Pre-Treatment with Ten-Minute Carbon Dioxide Inhalation Prevents Lipopolysaccharide-Induced Lung Injury in Mice via Down-Regulation of Toll-Like Receptor 4 Expression

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
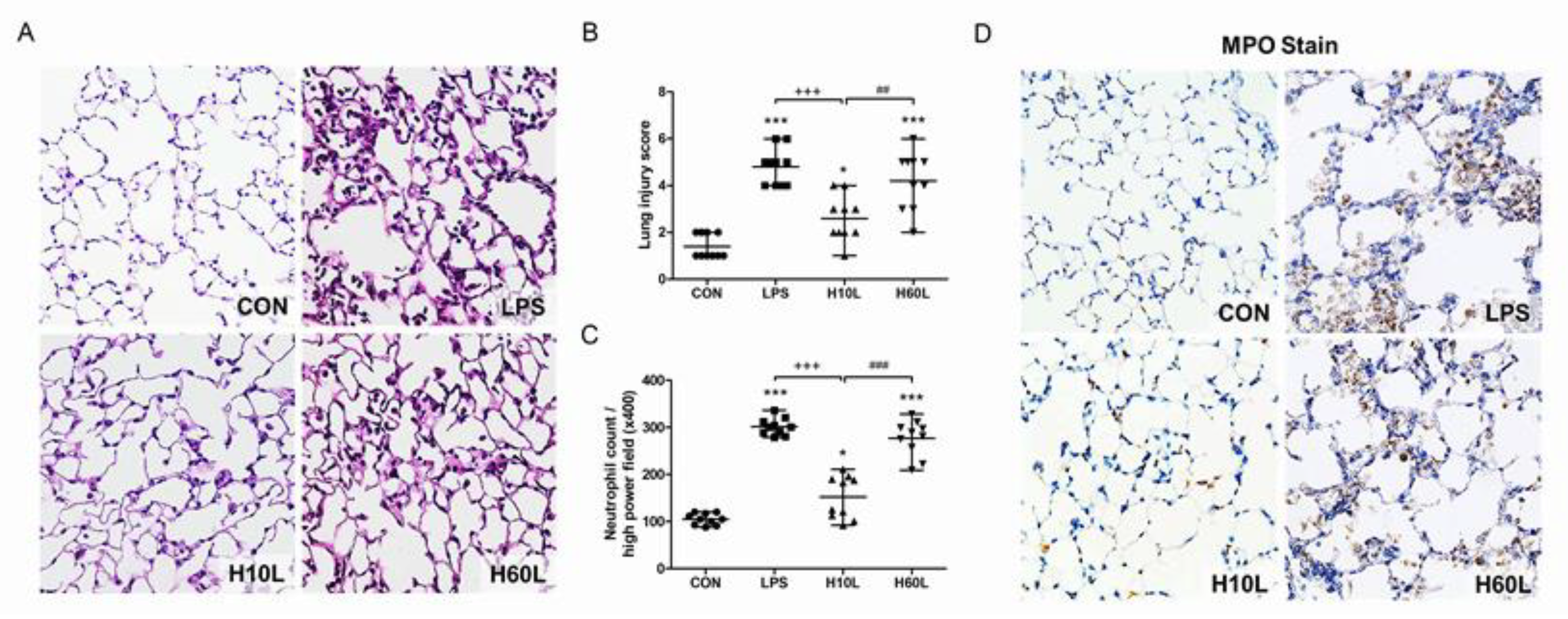
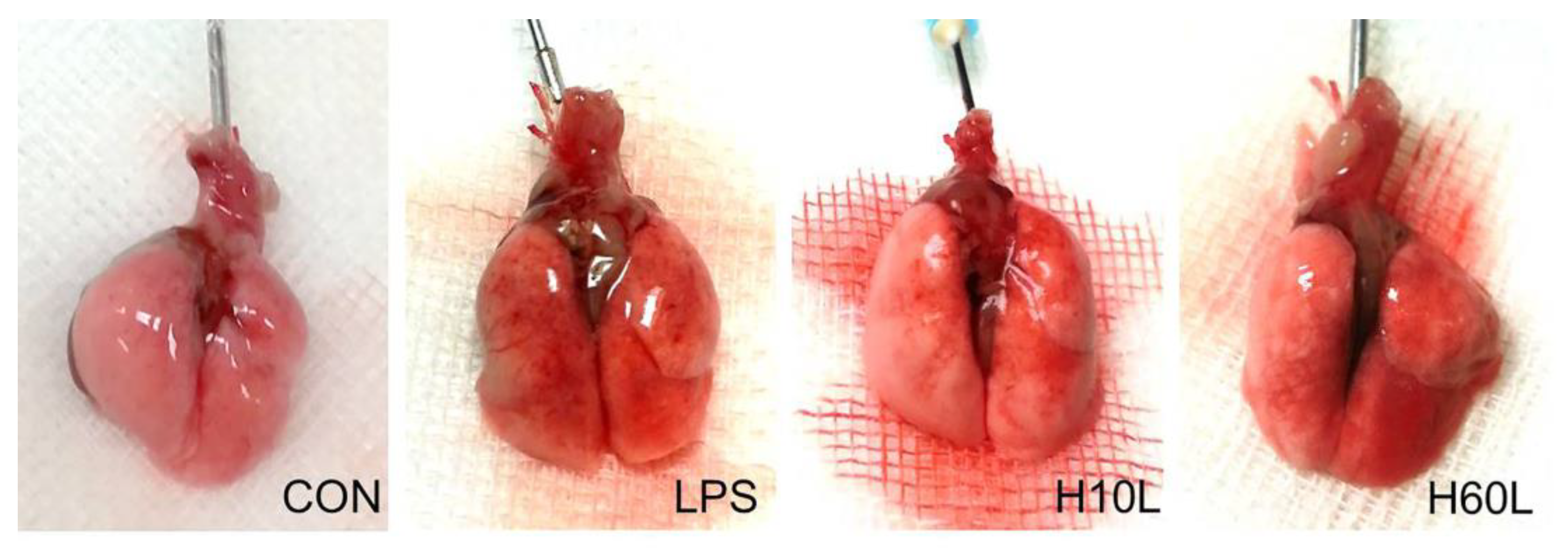
2.1. Pre-Treatment with Inhaled Carbon Dioxide Suppressed LPS-Induced Weight Loss, Lung Injury, and Indicators of Oxidative Stress
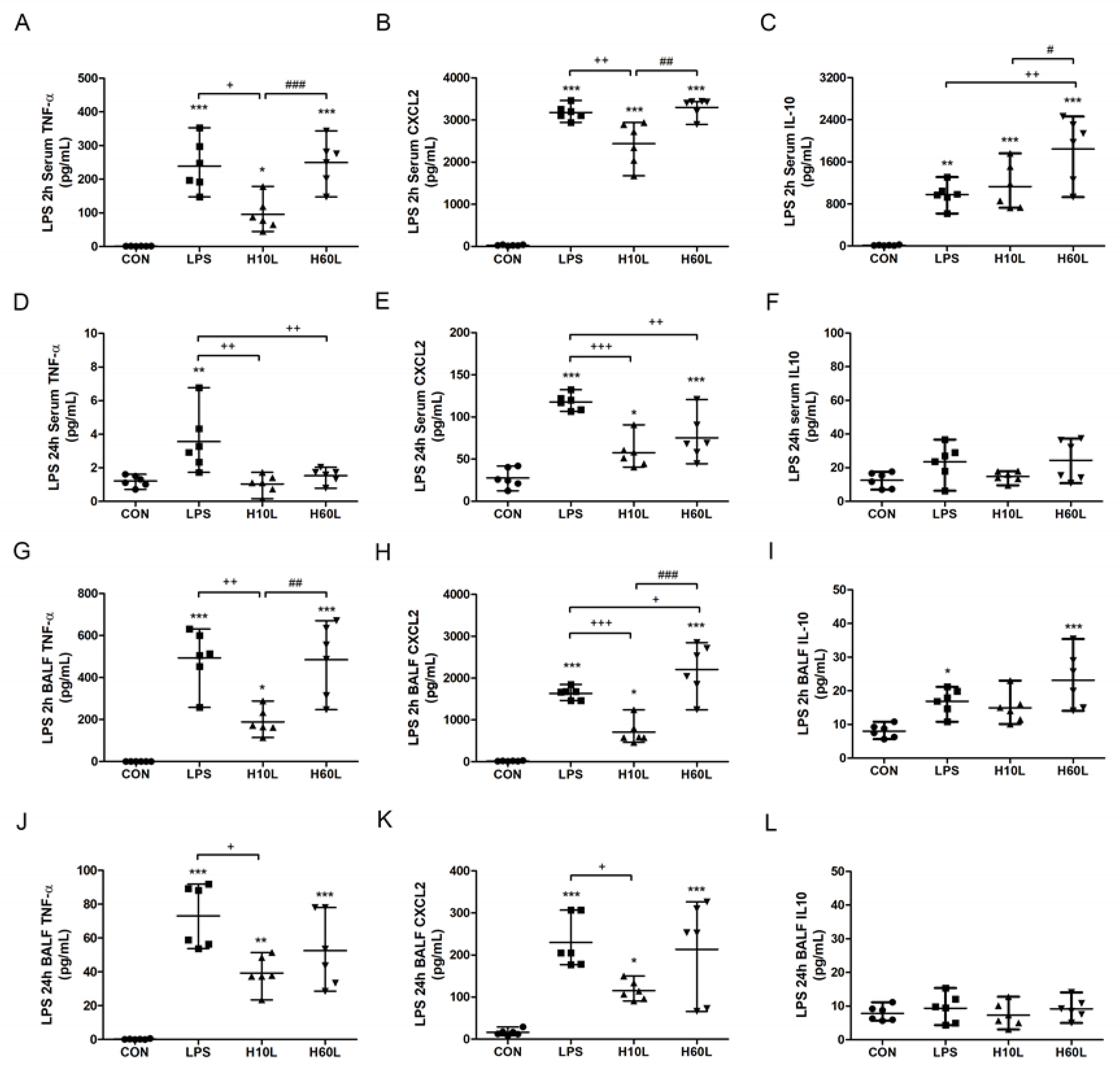
2.2. Pre-Treatment with Inhaled Carbon Dioxide Reduced LPS-Induced Inflammatory Cytokine Levels
2.3. Pre-Treatment with Inhaled Carbon Dioxide Suppressed LPS-Induced Tissue Damage and Inflammatory Cell Infiltration
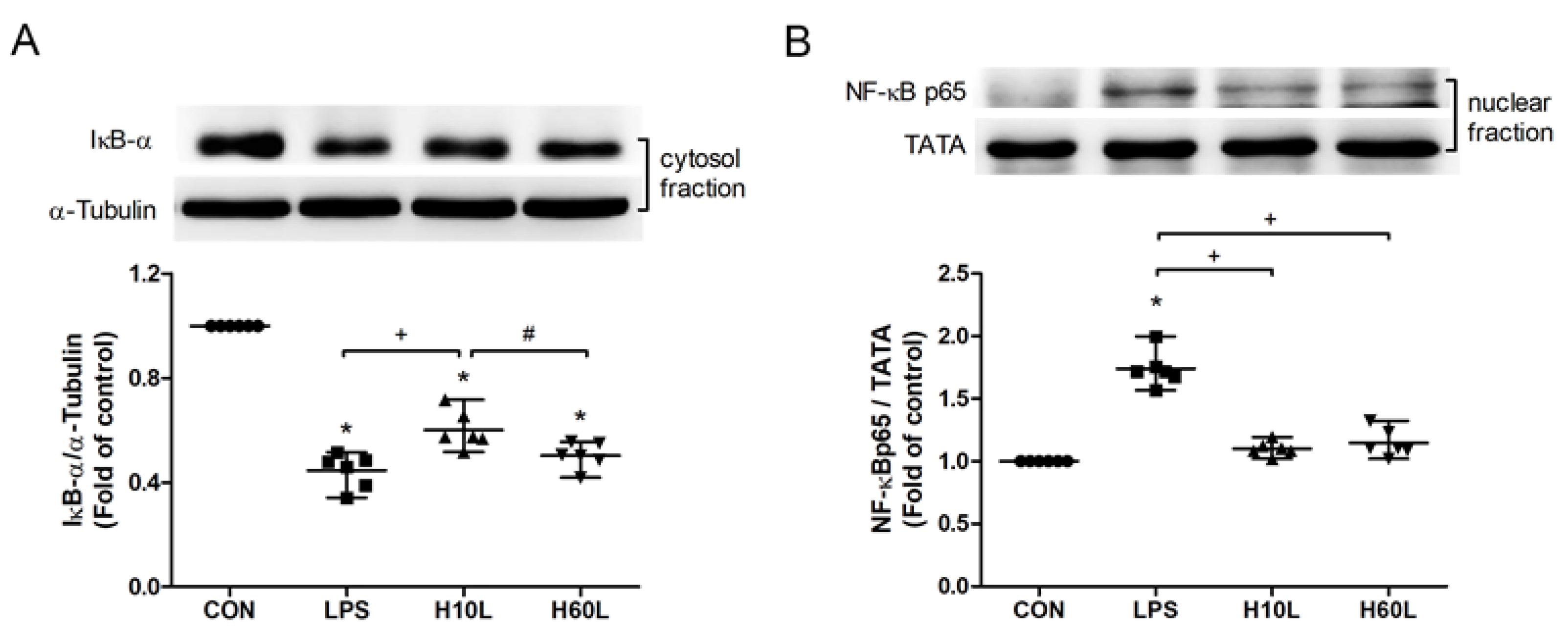
2.4. Inhaled Carbon Dioxide Pre-Treatment Inhibited LPS-Induced NF-κB Activation
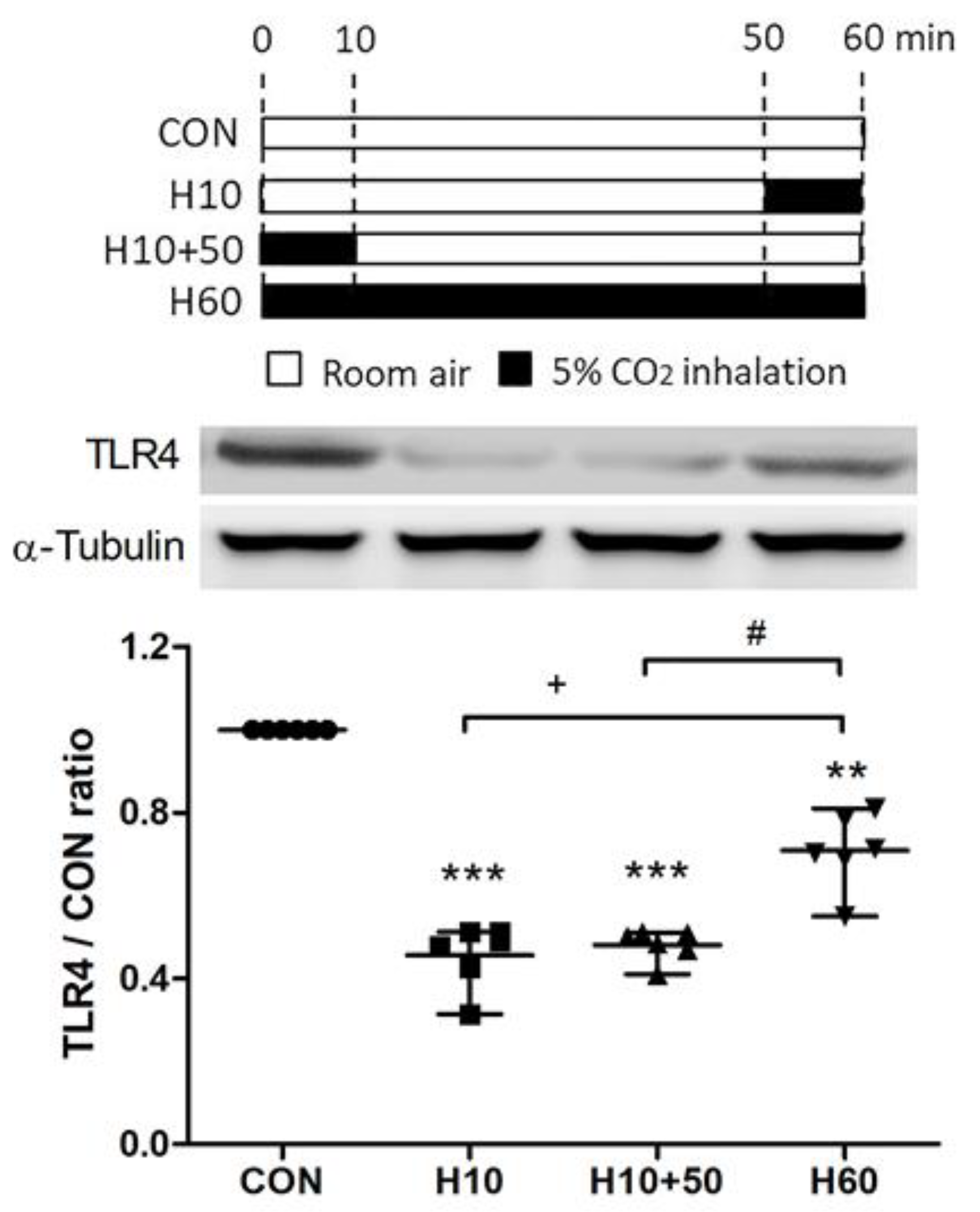
2.5. Inhaled Carbon Dioxide Pre-Treatment and LPS Affected TLR4 Surface Expression
2.6. Inhaled Carbon Dioxide Affected TLR4 Surface Expression
3. Discussion
4. Materials and Methods
4.1. Animals
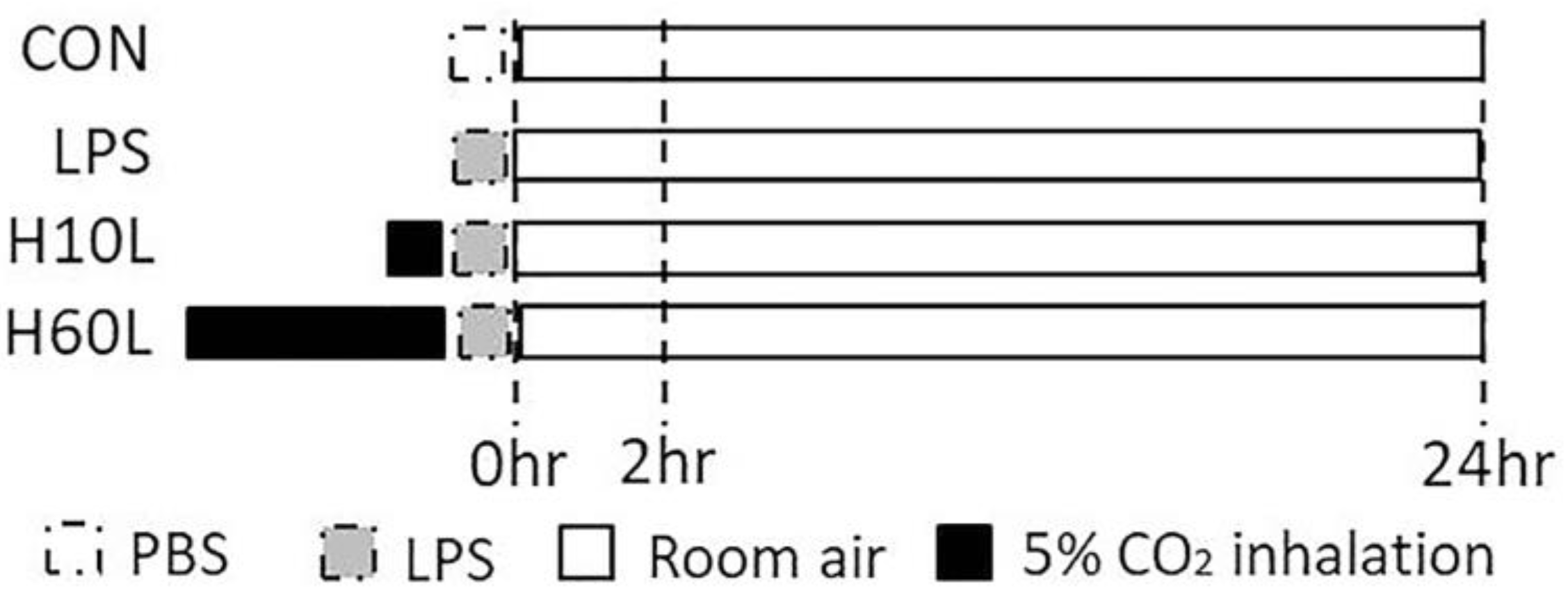
4.2. Experimental Protocols
4.3. Bronchoalveolar Lavage Fluid (BALF) Analysis
4.4. Protein Carbonyl Levels, Cytokine, and Chemokine Levels
4.5. Histopathological Analysis
4.6. Immunohistochemistry Staining for Myeloperoxidase (MPO) and Toll-Like Receptor 4 (TLR4)
4.7. Immunoblotting
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Umbrello, M.; Formenti, P.; Bolgiaghi, L.; Chiumello, D. Current Concepts of ARDS: A Narrative Review. Int. J. Mol. Sci. 2017, 18, 64. [Google Scholar] [CrossRef] [PubMed]
- Zilberberg, M.D.; Epstein, S.K. Acute lung injury in the medical ICU: Comorbid conditions, age, etiology, and hospital outcome. Am. J. Respir. Crit. Care Med. 1998, 157, 1159–1164. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.A.; Silverman, H.J. Gram-negative sepsis and the adult respiratory distress syndrome. Clin. Infect. Dis. 1992, 14, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- De Haro, C.; Martin-Loeches, I.; Torrents, E.; Artigas, A. Acute respiratory distress syndrome: Prevention and early recognition. Ann. Intensive Care 2013, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Levitt, J.E.; Matthay, M.A. Clinical review: Early treatment of acute lung injury—Paradigm shift toward prevention and treatment prior to respiratory failure. Crit. Care 2012, 16, 223. [Google Scholar] [CrossRef] [PubMed]
- Morales-Quinteros, L.; Camprubi-Rimblas, M.; Bringue, J.; Bos, L.D.; Schultz, M.J.; Artigas, A. The role of hypercapnia in acute respiratory failure. Intensive Care Med. Exp. 2019, 7, 39–50. [Google Scholar] [CrossRef]
- Morales Quinteros, L.; Bringue Roque, J.; Kaufman, D.; Artigas Raventos, A. Importance of carbon dioxide in the critical patient: Implications at the cellular and clinical levels. Med. Intensiva 2019, 43, 234–242. [Google Scholar] [CrossRef]
- Hickling, K.G.; Henderson, S.J.; Jackson, R. Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intensive Care Med. 1990, 16, 372–377. [Google Scholar] [CrossRef]
- Hickling, K.G.; Walsh, J.; Henderson, S.; Jackson, R. Low mortality rate in adult respiratory distress syndrome using low-volume, pressure-limited ventilation with permissive hypercapnia: A prospective study. Crit. Care Med. 1994, 22, 1568–1578. [Google Scholar] [CrossRef]
- Amato, M.B.P.; Barbas, C.S.V.; Medeiros, D.M.; Magaldi, R.B.; Schettino, G.P.; Lorenzi-Filho, G.; Kairalla, R.A.; Deheinzelin, D.; Munoz, C.; Oliveira, R.; et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N. Engl. J. Med. 1998, 338, 347–354. [Google Scholar] [CrossRef]
- Acute Respiratory Distress Syndrome Network; Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar]
- Shibata, K.; Cregg, N.; Engelberts, D.; Takeuchi, A.; Fedorko, L.; Kavanagh, B.P. Hypercapnic acidosis may attenuate acute lung injury by inhibition of endogenous xanthine oxidase. Am. J. Respir. Crit. Care Med. 1998, 158, 1578–1584. [Google Scholar] [CrossRef] [PubMed]
- Laffey, J.G.; Honan, D.; Hopkins, N.; Hyvelin, J.M.; Boylan, J.F.; McLoughlin, P. Hypercapnic acidosis attenuates endotoxin-induced acute lung injury. Am. J. Respir. Crit. Care Med. 2004, 169, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Ni Chonghaile, M.; Higgins, B.D.; Costello, J.F.; Laffey, J.G. Hypercapnic acidosis attenuates severe acute bacterial pneumonia-induced lung injury by a neutrophil-independent mechanism. Crit. Care Med. 2008, 36, 3135–3144. [Google Scholar] [CrossRef] [PubMed]
- Chonghaile, M.N.; Higgins, B.D.; Costello, J.; Laffey, J.G. Hypercapnic acidosis attenuates lung injury induced by established bacterial pneumonia. Anesthesiology. 2008, 109, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.; Higgins, B.; Contreras, M.; Chonghaile, M.N.; Hassett, P.; O’Toole, D.; Laffey, J.G. Hypercapnic acidosis attenuates shock and lung injury in early and prolonged systemic sepsis. Crit. Care Med. 2009, 37, 2412–2420. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Su, F.; Bruhn, A.; Yang, X.; Vincent, J.L. Acute hypercapnia improves indices of tissue oxygenation more than dobutamine in septic shock. Am. J. Respir. Crit. Care Med. 2008, 177, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Laffey, J.G.; Tanaka, M.; Engelberts, D.; Luo, X.; Yuan, S.; Tanswell, A.K.; Post, M.; Lindsay, T.; Kavanagh, B.P. Therapeutic hypercapnia reduces pulmonary and systemic injury following in vivo lung reperfusion. Am. J. Respir. Crit. Care Med. 2000, 162, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Laffey, J.G.; Jankov, R.P.; Engelberts, D.; Tanswell, A.K.; Post, M.; Lindsay, T.; Mullen, J.B.; Romaschin, A.; Stephens, D.; McKerlie, C.; et al. Effects of therapeutic hypercapnia on mesenteric ischemia-reperfusion injury. Am. J. Respir. Crit. Care Med. 2003, 168, 1383–1390. [Google Scholar] [CrossRef]
- Wu, S.Y.; Wu, C.P.; Kang, B.H.; Li, M.H.; Chu, S.J.; Huang, K.L. Hypercapnic acidosis attenuates reperfusion injury in isolated and perfused rat lungs. Crit. Care Med. 2012, 40, 553–559. [Google Scholar] [CrossRef]
- Wu, S.Y.; Li, M.H.; Ko, F.C.; Wu, G.C.; Huang, K.L.; Chu, S.J. Protective effect of hypercapnic acidosis in ischemia-reperfusion lung injury is attributable to upregulation of heme oxygenase-1. PLoS ONE 2013, 8, e74742. [Google Scholar] [CrossRef] [PubMed]
- Laffey, J.G.; Engelberts, D.; Duggan, M.; Veldhuizen, R.; Lewis, J.F.; Kavanagh, B.P. Carbon dioxide attenuates pulmonary impairment resulting from hyperventilation. Crit. Care Med. 2003, 31, 2634–2640. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.C.; Song, C.Y.; Wang, N.; Zhang, L.L.; Yue, Z.Y.; Cui, X.G.; Zhou, H.C. Hypercapnic acidosis confers antioxidant and anti-apoptosis effects against ventilator-induced lung injury. Lab. Investig. 2013, 93, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Contreras, M.; Ansari, B.; Curley, G.; Higgins, B.D.; Hassett, P.; O’Toole, D.; Laffey, J.G. Hypercapnic acidosis attenuates ventilation-induced lung injury by a nuclear factor-kappaB-dependent mechanism. Crit. Care Med. 2012, 40, 2622–2630. [Google Scholar] [CrossRef] [PubMed]
- O’Croinin, D.F.; Nichol, A.D.; Hopkins, N.; Boylan, J.; O’Brien, S.; O’Connor, C.; Laffey, J.G.; McLoughlin, P. Sustained hypercapnic acidosis during pulmonary infection increases bacterial load and worsens lung injury. Crit. Care Med. 2008, 36, 2128–2135. [Google Scholar] [CrossRef]
- Dong, L.; Krewson, E.A.; Yang, L.V. Acidosis Activates Endoplasmic Reticulum Stress Pathways through GPR4 in Human Vascular Endothelial Cells. Int. J. Mol. Sci. 2017, 18, 278. [Google Scholar] [CrossRef]
- Doerr, C.H.; Gajic, O.; Berrios, J.C.; Caples, S.; Abdel, M.; Lymp, J.F.; Hubmayr, R.D. Hypercapnic acidosis impairs plasma membrane wound resealing in ventilator-injured lungs. Am. J. Respir. Crit. Care Med. 2005, 171, 1371–1377. [Google Scholar] [CrossRef]
- Nin, N.; Muriel, A.; Penuelas, O.; Brochard, L.; Lorente, J.A.; Ferguson, N.D.; Raymondos, K.; Rios, F.; Violi, D.A.; Thille, A.W.; et al. Severe hypercapnia and outcome of mechanically ventilated patients with moderate or severe acute respiratory distress syndrome. Intensive Care Med. 2017, 43, 200–208. [Google Scholar] [CrossRef]
- Tiruvoipati, R.; Pilcher, D.; Buscher, H.; Botha, J.; Bailey, M. Effects of Hypercapnia and Hypercapnic Acidosis on Hospital Mortality in Mechanically Ventilated Patients. Crit. Care Med. 2017, 45, e649–e656. [Google Scholar] [CrossRef]
- Nemzek, J.A.; Call, D.R.; Ebong, S.J.; Newcomb, D.E.; Bolgos, G.L.; Remick, D.G. Immunopathology of a two-hit murine model of acid aspiration lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L512–L520. [Google Scholar] [CrossRef]
- Pavord, I.D.; Birring, S.S.; Berry, M.; Green, R.H.; Brightling, C.E.; Wardlaw, A.J. Multiple inflammatory hits and the pathogenesis of severe airway disease. Eur. Respir. J. 2006, 27, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Szleifer, I.; Beitel, G.J.; Sznajder, J.I.; Sporn, P.H.S. Hypercapnia Alters Expression of Immune Response, Nucleosome Assembly and Lipid Metabolism Genes in Differentiated Human Bronchial Epithelial Cells. Sci. Rep. 2018, 8, 13508–13518. [Google Scholar]
- Wang, N.; Gates, K.L.; Trejo, H.; Favoreto, S., Jr.; Schleimer, R.P.; Sznajder, J.I.; Beitel, G.J.; Sporn, P.H.S. Elevated CO2 selectively inhibits interleukin-6 and tumor necrosis factor expression and decreases phagocytosis in the macrophage. FASEB J. 2010, 24, 2178–2190. [Google Scholar] [CrossRef] [PubMed]
- Gates, K.L.; Howell, H.A.; Nair, A.; Vohwinkel, C.U.; Welch, L.C.; Beitel, G.J.; Hauser, A.R.; Sznajder, J.I.; Sporn, P.H. Hypercapnia impairs lung neutrophil function and increases mortality in murine pseudomonas pneumonia. Am. J. Respir. Cell. Mol. Biol. 2013, 49, 821–828. [Google Scholar] [CrossRef] [PubMed]
- O’Croinin, D.F.; Hopkins, N.O.; Moore, M.M.; Boylan, J.F.; McLoughlin, P.; Laffey, J.G. Hypercapnic acidosis does not modulate the severity of bacterial pneumonia-induced lung injury. Crit. Care Med. 2005, 33, 2606–2612. [Google Scholar] [CrossRef]
- Abolhassani, M.; Guais, A.; Chaumet-Riffaud, P.; Sasco, A.J.; Schwartz, L. Carbon dioxide inhalation causes pulmonary inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L657–L665. [Google Scholar] [CrossRef]
- Laserna, E.; Sibila, O.; Aguilar, P.R.; Mortensen, E.M.; Anzueto, A.; Blanquer, J.M.; Sanz, F.; Rello, J.; Marcos, P.J.; Velez, M.I.; et al. Hypocapnia and hypercapnia are predictors for ICU admission and mortality in hospitalized patients with community-acquired pneumonia. Chest 2012, 142, 1193–1199. [Google Scholar] [CrossRef]
- Minet, C.; Vivodtzev, I.; Tamisier, R.; Arbib, F.; Wuyam, B.; Timsit, J.F.; Monneret, D.; Borel, J.C.; Baguet, J.P.; Lévy, P.; et al. Reduced six-minute walking distance, high fat-free-mass index and hypercapnia are associated with endothelial dysfunction in COPD. Respir. Physiol. Neurobiol. 2012, 183, 128–134. [Google Scholar] [CrossRef]
- Costello, R.; Deegan, P.; Fitzpatrick, M.; McNicholas, W.T. Reversible hypercapnia in chronic obstructive pulmonary disease: A distinct pattern of respiratory failure with a favorable prognosis. Am. J. Med. 1997, 102, 239–244. [Google Scholar] [CrossRef]
- Connors, A.F., Jr.; Dawson, N.V.; Thomas, C.; Harrell, F.E., Jr.; Desbiens, N.; Fulkerson, W.J.; Kussin, P.; Bellamy, P.; Goldman, L.; Knaus, W.A. Outcomes following acute exacerbation of severe chronic obstructive lung disease. The SUPPORT investigators (Study to Understand Prognoses and Preferences for Outcomes and Risks of Treatments). Am. J. Respir. Crit. Care Med. 1996, 154, 959–967. [Google Scholar] [CrossRef]
- Bezemer, G.F.; Sagar, S.; Van Bergenhenegouwen, J.; Georgiou, N.A.; Garssen, J.; Kraneveld, A.D.; Folkerts, G. Dual role of Toll-like receptors in asthma and chronic obstructive pulmonary disease. Pharmacol. Rev. 2012, 64, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, T.J.; Paik, J.H.; Chung, J.H.; Jheon, S.; Huh, J.W.; Lee, J.H.; Lee, C.T. The association of down-regulated toll-like receptor 4 expression with airflow limitation and emphysema in smokers. Respir. Res. 2012, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi-Filho, G.; Rankin, F.; Bies, I.; Douglas Bradley, T. Effects of inhaled carbon dioxide and oxygen on cheyne-stokes respiration in patients with heart failure. Am. J. Respir. Crit. Care Med. 1999, 159, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Badr, M.S.; Grossman, J.E.; Weber, S.A. Treatment of refractory sleep apnea with supplemental carbon dioxide. Am. J. Respir. Crit. Care Med. 1994, 150, 561–564. [Google Scholar] [CrossRef]
- Wan, Z.H.; Wen, F.J.; Hu, K. Dynamic CO2 inhalation: A novel treatment for CSR-CSA associated with CHF. Sleep Breath. 2013, 17, 487–493. [Google Scholar] [CrossRef]
- Mebrate, Y.; Willson, K.; Manisty, C.H.; Baruah, R.; Mayet, J.; Hughes, A.D.; Parker, K.H.; Francis, D.P. Dynamic CO2 therapy in periodic breathing: A modeling study to determine optimal timing and dosage regimes. J. Appl. Physiol. 2009, 107, 696–706. [Google Scholar] [CrossRef][Green Version]
- Sakata, D.J.; Gopalakrishnan, N.A.; Orr, J.A.; White, J.L.; Westenskow, D.R. Hypercapnic hyperventilation shortens emergence time from isoflurane anesthesia. Anesth. Analg. 2007, 104, 587–591. [Google Scholar] [CrossRef]
- Ohlraun, S.; Wollersheim, T.; Weiß, C.; Martus, P.; Weber-Carstens, S.; Schmitz, D.; Schuelke, M. CARbon DIoxide for the treatment of Febrile seizures: Rationale, feasibility, and design of the CARDIF-study. J. Transl. Med. 2013, 11, 157. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, S.-E.; Wu, S.-Y.; Chu, S.-J.; Tzeng, Y.-S.; Peng, C.-K.; Lan, C.-C.; Perng, W.-C.; Wu, C.-P.; Huang, K.-L. Pre-Treatment with Ten-Minute Carbon Dioxide Inhalation Prevents Lipopolysaccharide-Induced Lung Injury in Mice via Down-Regulation of Toll-Like Receptor 4 Expression. Int. J. Mol. Sci. 2019, 20, 6293. https://doi.org/10.3390/ijms20246293
Tang S-E, Wu S-Y, Chu S-J, Tzeng Y-S, Peng C-K, Lan C-C, Perng W-C, Wu C-P, Huang K-L. Pre-Treatment with Ten-Minute Carbon Dioxide Inhalation Prevents Lipopolysaccharide-Induced Lung Injury in Mice via Down-Regulation of Toll-Like Receptor 4 Expression. International Journal of Molecular Sciences. 2019; 20(24):6293. https://doi.org/10.3390/ijms20246293
Chicago/Turabian StyleTang, Shih-En, Shu-Yu Wu, Shi-Jye Chu, Yuan-Sheng Tzeng, Chung-Kan Peng, Chou-Chin Lan, Wann-Cherng Perng, Chin-Pyng Wu, and Kun-Lun Huang. 2019. "Pre-Treatment with Ten-Minute Carbon Dioxide Inhalation Prevents Lipopolysaccharide-Induced Lung Injury in Mice via Down-Regulation of Toll-Like Receptor 4 Expression" International Journal of Molecular Sciences 20, no. 24: 6293. https://doi.org/10.3390/ijms20246293
APA StyleTang, S.-E., Wu, S.-Y., Chu, S.-J., Tzeng, Y.-S., Peng, C.-K., Lan, C.-C., Perng, W.-C., Wu, C.-P., & Huang, K.-L. (2019). Pre-Treatment with Ten-Minute Carbon Dioxide Inhalation Prevents Lipopolysaccharide-Induced Lung Injury in Mice via Down-Regulation of Toll-Like Receptor 4 Expression. International Journal of Molecular Sciences, 20(24), 6293. https://doi.org/10.3390/ijms20246293