The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process


Abstract
:1. Introduction
2. Plasma Membrane Ca2+-ATPase in Neuronal Cells
3. Differentiated PC12 Cells as a Model of Aging Neuron
4. The Interplay between PMCA, Calcium, and Mitochondrial Function during Aging
5. PMCA and Ca2+-Regulated Proteins—CaM, GAP43, CaN in Ageing
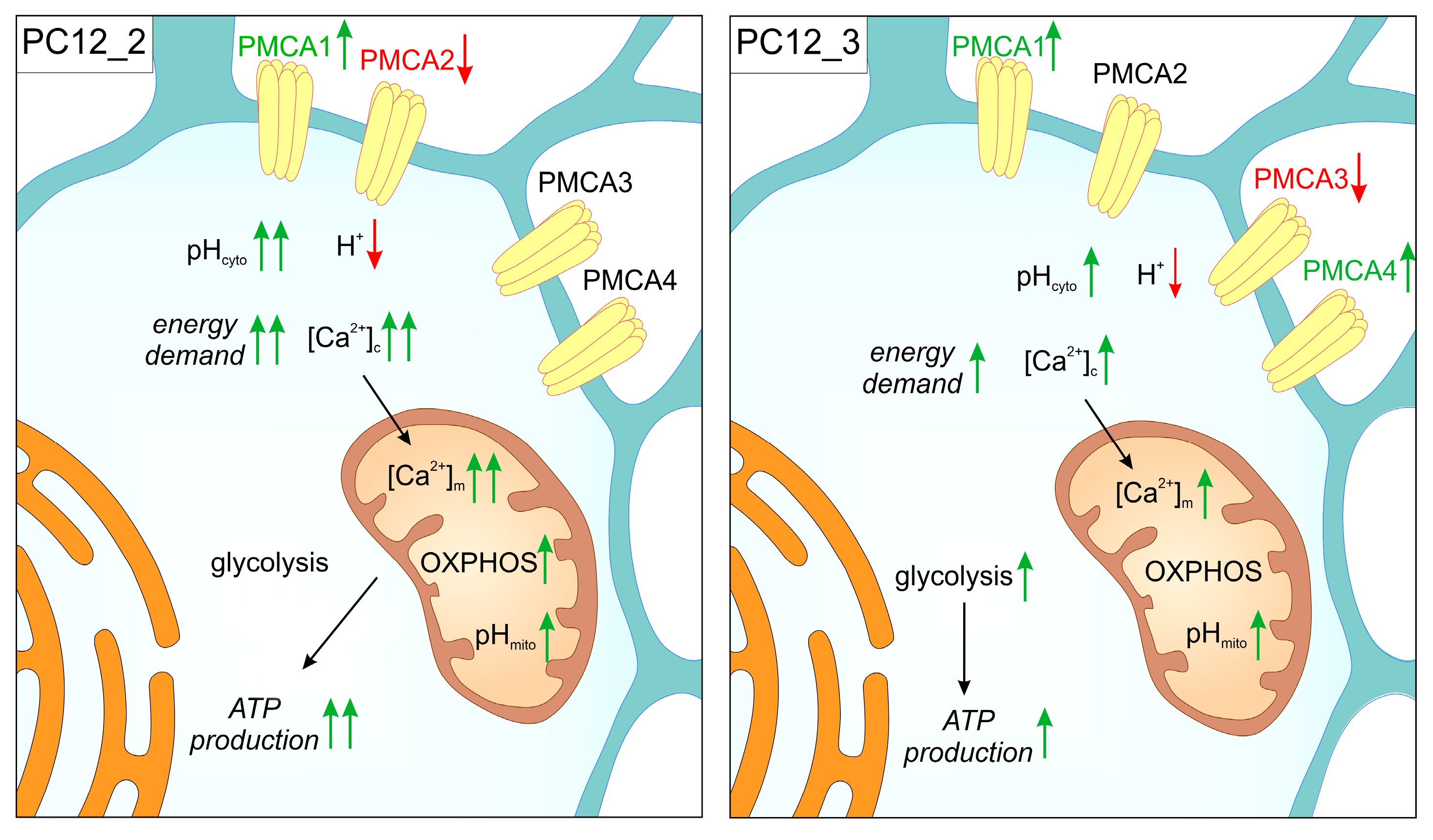
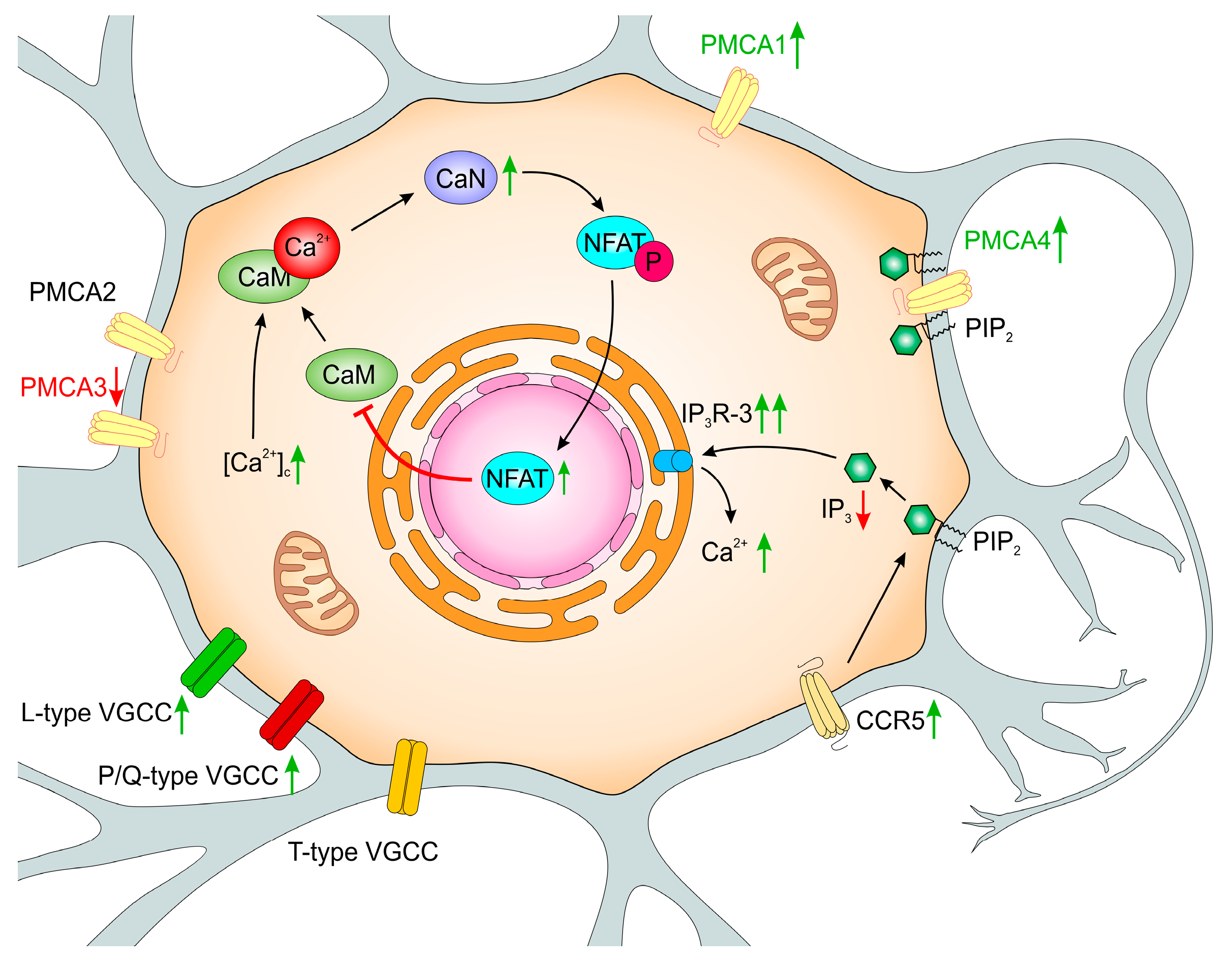
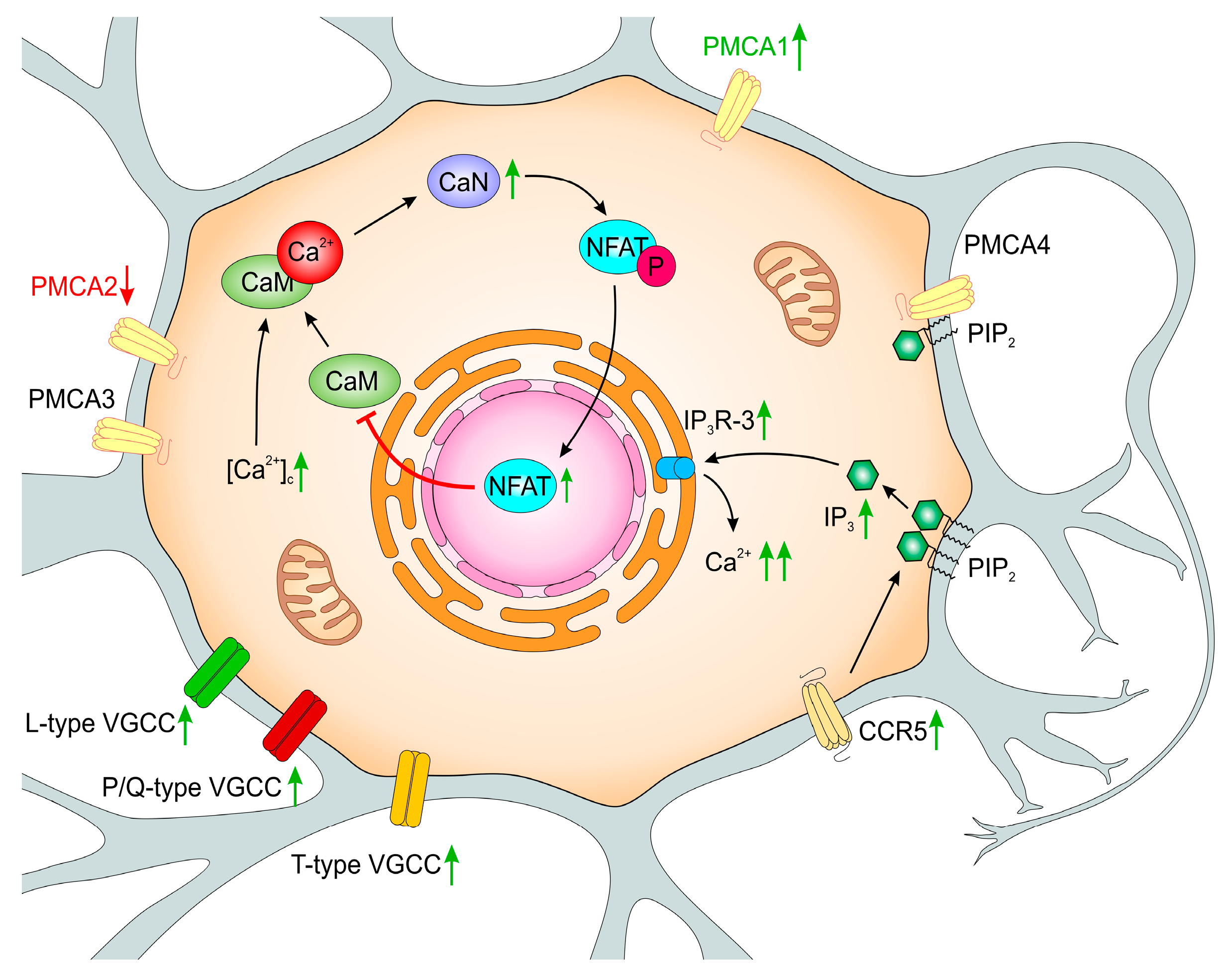
6. PMCA-Deficient PC12 Cells as a Tool for Modeling Functional Adaptive Strategies in Neurons during Aging
7. Conclusions
- intracellular pH regulation and changes in the mitochondrial bioenergetic processes,
- adaptive functional changes in the VGCC and in Ca2+ transport systems to the ER,
- regulation of CaM availability by Ca2+/CaN-dependent complex formation with GAP43,
- reduced CaM level and repressive regulation of Calm2 and Calm3 genes by NFATc2,
- altered expression of CCL5-sensitive receptors—CCR1, CCR3, and CCR5,
- altered expression of IP3 receptors.
Author Contributions
Funding
Conflicts of Interest
References
- Chandran, R.; Kumar, M.; Kesavan, L.; Jacob, R.S.; Gunasekaran, S.; Lakshmi, S.; Sadasivan, C.; Omkumar, R.V. Cellular calcium signaling in the aging brain. J. Chem. Neuroanat. 2019, 95, 95–114. [Google Scholar] [CrossRef] [PubMed]
- Squier, T.C.; Bigelow, D.J. Protein oxidation and age-dependent alterations in calcium homeostasis. Front. Biosci. 2000, 5, D504–D526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toescu, E.C.; Vreugdenhil, M. Calcium and normal brain ageing. Cell Calcium 2010, 47, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Gladyshev, V.N. Role of reactive oxygen species-mediated signaling in aging. Antioxid. Redox. Signal. 2013, 19, 1362–1372. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Yegla, B.; Foster, T.C. Redox Signaling in Neurotransmission and Cognition During Aging. Antioxid. Redox. Signal. 2018, 28, 1724–1745. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A. Calcium Signaling During Brain Aging and Its Influence on the Hippocampal Synaptic Plasticity. Adv. Exp. Med. Biol. 2020, 1131, 985–1012. [Google Scholar]
- Nikoletopoulou, V.; Tavernarakis, N. Calcium homeostasis in aging neurons. Front. Genet. 2012, 3, 200. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Bodhinathan, K.; Foster, T.C. Susceptibility to Calcium Dysregulation during Brain Aging. Front. Aging. Neurosci. 2009, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, E.M.; Vivar, C.; Camandola, S. Physiology and pathology of calcium signaling in the brain. Front. Pharmacol. 2012, 3, 61. [Google Scholar] [CrossRef] [Green Version]
- Lopreiato, R.; Giacomello, M.; Carafoli, E. The plasma membrane calcium pump: New ways to look at an old enzyme. J. Biol. Chem. 2014, 289, 10261–10268. [Google Scholar] [CrossRef] [Green Version]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E.J. The Plasma Membrane Calcium ATPases and Their Role as Major New Players in Human Disease. Physiol. Rev. 2017, 97, 1089–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunter, T.E.; Buntinas, L.; Sparagna, G.; Eliseev, R.; Gunter, K. Mitochondrial calcium transport: Mechanisms and functions. Cell Calcium 2000, 28, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Frazier, H.N.; Maimaiti, S.; Anderson, K.L.; Brewer, L.D.; Gant, J.C.; Porter, N.M.; Thibault, O. Calcium’s role as nuanced modulator of cellular physiology in the brain. Biochem. Biophys. Res. Commun. 2017, 483, 981–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Court, F.A.; Cardenas, C. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated With Aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- Akbar, M.; Essa, M.M.; Daradkeh, G.; Abdelmegeed, M.A.; Choi, Y.; Mahmood, L.; Song, B.J. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 2016, 1637, 34–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Wu, S.B.; Wu, Y.T.; Wei, Y.H. Oxidative stress response elicited by mitochondrial dysfunction: Implication in the pathophysiology of aging. Exp. Biol. Med. 2013, 238, 450–460. [Google Scholar] [CrossRef]
- Barja, G. Updating the mitochondrial free radical theory of aging: An integrated view, key aspects, and confounding concepts. Antioxid. Redox. Signal. 2013, 19, 1420–1445. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. The Free Radical Theory of Aging: Effect of Age on Serum Copper Levels. J. Gerontol. 1965, 20, 151–153. [Google Scholar] [CrossRef]
- Strehler, E.E.; Zacharias, D.A. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 2001, 81, 21–50. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Carafoli, E. Calcium pumps in health and disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef] [Green Version]
- Burette, A.; Rockwood, J.M.; Strehler, E.E.; Weinberg, R.J. Isoform-specific distribution of the plasma membrane Ca2+ ATPase in the rat brain. J. Comp. Neurol. 2003, 467, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, F.; Domi, T.; Fedrizzi, L.; Lim, D.; Carafoli, E. The plasma membrane Ca2+ ATPase of animal cells: Structure, function and regulation. Arch. Biochem. Biophys. 2008, 476, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E. The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb. Perspect. Biol. 2011, 3, a004168. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, T.P.; Hilfiker, H.; Carafoli, E.; Strehler, E.E. Quantitative analysis of alternative splicing options of human plasma membrane calcium pump genes. J. Biol. Chem. 1993, 268, 25993–26003. [Google Scholar]
- Stauffer, T.P.; Guerini, D.; Carafoli, E. Tissue distribution of the four gene products of the plasma membrane Ca2+ pump. A study using specific antibodies. J. Biol. Chem. 1995, 270, 12184–12190. [Google Scholar] [CrossRef] [Green Version]
- Zacharias, D.A.; Kappen, C. Developmental expression of the four plasma membrane calcium ATPase (PMCA) genes in the mouse. Biochim. Biophys. Acta 1999, 1428, 397–405. [Google Scholar] [CrossRef]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life. Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Sepulveda, M.R.; Hidalgo-Sanchez, M.; Marcos, D.; Mata, A.M. Developmental distribution of plasma membrane Ca2+-ATPase isoforms in chick cerebellum. Dev. Dyn. 2007, 236, 1227–1236. [Google Scholar] [CrossRef]
- Kip, S.N.; Gray, N.W.; Burette, A.; Canbay, A.; Weinberg, R.J.; Strehler, E.E. Changes in the expression of plasma membrane calcium extrusion systems during the maturation of hippocampal neurons. Hippocampus 2006, 16, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Bechtel, M.D.; Galeva, N.A.; Williams, T.D.; Michaelis, E.K.; Michaelis, M.L. Decreases in plasma membrane Ca2+-ATPase in brain synaptic membrane rafts from aged rats. J. Neurochem. 2012, 123, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, A.; Gao, J.; Squier, T.C.; Michaelis, M.L. Age-related decrease in brain synaptic membrane Ca2+-ATPase in F344/BNF1 rats. Neurobiol. Aging. 1998, 19, 487–495. [Google Scholar] [CrossRef]
- Michaelis, M.L.; Bigelow, D.J.; Schoneich, C.; Williams, T.D.; Ramonda, L.; Yin, D.; Huhmer, A.F.; Yao, Y.; Gao, J.; Squier, T.C. Decreased plasma membrane calcium transport activity in aging brain. Life Sci. 1996, 59, 405–412. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Chen, X.; Krainev, A.G.; Michaelis, E.K.; Bigelow, D.J. Protein half-lives of calmodulin and the plasma membrane Ca-ATPase in rat brain. Biochem. Biophys. Res. Commun. 1997, 237, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A. Plasma membrane Ca-ATPases: Targets of oxidative stress in brain aging and neurodegeneration. World J. Biol. Chem. 2010, 1, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.; Michaelis, M.L. Effects of reactive oxygen species on brain synaptic plasma membrane Ca2+-ATPase. Free Radic. Biol. Med. 1999, 27, 810–821. [Google Scholar] [CrossRef]
- Kip, S.N.; Strehler, E.E. Rapid downregulation of NCX and PMCA in hippocampal neurons following H2O2 oxidative stress. Ann. N. Y. Acad. Sci. 2007, 1099, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, M.; De Mario, A.; Lopreiato, R.; Primerano, S.; Campeol, M.; Brini, M.; Carafoli, E. Mutations in PMCA2 and hereditary deafness: A molecular analysis of the pump defect. Cell Calcium 2011, 50, 569–576. [Google Scholar] [CrossRef]
- Cali, T.; Lopreiato, R.; Shimony, J.; Vineyard, M.; Frizzarin, M.; Zanni, G.; Zanotti, G.; Brini, M.; Shinawi, M.; Carafoli, E. A Novel Mutation in Isoform 3 of the Plasma Membrane Ca2+ Pump Impairs Cellular Ca2+ Homeostasis in a Patient with Cerebellar Ataxia and Laminin Subunit 1α Mutations. J. Biol. Chem. 2015, 290, 16132–16141. [Google Scholar] [CrossRef] [Green Version]
- Zanni, G.; Cali, T.; Kalscheuer, V.M.; Ottolini, D.; Barresi, S.; Lebrun, N.; Montecchi-Palazzi, L.; Hu, H.; Chelly, J.; Bertini, E.; et al. Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 14514–14519. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, M.; Marcos, D.; Sepulveda, M.R.; Perez, M.; Avila, J.; Mata, A.M. Altered Ca2+ dependence of synaptosomal plasma membrane Ca2+-ATPase in human brain affected by Alzheimer’s disease. FASEB J. 2009, 23, 1826–1834. [Google Scholar] [CrossRef]
- Berrocal, M.; Sepulveda, M.R.; Vazquez-Hernandez, M.; Mata, A.M. Calmodulin antagonizes amyloid-β peptides-mediated inhibition of brain plasma membrane Ca2+-ATPase. Biochim. Biophys. Acta 2012, 1822, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, M.; Corbacho, I.; Vazquez-Hernandez, M.; Avila, J.; Sepulveda, M.R.; Mata, A.M. Inhibition of PMCA activity by tau as a function of aging and Alzheimer’s neuropathology. Biochim. Biophys. Acta 2015, 1852, 1465–1476. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, M.; Corbacho, I.; Sepulveda, M.R.; Gutierrez-Merino, C.; Mata, A.M. Phospholipids and calmodulin modulate the inhibition of PMCA activity by tau. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 1028–1035. [Google Scholar] [CrossRef]
- Greene, L.A.; Tischler, A.S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef] [Green Version]
- Keller, D.; Grover, A.K. Nerve growth factor treatment alters Ca2+ pump levels in PC12 cells. Neuroreport 2000, 11, 65–68. [Google Scholar] [CrossRef]
- Lambeng, N.; Michel, P.P.; Agid, Y.; Ruberg, M. The relationship between differentiation and survival in PC12 cells treated with cyclic adenosine monophosphate in the presence of epidermal growth factor or nerve growth factor. Neurosci. Lett. 2001, 297, 133–136. [Google Scholar] [CrossRef]
- Hammes, A.; Oberdorf, S.; Strehler, E.E.; Stauffer, T.; Carafoli, E.; Vetter, H.; Neyses, L. Differentiation-specific isoform mRNA expression of the calmodulin-dependent plasma membrane Ca2+-ATPase. FASEB J. 1994, 8, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.L.; Usachev, Y.M.; Thayer, S.A.; Strehler, E.E.; Windebank, A.J. Plasma membrane calcium ATPase plays a role in reducing Ca2+-mediated cytotoxicity in PC12 cells. J. Neurosci. Res. 2001, 64, 661–669. [Google Scholar] [CrossRef]
- Boczek, T.; Lisek, M.; Kowalski, A.; Pikula, S.; Niewiarowska, J.; Wiktorska, M.; Zylinska, L. Downregulation of PMCA2 or PMCA3 reorganizes Ca2+ handling systems in differentiating PC12 cells. Cell Calcium 2012, 52, 433–444. [Google Scholar] [CrossRef]
- Luo, L.; O’Leary, D.D. Axon retraction and degeneration in development and disease. Annu. Rev. Neurosci. 2005, 28, 127–156. [Google Scholar] [CrossRef] [Green Version]
- Baranov, S.V.; Baranova, O.V.; Yablonska, S.; Suofu, Y.; Vazquez, A.L.; Kozai, T.D.Y.; Cui, X.T.; Ferrando, L.M.; Larkin, T.M.; Tyurina, Y.Y.; et al. Mitochondria modulate programmed neuritic retraction. Proc. Natl. Acad. Sci. USA 2019, 116, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, D.; Zaidi, A.; Bean, J.; Hui, D.; Michaelis, M.L. RNAi—Induced silencing of the plasma membrane Ca2+-ATPase 2 in neuronal cells: Effects on Ca2+ homeostasis and cell viability. J. Neurochem. 2007, 102, 454–465. [Google Scholar] [CrossRef]
- Kurnellas, M.P.; Li, H.; Jain, M.R.; Giraud, S.N.; Nicot, A.B.; Ratnayake, A.; Heary, R.F.; Elkabes, S. Reduced expression of plasma membrane calcium ATPase 2 and collapsin response mediator protein 1 promotes death of spinal cord neurons. Cell. Death Differ. 2010, 17, 1501–1510. [Google Scholar] [CrossRef]
- Empson, R.M.; Turner, P.R.; Nagaraja, R.Y.; Beesley, P.W.; Knopfel, T. Reduced expression of the Ca2+ transporter protein PMCA2 slows Ca2+ dynamics in mouse cerebellar Purkinje neurones and alters the precision of motor coordination. J. Physiol. 2010, 588, 907–922. [Google Scholar] [CrossRef]
- Raza, M.; Deshpande, L.S.; Blair, R.E.; Carter, D.S.; Sombati, S.; DeLorenzo, R.J. Aging is associated with elevated intracellular calcium levels and altered calcium homeostatic mechanisms in hippocampal neurons. Neurosci. Lett. 2007, 418, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Mata, A.M.; Sepulveda, M.R. Plasma membrane Ca-ATPases in the nervous system during development and ageing. World J. Biol. Chem. 2010, 1, 229–234. [Google Scholar] [CrossRef]
- Strehler, E.E.; Thayer, S.A. Evidence for a role of plasma membrane calcium pumps in neurodegenerative disease: Recent developments. Neurosci. Lett. 2018, 663, 39–47. [Google Scholar] [CrossRef]
- Carafoli, E.; Brini, M. Calcium pumps: Structural basis for and mechanism of calcium transmembrane transport. Curr. Opin. Chem. Biol. 2000, 4, 152–161. [Google Scholar] [CrossRef]
- Mata, A.M.; Sepulveda, M.R. Calcium pumps in the central nervous system. Brain Res. Rev. 2005, 49, 398–405. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Rice, W.J.; Green, N.M. The mechanism of Ca2+ transport by sarco(endo)plasmic reticulum Ca2+-ATPases. J. Biol. Chem. 1997, 272, 28815–28818. [Google Scholar] [CrossRef] [Green Version]
- Janigro, D.; Maccaferri, G.; Meldolesi, J. Calcium channels in undifferentiated PC12 rat pheochromocytoma cells. FEBS Lett 1989, 255, 398–400. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Felix, R.; Gurnett, C.A.; De Waard, M.; Witcher, D.R.; Campbell, K.P. Expression and subunit interaction of voltage-dependent Ca2+ channels in PC12 cells. J. Neurosci. 1996, 16, 7557–7565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickland, M.; Yacoubi-Loueslati, B.; Bouhaouala-Zahar, B.; Pender, S.L.F.; Larbi, A. Relationships between Ion Channels, Mitochondrial Functions and Inflammation in Human Aging. Front. Physiol. 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, O.; Landfield, P.W. Increase in single L-type calcium channels in hippocampal neurons during aging. Science 1996, 272, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.W.; Hao, S.Y.; Thibault, O.; Blalock, E.M.; Landfield, P.W. Aging changes in voltage-gated calcium currents in hippocampal CA1 neurons. J. Neurosci. 1996, 16, 6286–6295. [Google Scholar] [CrossRef]
- Brewer, L.D.; Thibault, O.; Staton, J.; Thibault, V.; Rogers, J.T.; Garcia-Ramos, G.; Kraner, S.; Landfield, P.W.; Porter, N.M. Increased vulnerability of hippocampal neurons with age in culture: Temporal association with increases in NMDA receptor current, NR2A subunit expression and recruitment of L-type calcium channels. Brain Res. 2007, 1151, 20–31. [Google Scholar] [CrossRef]
- Thibault, O.; Gant, J.C.; Landfield, P.W. Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: Minding the store. Aging Cell 2007, 6, 307–317. [Google Scholar] [CrossRef]
- Navakkode, S.; Liu, C.; Soong, T.W. Altered function of neuronal L-type calcium channels in ageing and neuroinflammation: Implications in age-related synaptic dysfunction and cognitive decline. Ageing Res. Rev. 2018, 42, 86–99. [Google Scholar] [CrossRef]
- Thibault, O.; Hadley, R.; Landfield, P.W. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: Relationship to impaired synaptic plasticity. J. Neurosci. 2001, 21, 9744–9756. [Google Scholar] [CrossRef] [Green Version]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Rice, R.A.; Berchtold, N.C.; Cotman, C.W.; Green, K.N. Age-related downregulation of the CaV3.1 T-type calcium channel as a mediator of amyloid beta production. Neurobiol. Aging 2014, 35, 1002–1011. [Google Scholar] [CrossRef] [Green Version]
- Del Toro, R.; Levitsky, K.L.; Lopez-Barneo, J.; Chiara, M.D. Induction of T-type calcium channel gene expression by chronic hypoxia. J. Biol. Chem. 2003, 278, 22316–22324. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Peleg, S. Biphasic Modeling of Mitochondrial Metabolism Dysregulation during Aging. Trends Biochem. Sci. 2017, 42, 702–711. [Google Scholar] [CrossRef] [Green Version]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, D.V.; Wiley, C.D.; Velarde, M.C. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell 2015, 14, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lopes, G.S.; Ferreira, A.T.; Oshiro, M.E.; Vladimirova, I.; Jurkiewicz, N.H.; Jurkiewicz, A.; Smaili, S.S. Aging-related changes of intracellular Ca2+ stores and contractile response of intestinal smooth muscle. Exp. Gerontol. 2006, 41, 55–62. [Google Scholar] [CrossRef]
- Buchholz, J.N.; Behringer, E.J.; Pottorf, W.J.; Pearce, W.J.; Vanterpool, C.K. Age-dependent changes in Ca2+ homeostasis in peripheral neurones: Implications for changes in function. Aging Cell 2007, 6, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Friel, D.D. Mitochondria as regulators of stimulus-evoked calcium signals in neurons. Cell Calcium 2000, 28, 307–316. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Toescu, E.C. Calcium and neuronal ageing. Trends Neurosci. 1998, 21, 2–7. [Google Scholar] [CrossRef]
- Martinez-Serrano, A.; Blanco, P.; Satrustegui, J. Calcium binding to the cytosol and calcium extrusion mechanisms in intact synaptosomes and their alterations with aging. J. Biol. Chem. 1992, 267, 4672–4679. [Google Scholar]
- Kirischuk, S.; Verkhratsky, A. Calcium homeostasis in aged neurones. Life Sci. 1996, 59, 451–459. [Google Scholar] [CrossRef]
- Denton, R.M.; Randle, P.J.; Bridges, B.J.; Cooper, R.H.; Kerbey, A.L.; Pask, H.T.; Severson, D.L.; Stansbie, D.; Whitehouse, S. Regulation of mammalian pyruvate dehydrogenase. Mol. Cell. Biochem. 1975, 9, 27–53. [Google Scholar] [CrossRef]
- McKenzie, M.; Lim, S.C.; Duchen, M.R. Simultaneous Measurement of Mitochondrial Calcium and Mitochondrial Membrane Potential in Live Cells by Fluorescent Microscopy. J. Vis. Exp. 2017, 119, e55166. [Google Scholar] [CrossRef] [Green Version]
- Madreiter-Sokolowski, C.T.; Sokolowski, A.A.; Graier, W.F. Dosis Facit Sanitatem—Concentration-Dependent Effects of Resveratrol on Mitochondria. Nutrients 2017, 9, 1117. [Google Scholar] [CrossRef] [Green Version]
- Gauba, E.; Guo, L.; Du, H. Cyclophilin D Promotes Brain Mitochondrial F1FO ATP Synthase Dysfunction in Aging Mice. J. Alzheimers Dis. 2017, 55, 1351–1362. [Google Scholar] [CrossRef] [Green Version]
- Panel, M.; Ghaleh, B.; Morin, D. Mitochondria and aging: A role for the mitochondrial transition pore? Aging Cell 2018, 17, e12793. [Google Scholar] [CrossRef]
- Scorrano, L.; Petronilli, V.; Bernardi, P. On the voltage dependence of the mitochondrial permeability transition pore. A critical appraisal. J. Biol. Chem. 1997, 272, 12295–12299. [Google Scholar] [CrossRef] [Green Version]
- Boczek, T.; Kozaczuk, A.; Taha, J.; Ferenc, B.; Zylinska, L. Adaptation of microsomal glutathione transferase 1 in PC12 cells with modified PMCA isoforms composition. Indian J. Biochem. Biophys. 2010, 47, 265–271. [Google Scholar]
- Marques-Aleixo, I.; Rocha-Rodrigues, S.; Santos-Alves, E.; Coxito, P.M.; Passos, E.; Oliveira, P.J.; Magalhaes, J.; Ascensao, A. In vitro salicylate does not further impair aging-induced brain mitochondrial dysfunction. Toxicology 2012, 302, 51–59. [Google Scholar] [CrossRef]
- Krestinina, O.; Azarashvili, T.; Baburina, Y.; Galvita, A.; Grachev, D.; Stricker, R.; Reiser, G. In aging, the vulnerability of rat brain mitochondria is enhanced due to reduced level of 2′,3′-cyclic nucleotide-3′-phosphodiesterase (CNP) and subsequently increased permeability transition in brain mitochondria in old animals. Neurochem. Int. 2015, 80, 41–50. [Google Scholar] [CrossRef]
- Pandya, J.D.; Grondin, R.; Yonutas, H.M.; Haghnazar, H.; Gash, D.M.; Zhang, Z.; Sullivan, P.G. Decreased mitochondrial bioenergetics and calcium buffering capacity in the basal ganglia correlates with motor deficits in a nonhuman primate model of aging. Neurobiol. Aging 2015, 36, 1903–1913. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 2012, 942, 3–37. [Google Scholar] [PubMed]
- Naghdi, S.; Waldeck-Weiermair, M.; Fertschai, I.; Poteser, M.; Graier, W.F.; Malli, R. Mitochondrial Ca2+ uptake and not mitochondrial motility is required for STIM1-Orai1-dependent store-operated Ca2+ entry. J. Cell Sci. 2010, 123, 2553–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldeck-Weiermair, M.; Alam, M.R.; Khan, M.J.; Deak, A.T.; Vishnu, N.; Karsten, F.; Imamura, H.; Graier, W.F.; Malli, R. Spatiotemporal correlations between cytosolic and mitochondrial Ca2+ signals using a novel red-shifted mitochondrial targeted cameleon. PLoS ONE 2012, 7, e45917. [Google Scholar] [CrossRef] [PubMed]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 2013, 1833, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Kruger, W.A.; Monteith, G.R.; Poronnik, P. The plasma membrane Ca2+-ATPase: Regulation by PSD-95/Dlg/Zo-1 scaffolds. Int. J. Biochem. Cell Biol. 2010, 42, 805–808. [Google Scholar] [CrossRef]
- Garside, M.L.; Turner, P.R.; Austen, B.; Strehler, E.E.; Beesley, P.W.; Empson, R.M. Molecular interactions of the plasma membrane calcium ATPase 2 at pre- and post-synaptic sites in rat cerebellum. Neuroscience 2009, 162, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.; Ramsden, D.B. Huntington’s disease. Mol. Pathol. 2001, 54, 409–413. [Google Scholar] [CrossRef]
- Lisek, M.; Ferenc, B.; Studzian, M.; Pulaski, L.; Guo, F.; Zylinska, L.; Boczek, T. Glutamate Deregulation in Ketamine-Induced Psychosis-A Potential Role of PSD95, NMDA Receptor and PMCA Interaction. Front. Cell. Neurosci. 2017, 11, 181. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, D.G. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim. Biophys. Acta 2009, 1787, 1416–1424. [Google Scholar] [CrossRef] [Green Version]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchen, M.R. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers. Arch. 2012, 464, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Tan, Y.W.; Hagenston, A.M.; Martel, M.A.; Kneisel, N.; Skehel, P.A.; Wyllie, D.J.; Bading, H.; Hardingham, G.E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013, 4, 2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochim. Biophys. Acta 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Li, V.; Wang, Y.T. Molecular mechanisms of NMDA receptor-mediated excitotoxicity: Implications for neuroprotective therapeutics for stroke. Neural Regen. Res. 2016, 11, 1752–1753. [Google Scholar]
- Zhou, Q.; Sheng, M. NMDA receptors in nervous system diseases. Neuropharmacology 2013, 74, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [Green Version]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell. Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef]
- Kamer, K.J.; Jiang, W.; Kaushik, V.K.; Mootha, V.K.; Grabarek, Z. Crystal structure of MICU2 and comparison with MICU1 reveal insights into the uniporter gating mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 3546–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallilankaraman, K.; Doonan, P.; Cardenas, C.; Chandramoorthy, H.C.; Muller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgo, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csordas, G.; Golenar, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; de la Fuente Perez, S.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabo, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell. 2014, 53, 726–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.C.; Liu, J.; Holmstrom, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, T.P.; Wu, Y.; Joiner, M.L.; Koval, O.M.; Wilson, N.R.; Luczak, E.D.; Wang, Q.; Chen, B.; Gao, Z.; Zhu, Z.; et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl. Acad. Sci. USA 2015, 112, 9129–9134. [Google Scholar] [CrossRef] [Green Version]
- Konig, T.; Troder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Muhlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell. 2016, 64, 148–162. [Google Scholar] [CrossRef] [Green Version]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef] [Green Version]
- Markus, N.M.; Hasel, P.; Qiu, J.; Bell, K.F.; Heron, S.; Kind, P.C.; Dando, O.; Simpson, T.I.; Hardingham, G.E. Expression of mRNA Encoding Mcu and Other Mitochondrial Calcium Regulatory Genes Depends on Cell Type, Neuronal Subtype, and Ca2+ Signaling. PLoS ONE 2016, 11, e0148164. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Rodriguez, M.; Garcia-Durillo, M.; Villalobos, C.; Nunez, L. In vitro aging promotes endoplasmic reticulum (ER)-mitochondria Ca2+ cross talk and loss of store-operated Ca2+ entry (SOCE) in rat hippocampal neurons. Biochim. Biophys. Acta 2016, 1863, 2637–2649. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Waldeck-Weiermair, M.; Bourguignon, M.P.; Villeneuve, N.; Gottschalk, B.; Klec, C.; Stryeck, S.; Radulovic, S.; Parichatikanond, W.; Frank, S.; et al. Enhanced inter-compartmental Ca2+ flux modulates mitochondrial metabolism and apoptotic threshold during aging. Redox Biol. 2019, 20, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Sokolowski, A.A.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Targeting Mitochondria to Counteract Age-Related Cellular Dysfunction. Genes 2018, 9, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Jung, Y.; Park, B.S.; Partovi, D.; Reddy, P.H. Time-course of mitochondrial gene expressions in mice brains: Implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J. Neurochem. 2005, 92, 494–504. [Google Scholar] [CrossRef]
- Itoh, K.; Weis, S.; Mehraein, P.; Muller-Hocker, J. Cytochrome c oxidase defects of the human substantia nigra in normal aging. Neurobiol. Aging 1996, 17, 843–848. [Google Scholar] [CrossRef]
- Bertoni-Freddari, C.; Fattoretti, P.; Giorgetti, B.; Solazzi, M.; Balietti, M.; Casoli, T.; Di Stefano, G. Cytochrome oxidase activity in hippocampal synaptic mitochondria during aging: A quantitative cytochemical investigation. Ann. N. Y. Acad. Sci. 2004, 1019, 33–36. [Google Scholar] [CrossRef]
- Poburko, D.; Santo-Domingo, J.; Demaurex, N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J. Biol. Chem. 2011, 286, 11672–11684. [Google Scholar] [CrossRef] [Green Version]
- Balut, C.; vandeVen, M.; Despa, S.; Lambrichts, I.; Ameloot, M.; Steels, P.; Smets, I. Measurement of cytosolic and mitochondrial pH in living cells during reversible metabolic inhibition. Kidney Int. 2008, 73, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Bolshakov, A.P.; Mikhailova, M.M.; Szabadkai, G.; Pinelis, V.G.; Brustovetsky, N.; Rizzuto, R.; Khodorov, B.I. Measurements of mitochondrial pH in cultured cortical neurons clarify contribution of mitochondrial pore to the mechanism of glutamate-induced delayed Ca2+ deregulation. Cell Calcium 2008, 43, 602–614. [Google Scholar] [CrossRef]
- Malli, R.; Frieden, M.; Osibow, K.; Zoratti, C.; Mayer, M.; Demaurex, N.; Graier, W.F. Sustained Ca2+ transfer across mitochondria is Essential for mitochondrial Ca2+ buffering, sore-operated Ca2+ entry, and Ca2+ store refilling. J. Biol. Chem. 2003, 278, 44769–44779. [Google Scholar] [CrossRef] [Green Version]
- Abad, M.F.; Di Benedetto, G.; Magalhaes, P.J.; Filippin, L.; Pozzan, T. Mitochondrial pH monitored by a new engineered green fluorescent protein mutant. J. Biol. Chem. 2004, 279, 11521–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petronilli, V.; Cola, C.; Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. II. The minimal requirements for pore induction underscore a key role for transmembrane electrical potential, matrix pH, and matrix Ca2+. J. Biol. Chem. 1993, 268, 1011–1016. [Google Scholar] [PubMed]
- Selivanov, V.A.; Zeak, J.A.; Roca, J.; Cascante, M.; Trucco, M.; Votyakova, T.V. The role of external and matrix pH in mitochondrial reactive oxygen species generation. J. Biol. Chem. 2008, 283, 29292–29300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behringer, E.J.; Segal, S.S. Impact of Aging on Calcium Signaling and Membrane Potential in Endothelium of Resistance Arteries: A Role for Mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1627–1637. [Google Scholar] [CrossRef]
- Gunter, T.E.; Pfeiffer, D.R. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990, 258, C755–C786. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Luciani, D.S.; Misler, S.; Polonsky, K.S. Ca2+ controls slow NAD(P)H oscillations in glucose-stimulated mouse pancreatic islets. J. Physiol. 2006, 572, 379–392. [Google Scholar] [CrossRef]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Robb-Gaspers, L.D.; Burnett, P.; Rutter, G.A.; Denton, R.M.; Rizzuto, R.; Thomas, A.P. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998, 17, 4987–5000. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.A.; Das, A.M. Control of mitochondrial ATP synthesis in the heart. Biochem. J. 1991, 280, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Scholz, T.D.; Balaban, R.S. Mitochondrial F1-ATPase activity of canine myocardium: Effects of hypoxia and stimulation. Am. J. Physiol. 1994, 266, H2396–H2403. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, M.J.; McHugh, N.J. Mitochondrial ATP synthase F1-β-subunit is a calcium-binding protein. FEBS Lett. 1996, 391, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boerries, M.; Most, P.; Gledhill, J.R.; Walker, J.E.; Katus, H.A.; Koch, W.J.; Aebi, U.; Schoenenberger, C.A. Ca2+ -dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol. Cell. Biol. 2007, 27, 4365–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padanyi, R.; Paszty, K.; Hegedus, L.; Varga, K.; Papp, B.; Penniston, J.T.; Enyedi, A. Multifaceted plasma membrane Ca2+ pumps: From structure to intracellular Ca2+ handling and cancer. Biochim. Biophys. Acta 2016, 1863, 1351–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cali, T.; Brini, M.; Carafoli, E. Regulation of Cell Calcium and Role of Plasma Membrane Calcium ATPases. Int. Rev. Cell. Mol. Biol. 2017, 332, 259–296. [Google Scholar]
- Elwess, N.L.; Filoteo, A.G.; Enyedi, A.; Penniston, J.T. Plasma membrane Ca2+ pump isoforms 2a and 2b are unusually responsive to calmodulin and Ca2+. J. Biol. Chem. 1997, 272, 17981–17986. [Google Scholar] [CrossRef] [Green Version]
- Strehler, E.E. Plasma membrane calcium ATPases: From generic Ca2+ sump pumps to versatile systems for fine-tuning cellular Ca2+. Biochem. Biophys. Res. Commun. 2015, 460, 26–33. [Google Scholar] [CrossRef]
- Toutenhoofd, S.L.; Strehler, E.E. The calmodulin multigene family as a unique case of genetic redundancy: Multiple levels of regulation to provide spatial and temporal control of calmodulin pools? Cell Calcium 2000, 28, 83–96. [Google Scholar] [CrossRef]
- Shen, X.; Valencia, C.A.; Szostak, J.W.; Dong, B.; Liu, R. Scanning the human proteome for calmodulin-binding proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 5969–5974. [Google Scholar] [CrossRef] [Green Version]
- Bigelow, D.J.; Squier, T.C. Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim. Biophys. Acta 2005, 1703, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Davidkova, G.; Zhang, S.P.; Nichols, R.A.; Weiss, B. Reduced level of calmodulin in PC12 cells induced by stable expression of calmodulin antisense RNA inhibits cell proliferation and induces neurite outgrowth. Neuroscience 1996, 75, 1003–1019. [Google Scholar] [CrossRef]
- Boczek, T.; Lisek, M.; Ferenc, B.; Zylinska, L. Cross talk among PMCA, calcineurin and NFAT transcription factors in control of calmodulin gene expression in differentiating PC12 cells. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, L.I.; Routtenberg, A. GAP-43: An intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997, 20, 84–91. [Google Scholar] [CrossRef]
- Denny, J.B. Molecular mechanisms, biological actions, and neuropharmacology of the growth-associated protein GAP-43. Curr. Neuropharmacol. 2006, 4, 293–304. [Google Scholar] [CrossRef] [Green Version]
- Holahan, M.R. A Shift from a Pivotal to Supporting Role for the Growth-Associated Protein (GAP-43) in the Coordination of Axonal Structural and Functional Plasticity. Front. Cell. Neurosci. 2017, 11, 266. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, C.J.; Park, M.; Spillane, M.; Yoo, S.; Pacheco, A.; Gomes, C.; Vuppalanchi, D.; McDonald, M.; Kim, H.H.; Merianda, T.T.; et al. Axonally synthesized β-actin and GAP-43 proteins support distinct modes of axonal growth. J. Neurosci. 2013, 33, 3311–3322. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.; Kim, H.H.; Kim, P.; Donnelly, C.J.; Kalinski, A.L.; Vuppalanchi, D.; Park, M.; Lee, S.J.; Merianda, T.T.; Perrone-Bizzozero, N.I.; et al. A HuD-ZBP1 ribonucleoprotein complex localizes GAP-43 mRNA into axons through its 3′ untranslated region AU-rich regulatory element. J. Neurochem. 2013, 126, 792–804. [Google Scholar] [CrossRef] [Green Version]
- Neve, R.L.; Finch, E.A.; Bird, E.D.; Benowitz, L.I. Growth-associated protein GAP-43 is expressed selectively in associative regions of the adult human brain. Proc. Natl. Acad. Sci. USA 1988, 85, 3638–3642. [Google Scholar] [CrossRef] [Green Version]
- Latchney, S.E.; Masiulis, I.; Zaccaria, K.J.; Lagace, D.C.; Powell, C.M.; McCasland, J.S.; Eisch, A.J. Developmental and adult GAP-43 deficiency in mice dynamically alters hippocampal neurogenesis and mossy fiber volume. Dev. Neurosci. 2014, 36, 44–63. [Google Scholar] [CrossRef] [Green Version]
- Schmoll, H.; Ramboiu, S.; Platt, D.; Herndon, J.G.; Kessler, C.; Popa-Wagner, A. Age influences the expression of GAP-43 in the rat hippocampus following seizure. Gerontology 2005, 51, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.A.; Mizumori, S.J.; Lovinger, D.M.; Sheu, F.S.; Murakami, K.; Chan, S.Y.; Linden, D.J.; Nelson, R.B.; Routtenberg, A. Selective decline in protein F1 phosphorylation in hippocampus of senescent rats. Neurobiol. Aging 1988, 9, 393–398. [Google Scholar] [CrossRef]
- Casoli, T.; Di Stefano, G.; Gracciotti, N.; Giovagnetti, S.; Fattoretti, P.; Solazzi, M.; Bertoni-Freddari, C. Cellular distribution of GAP-43 mRNA in hippocampus and cerebellum of adult rat brain by in situ RT-PCR. J. Histochem. Cytochem. 2001, 49, 1195–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casoli, T.; Spagna, C.; Fattoretti, P.; Gesuita, R.; Bertoni-Freddari, C. Neuronal plasticity in aging: A quantitative immunohistochemical study of GAP-43 distribution in discrete regions of the rat brain. Brain Res. 1996, 714, 111–117. [Google Scholar] [CrossRef]
- Casoli, T.; Stefano, G.D.; Fattoretti, P.; Solazzi, M.; Delfino, A.; Biagini, G.; Bertoni-Freddari, C. GAP-43 mRNA detection by in situ hybridization, direct and indirect in situ RT-PCR in hippocampal and cerebellar tissue sections of adult rat brain. Micron 2003, 34, 415–422. [Google Scholar] [CrossRef]
- Webster, M.J.; Elashoff, M.; Weickert, C.S. Molecular evidence that cortical synaptic growth predominates during the first decade of life in humans. Int. J. Dev. Neurosci. 2011, 29, 225–236. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Ng, S.C.; Hsu, D.W. Aberrant GAP-43 gene expression in Alzheimer’s disease. Am. J. Pathol. 1995, 147, 934–946. [Google Scholar]
- Rekart, J.L.; Quinn, B.; Mesulam, M.M.; Routtenberg, A. Subfield-specific increase in brain growth protein in postmortem hippocampus of Alzheimer’s patients. Neuroscience 2004, 126, 579–584. [Google Scholar] [CrossRef]
- Boczek, T.; Ferenc, B.; Lisek, M.; Zylinska, L. Regulation of GAP43/calmodulin complex formation via calcineurin-dependent mechanism in differentiated PC12 cells with altered PMCA isoforms composition. Mol. Cell. Biochem. 2015, 407, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Kipanyula, M.J.; Kimaro, W.H.; Seke Etet, P.F. The Emerging Roles of the Calcineurin-Nuclear Factor of Activated T-Lymphocytes Pathway in Nervous System Functions and Diseases. J. Aging Res. 2016, 2016, 5081021. [Google Scholar] [CrossRef] [Green Version]
- Abdul, H.M.; Furman, J.L.; Sama, M.A.; Mathis, D.M.; Norris, C.M. NFATs and Alzheimer’s Disease. Mol. Cell. Pharmacol. 2010, 2, 7–14. [Google Scholar] [PubMed]
- Norris, C.M.; Kadish, I.; Blalock, E.M.; Chen, K.C.; Thibault, V.; Porter, N.M.; Landfield, P.W.; Kraner, S.D. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J. Neurosci. 2005, 25, 4649–4658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Rao, A.; Hogan, P.G. Interaction of calcineurin with substrates and targeting proteins. Trends Cell. Biol. 2011, 21, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, M.C.; Luk, N.A.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Distinct regulation of cytoplasmic calcium signals and cell death pathways by different plasma membrane calcium ATPase isoforms in MDA-MB-231 breast cancer cells. J. Biol. Chem. 2012, 287, 28598–28608. [Google Scholar] [CrossRef] [Green Version]
- Holton, M.; Yang, D.; Wang, W.; Mohamed, T.M.; Neyses, L.; Armesilla, A.L. The interaction between endogenous calcineurin and the plasma membrane calcium-dependent ATPase is isoform specific in breast cancer cells. FEBS Lett. 2007, 581, 4115–4119. [Google Scholar] [CrossRef]
- Wu, X.; Chang, B.; Blair, N.S.; Sargent, M.; York, A.J.; Robbins, J.; Shull, G.E.; Molkentin, J.D. Plasma membrane Ca2+-ATPase isoform 4 antagonizes cardiac hypertrophy in association with calcineurin inhibition in rodents. J. Clin. Investig. 2009, 119, 976–985. [Google Scholar] [CrossRef] [Green Version]
- Baggott, R.R.; Mohamed, T.M.; Oceandy, D.; Holton, M.; Blanc, M.C.; Roux-Soro, S.C.; Brown, S.; Brown, J.E.; Cartwright, E.J.; Wang, W.; et al. Disruption of the interaction between PMCA2 and calcineurin triggers apoptosis and enhances paclitaxel-induced cytotoxicity in breast cancer cells. Carcinogenesis 2012, 33, 2362–2368. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.C.; Sharrow, K.M.; Masse, J.R.; Norris, C.M.; Kumar, A. Calcineurin links Ca2+ dysregulation with brain aging. J. Neurosci. 2001, 21, 4066–4073. [Google Scholar] [CrossRef] [Green Version]
- Sommerer, C.; Meuer, S.; Zeier, M.; Giese, T. Calcineurin inhibitors and NFAT-regulated gene expression. Clin. Chim. Acta 2012, 413, 1379–1386. [Google Scholar] [CrossRef]
- Muller, M.R.; Rao, A. NFAT, immunity and cancer: A transcription factor comes of age. Nat. Rev. Immunol. 2010, 10, 645–656. [Google Scholar] [CrossRef]
- Teolato, S.; Calderini, G.; Bonetti, A.C.; Toffano, G. Calmodulin content in different brain areas of aging rats. Neurosci. Lett. 1983, 38, 57–60. [Google Scholar] [CrossRef]
- Saimi, Y.; Kung, C. Calmodulin as an ion channel subunit. Annu. Rev. Physiol. 2002, 64, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Norris, C.M.; Blalock, E.M.; Chen, K.C.; Porter, N.M.; Landfield, P.W. Calcineurin enhances L-type Ca2+ channel activity in hippocampal neurons: Increased effect with age in culture. Neuroscience 2002, 110, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [Green Version]
- Decuypere, J.P.; Monaco, G.; Missiaen, L.; De Smedt, H.; Parys, J.B.; Bultynck, G. IP3 Receptors, Mitochondria, and Ca Signaling: Implications for Aging. J. Aging Res. 2011, 2011, 920178. [Google Scholar] [CrossRef] [Green Version]
- Vervloessem, T.; Yule, D.I.; Bultynck, G.; Parys, J.B. The type 2 inositol 1,4,5-trisphosphate receptor, emerging functions for an intriguing Ca2+-release channel. Biochim. Biophys. Acta 2015, 1853, 1992–2005. [Google Scholar] [CrossRef] [Green Version]
- Dent, M.A.; Raisman, G.; Lai, F.A. Expression of type 1 inositol 1,4,5-trisphosphate receptor during axogenesis and synaptic contact in the central and peripheral nervous system of developing rat. Development 1996, 122, 1029–1039. [Google Scholar]
- Mikoshiba, K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1446. [Google Scholar] [CrossRef]
- Kumar, A.; Gibbs, J.R.; Beilina, A.; Dillman, A.; Kumaran, R.; Trabzuni, D.; Ryten, M.; Walker, R.; Smith, C.; Traynor, B.J.; et al. Age-associated changes in gene expression in human brain and isolated neurons. Neurobiol. Aging 2013, 34, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.H.; Nucifora, F.C., Jr.; Blondel, O.; Sheppard, C.A.; Zhang, C.; Snyder, S.H.; Russell, J.T.; Ryugo, D.K.; Ross, C.A. Differential cellular expression of isoforms of inositol 1,4,5-triphosphate receptors in neurons and glia in brain. J. Comp. Neurol. 1999, 406, 207–220. [Google Scholar] [CrossRef]
- Simonyi, A.; Xia, J.; Igbavboa, U.; Wood, W.G.; Sun, G.Y. Age differences in the expression of metabotropic glutamate receptor 1 and inositol 1,4,5-trisphosphate receptor in mouse cerebellum. Neurosci. Lett. 1998, 244, 29–32. [Google Scholar] [CrossRef]
- Ivanova, H.; Vervliet, T.; Missiaen, L.; Parys, J.B.; De Smedt, H.; Bultynck, G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim. Biophys. Acta 2014, 1843, 2164–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, M.; Michikawa, T.; Bosanac, I.; Ikura, M.; Mikoshiba, K. Molecular basis of the isoform-specific ligand-binding affinity of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 2007, 282, 12755–12764. [Google Scholar] [CrossRef] [Green Version]
- Parys, J.B.; De Smedt, H. Inositol 1,4,5-trisphosphate and its receptors. Adv. Exp. Med. Biol. 2012, 740, 255–279. [Google Scholar]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [Green Version]
- Radzik, T.; Boczek, T.; Ferenc, B.; Studzian, M.; Pulaski, L.; Zylinska, L. Calcium Dyshomeostasis Alters CCL5 Signaling in Differentiated PC12 Cells. Biomed. Res. Int. 2019, 2019, 9616248. [Google Scholar] [CrossRef]
- Penniston, J.T.; Padanyi, R.; Paszty, K.; Varga, K.; Hegedus, L.; Enyedi, A. Apart from its known function, the plasma membrane Ca2+ ATPase can regulate Ca2+ signaling by controlling phosphatidylinositol 4,5-bisphosphate levels. J. Cell. Sci. 2014, 127, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Sama, D.M.; Norris, C.M. Calcium dysregulation and neuroinflammation: Discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev. 2013, 12, 982–995. [Google Scholar] [CrossRef] [Green Version]
- Marques, R.E.; Guabiraba, R.; Russo, R.C.; Teixeira, M.M. Targeting CCL5 in inflammation. Expert Opin. Ther. Targets 2013, 17, 1439–1460. [Google Scholar] [CrossRef] [PubMed]
- Pranzatelli, M.R. Advances in Biomarker-Guided Therapy for Pediatric- and Adult-Onset Neuroinflammatory Disorders: Targeting Chemokines/Cytokines. Front. Immunol. 2018, 9, 557. [Google Scholar] [CrossRef] [PubMed]
- Gamo, K.; Kiryu-Seo, S.; Konishi, H.; Aoki, S.; Matsushima, K.; Wada, K.; Kiyama, H. G-protein-coupled receptor screen reveals a role for chemokine receptor CCR5 in suppressing microglial neurotoxicity. J. Neurosci. 2008, 28, 11980–11988. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, Y.; Ransohoff, R.M. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balistreri, C.R.; Caruso, C.; Grimaldi, M.P.; Listi, F.; Vasto, S.; Orlando, V.; Campagna, A.M.; Lio, D.; Candore, G. CCR5 receptor: Biologic and genetic implications in age-related diseases. Ann. N. Y. Acad. Sci. 2007, 1100, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Mo, R.; Lescure, P.A.; Misek, D.E.; Hanash, S.; Rochford, R.; Hobbs, M.; Yung, R.L. Aging is associated with increased T-cell chemokine expression in C57BL/6 mice. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumova, E.; Ivanova, M.; Pawelec, G. Immunogenetics of ageing. Int. J. Immunogenet. 2011, 38, 373–381. [Google Scholar] [CrossRef]
- Yung, R.L.; Mo, R. Aging is associated with increased human T cell CC chemokine receptor gene expression. J. Interferon Cytokine Res. 2003, 23, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Tripathy, D.; Thirumangalakudi, L.; Grammas, P. Expression of macrophage inflammatory protein 1-α is elevated in Alzheimer’s vessels and is regulated by oxidative stress. J. Alzheimers Dis. 2007, 11, 447–455. [Google Scholar] [CrossRef]
- Zhu, M.; Allard, J.S.; Zhang, Y.; Perez, E.; Spangler, E.L.; Becker, K.G.; Rapp, P.R. Age-related brain expression and regulation of the chemokine CCL4/MIP-1beta in APP/PS1 double-transgenic mice. J. Neuropathol. Exp. Neurol. 2014, 73, 362–374. [Google Scholar] [CrossRef] [Green Version]
- Pomatto, L.C.D.; Davies, K.J.A. The role of declining adaptive homeostasis in ageing. J. Physiol. 2017, 595, 7275–7309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomatto, L.C.D.; Sun, P.Y.; Davies, K.J.A. To adapt or not to adapt: Consequences of declining Adaptive Homeostasis and Proteostasis with age. Mech. Ageing Dev. 2019, 177, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Janikiewicz, J.; Szymanski, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszynski, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell. Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Protein or mRNA | PC12_2 | PC12_3 |
|---|---|---|
| PMCA1 | increased | increased |
| PMCA2 | decreased | unchanged |
| PMCA3 | unchanged | decreased |
| PMCA4 | unchanged | increased |
| Total PMCA | unchanged | unchanged |
| SERCA2 | increased | increased |
| SERCA3 | increased | increased |
| CaM | decreased | decreased |
| CaN | increased | increased |
| L-type VGCC | increased | increased |
| P/Q-type VGCC | increased | increased |
| T-type VGCC | increased | unchanged |
| CCR1 | increased | unchanged |
| CCR3 | unchanged | increased |
| CCR5 | increased | increased |
| IP3R1 | decreased | decreased |
| IP3R2 | decreased | decreased |
| IP3R3 | increased | increased |
| Total IP3R | unchanged | increased |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boczek, T.; Radzik, T.; Ferenc, B.; Zylinska, L. The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process. Int. J. Mol. Sci. 2019, 20, 6338. https://doi.org/10.3390/ijms20246338
Boczek T, Radzik T, Ferenc B, Zylinska L. The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process. International Journal of Molecular Sciences. 2019; 20(24):6338. https://doi.org/10.3390/ijms20246338
Chicago/Turabian StyleBoczek, Tomasz, Tomasz Radzik, Bozena Ferenc, and Ludmila Zylinska. 2019. "The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process" International Journal of Molecular Sciences 20, no. 24: 6338. https://doi.org/10.3390/ijms20246338
APA StyleBoczek, T., Radzik, T., Ferenc, B., & Zylinska, L. (2019). The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process. International Journal of Molecular Sciences, 20(24), 6338. https://doi.org/10.3390/ijms20246338