam, P.; Kondepudi, K.K.; Sarma, S.M.; Boparai, R.K.; Podili, K. Functional food ingredients for the management of obesity and associated co-morbidities—A review. J. Funct. Foods 2013, 5, 997–1012. [Google Scholar] [CrossRef]
- Suárez, M.; Boqué, N.; Del Bas, J.M.; Mayneris-Perxachs, J.; Arola, L.; Caimari, A. Mediterranean Diet and Multi-Ingredient-Based Interventions for the Management of Non-Alcoholic Fatty Liver Disease. Nutrients 2017, 9, 1052. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, G.; Suárez, M.; Ardid-Ruiz, A.; Vinaixa, M.; Rodríguez, M.A.; Correig, X.; Arola, L.; Bladé, C. Dietary proanthocyanidins boost hepatic NAD+ metabolism and SIRT1 expression and activity in a dose-dependent manner in healthy rats. Sci. Rep. 2016, 6, 24977. [Google Scholar] [CrossRef]
- Aguirre, L.; Portillo, M.P.; Hijona, E.; Bujanda, L. Effects of resveratrol and other polyphenols in hepatic steatosis. World J. Gastroenterol. 2014, 20, 7366–7380. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Jiang, J.; Zhang, G.; Bu, Y.; Zhang, G.; Zhao, X. Resveratrol and caloric restriction prevent hepatic steatosis by regulating SIRT1-autophagy pathway and alleviating endoplasmic reticulum stress in high-fat diet-fed rats. PLoS ONE 2017, 12, e0183541. [Google Scholar] [CrossRef]
- Ardid-Ruiz, A.; Ibars, M.; Mena, P.; Del Rio, D.; Muguerza, B.; Bladé, C.; Arola, L.; Aragonès, G.; Suárez, M. Potential Involvement of Peripheral Leptin/STAT3 Signaling in the Effects of Resveratrol and Its Metabolites on Reducing Body Fat Accumulation. Nutrients 2018, 10, 1757. [Google Scholar] [CrossRef]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006, 87, 1–16. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Leptin in nonalcoholic fatty liver disease: A narrative review. Metabolism 2015, 64, 60–78. [Google Scholar] [CrossRef]
- Aragonès, G.; Ardid-Ruiz, A.; Ibars, M.; Suárez, M.; Bladé, C. Modulation of leptin resistance by food compounds. Mol. Nutr. Food. Res. 2016, 60, 1789–1803. [Google Scholar] [CrossRef] [PubMed]
- Unger, R.H. Minireview: Weapons of Lean Body Mass Destruction: The Role of Ectopic Lipids in the Metabolic Syndrome. Endocrinology 2003, 144, 5159–5165. [Google Scholar] [CrossRef] [PubMed]
- Brabant, G.; Müller, G.; Horn, R.; Anderwald, C.; Roden, M.; Nave, H. Hepatic leptin signaling in obesity. FASEB J. 2005, 19, 1048–1050. [Google Scholar] [CrossRef] [PubMed]
- Puri, P.; Sanyal, A.J. Sirtuins and the hepatic regulation of energy homeostasis. Hepatology 2009, 50, 1668–1670. [Google Scholar] [CrossRef] [PubMed]
- Schug, T.T.; Li, X. Sirtuin 1 in lipid metabolism and obesity. Ann. Med. 2011, 43, 198–211. [Google Scholar] [CrossRef]
- Aragonès, G.; Danesi, F.; del Rio, D.; Mena, P. The importance of studying cell metabolism when testing the bioactivity of phenolic compounds. Trends Food Sci. Technol. 2017, 69, 230–242. [Google Scholar] [CrossRef]
- Bates, S.H.; Kulkarni, R.N.; Seifert, M.; Myers, M.G., Jr. Roles for leptin receptor/STAT3-dependent and -independent signals in the regulation of glucose homeostasis. Cell Metab. 2005, 1, 169–178. [Google Scholar] [CrossRef]
- Franco, J.G.; Dias-Rocha, C.P.; Fernandes, T.P.; Albuquerque, M.L.; Lisboa, P.C.; Moura, E.G.; Pazos-Moura, C.C.; Trevenzoli, I.H. Resveratrol treatment rescues hyperleptinemia and improves hypothalamic leptin signaling programmed by maternal high-fat diet in rats. Eur. J. Nutr. 2016, 55, 601–610. [Google Scholar] [CrossRef]
- Ardid-Ruiz, A.; Ibars, M.; Bladé, C.; Aragonès, G.; Suárez, M.; Arola, L. Resveratrol rescues hepatic leptin signal transduction via STAT3 pathway in a cellular model of fat accumulation induced by palmitic acid. In Proceedings of the XXVIIIth International Conference on Polyphenols, Vienna, Austria, 15 November 2016. [Google Scholar]
- Hackl, M.T.; Fürnsinn, C.; Schuh, C.M.; Krssak, M.; Carli, F.; Guerra, S.; Freudenthaler, A.; Baumgartner-Parzer, S.; Helbich, T.H.; Luger, A.; et al. Brain leptin reduces liver lipids by increasing hepatic triglyceride secretion and lowering lipogenesis. Nat. Commun. 2019, 10, 2717. [Google Scholar] [CrossRef]
- Liu, J.F.; Ma, Y.; Wang, Y.; Du, Z.Y.; Shen, J.K.; Peng, H.L. Reduction of lipid accumulation in HepG2 Cells by luteolin is associated with activation of AMPK and Mitigation of oxidative stress. Phyther. Res. 2011, 25, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Chen, S.L.; Hu, K.Q. Quantification and mechanisms of oleic acid-induced steatosis in HepG2 cells. Am. J. Transl. Res. 2010, 2, 95–104. [Google Scholar] [PubMed]
- Zang, M.; Xu, S.; Maitland-Toolan, K.A.; Zuccollo, A.; Hou, X.; Jiang, B.; Wierzbicki, M.; Verbeuren, T.J.; Cohen, R.A. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes 2006, 55, 2180–2191. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Fu, Y.C.; Xu, W.C.; Feng, Y.Q.; Fang, S.R.; Zhou, X.H. Resveratrol inhibits the expression of SREBP1 in cell model of steatosis via Sirt1-FOXO1 signaling pathway. Biochem. Biophys. Res. Commun. 2009, 380, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Ardid-Ruiz, A.; Harazin, A.; Barna, L.; Walter, F.R.; Bladé, C.; Suárez, M.; Deli, M.A.; Aragonès, G. The effects of Vitis vinifera L. phenolic compounds on a blood-brain barrier culture model: Expression of leptin receptors and protection against cytokine-induced damage. J. Ethnopharmacol. 2020, 247, 112253. [Google Scholar] [CrossRef] [PubMed]
- Sefried, S.; Häring, H.U.; Weigert, C.; Eckstein, S.S. Suitability of hepatocyte cell lines HepG2, AML12 and THLE-2 for investigation of insulin signalling and hepatokine gene expression. Open Biol. 2018, 8, 180147. [Google Scholar] [CrossRef]
- Vitaglione, P.; Sforza, S.; Galaverna, G.; Ghidini, C.; Caporaso, N.; Vescovi, P.P.; Fogliano, V.; Marchelli, R. Bioavailability of trans-resveratrol from red wine in humans. Mol. Nutr. Food Res. 2005, 49, 495–504. [Google Scholar] [CrossRef]
- Walle, T.; Hsieh, F.; DeLegge, M.H.; Oatis, J.E., Jr.; Walle, U.K. High absorption but very low bioavailability of oral resveratrol in humans. Drug Metab. Dispos. 2004, 32, 1377–1382. [Google Scholar] [CrossRef]
- Lançon, A.; Hanet, N.; Jannin, B.; Delmas, D.; Heydel, J.M.; Lizard, G.; Chagnon, M.C.; Artur, Y.; Latruffe, N. Resveratrol in human hepatoma HepG2 cells: Metabolism and inducibility of detoxifying enzymes. Drug Metab. Dispos. 2007, 35, 699–703. [Google Scholar] [CrossRef]
- Fernández-Castillejo, S.; Macià, A.; Motilva, M.J.; Catalán, Ú.; Solà, R. Endothelial Cells Deconjugate Resveratrol Metabolites to Free Resveratrol: A Possible Role in Tissue Factor Modulation. Mol. Nutr. Food Res. 2019, 63, e1800715. [Google Scholar] [CrossRef]
- Rojas, C.; Pan-Castillo, B.; Valls, C.; Pujadas, G.; Garcia-Vallve, S.; Arola, L.; Mulero, M. Resveratrol enhances palmitate-induced ER stress and apoptosis in cancer cells. PLoS ONE 2014, 9, e113929. [Google Scholar] [CrossRef] [PubMed]
- Borenfreund, E.; Puerner, J.A. A simple quantitative procedure using monolayer cultures for cytotoxicity assays. J. Tissue Cult. Methods 1984, 9, 7–9. [Google Scholar] [CrossRef]
- Castell-Auví, A.; Cedó, L.; Pallarés, V.; Blay, M.T.; Pinent, M.; Motilva, M.J.; Arola, L.; Ardévol, A. Development of a coculture system to evaluate the bioactivity of plant extracts on pancreatic β-cells. Planta Med. 2010, 76, 1576–1581. [Google Scholar] [CrossRef] [PubMed]
- Plut, C.; Ribière, C.; Giudicelli, Y.; Dausse, J.P. Hypothalamic leptin receptor and signaling molecule expressions in cafeteria diet-fed rats. J. Pharmacol. Exp. Ther. 2003, 307, 544–549. [Google Scholar] [CrossRef]
- Sala, R.; Mena, P.; Savi, M.; Brighenti, F.; Crozier, A.; Miragoli, M.; Stilli, D.; del Rio, D. Urolithins at physiological concentrations affect the levels of pro-inflammatory cytokines and growth factor in cultured cardiac cells in hyperglucidic conditions. J. Funct. Foods. 2015, 15, 97–105. [Google Scholar] [CrossRef]
- Bresciani, L.; Calani, L.; Bocchi, L.; Delucchi, F.; Savi, M.; Ray, S.; Brighenti, F.; Stilli, D.; Del Rio, D. Bioaccumulation of resveratrol metabolites in myocardial tissue is dose-time dependent and related to cardiac hemodynamics in diabetic rats. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 408–415. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ardid-Ruiz, A.; Ibars, M.; Mena, P.; Del Rio, D.; Muguerza, B.; Arola, L.; Aragonès, G.; Suárez, M. Resveratrol Treatment Enhances the Cellular Response to Leptin by Increasing OBRb Content in Palmitate-Induced Steatotic HepG2 Cells. Int. J. Mol. Sci. 2019, 20, 6282. https://doi.org/10.3390/ijms20246282
Ardid-Ruiz A, Ibars M, Mena P, Del Rio D, Muguerza B, Arola L, Aragonès G, Suárez M. Resveratrol Treatment Enhances the Cellular Response to Leptin by Increasing OBRb Content in Palmitate-Induced Steatotic HepG2 Cells. International Journal of Molecular Sciences. 2019; 20(24):6282. https://doi.org/10.3390/ijms20246282
Chicago/Turabian StyleArdid-Ruiz, Andrea, Maria Ibars, Pedro Mena, Daniele Del Rio, Begoña Muguerza, Lluís Arola, Gerard Aragonès, and Manuel Suárez. 2019. "Resveratrol Treatment Enhances the Cellular Response to Leptin by Increasing OBRb Content in Palmitate-Induced Steatotic HepG2 Cells" International Journal of Molecular Sciences 20, no. 24: 6282. https://doi.org/10.3390/ijms20246282
APA StyleArdid-Ruiz, A., Ibars, M., Mena, P., Del Rio, D., Muguerza, B., Arola, L., Aragonès, G., & Suárez, M. (2019). Resveratrol Treatment Enhances the Cellular Response to Leptin by Increasing OBRb Content in Palmitate-Induced Steatotic HepG2 Cells. International Journal of Molecular Sciences, 20(24), 6282. https://doi.org/10.3390/ijms20246282