PARP-1/2 Inhibitor Olaparib Prevents or Partially Reverts EMT Induced by TGF-β in NMuMG Cells


Abstract
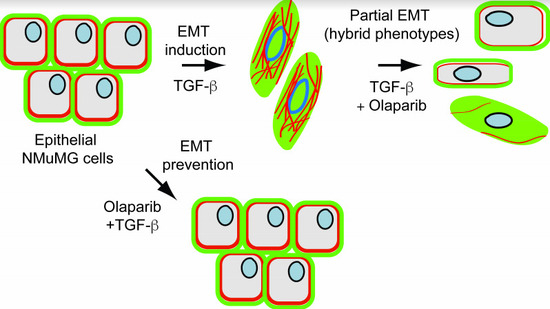
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
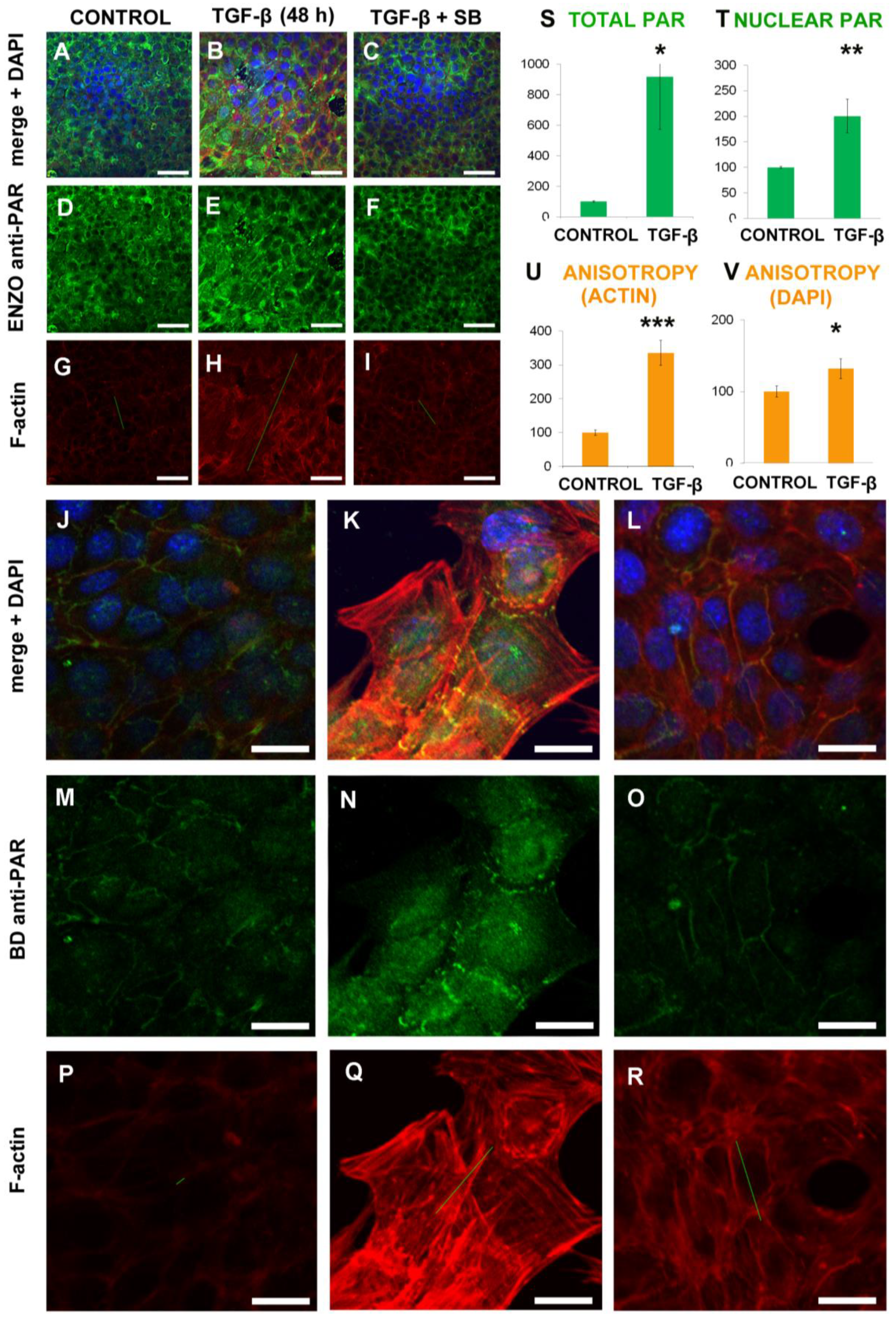
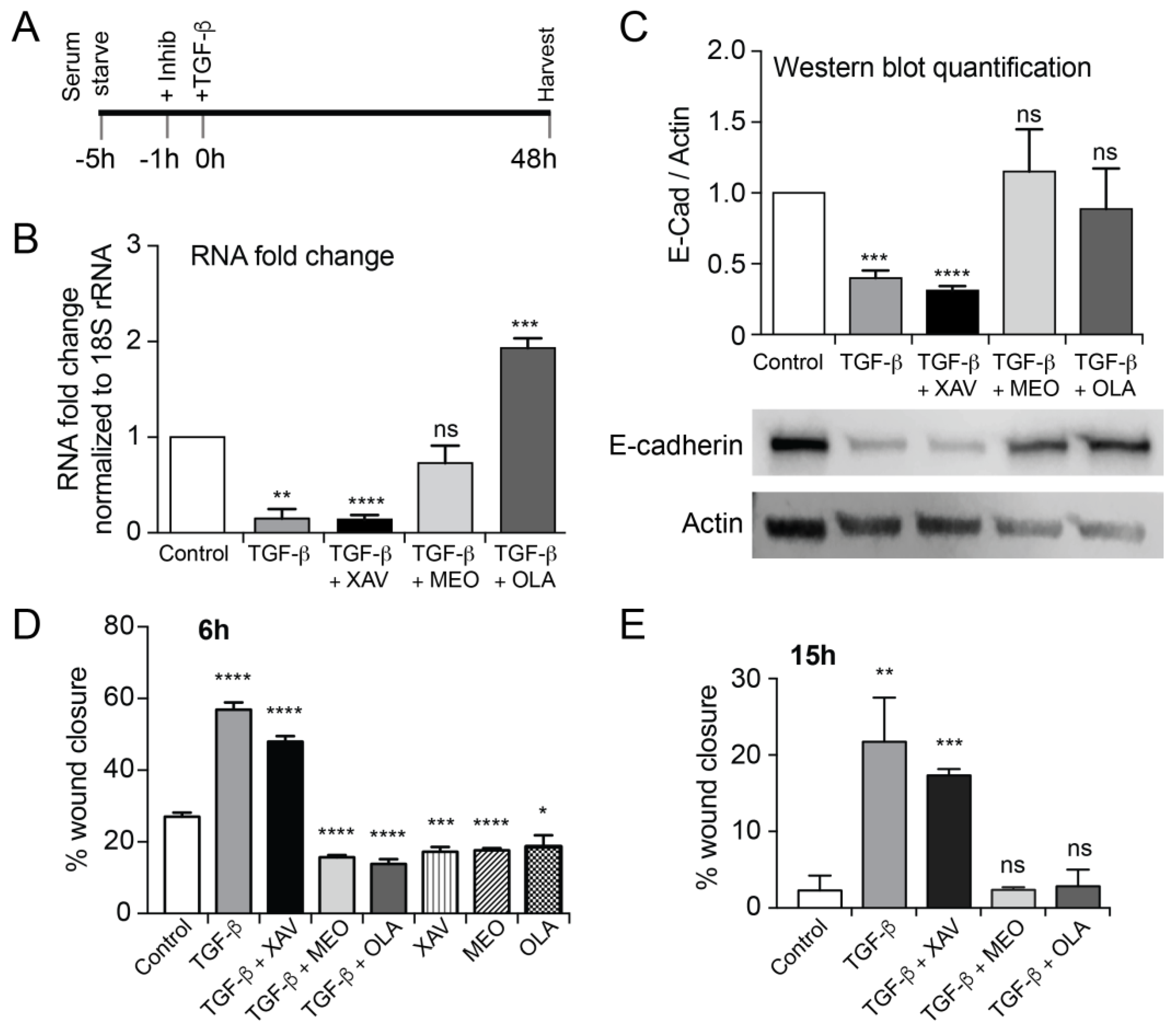
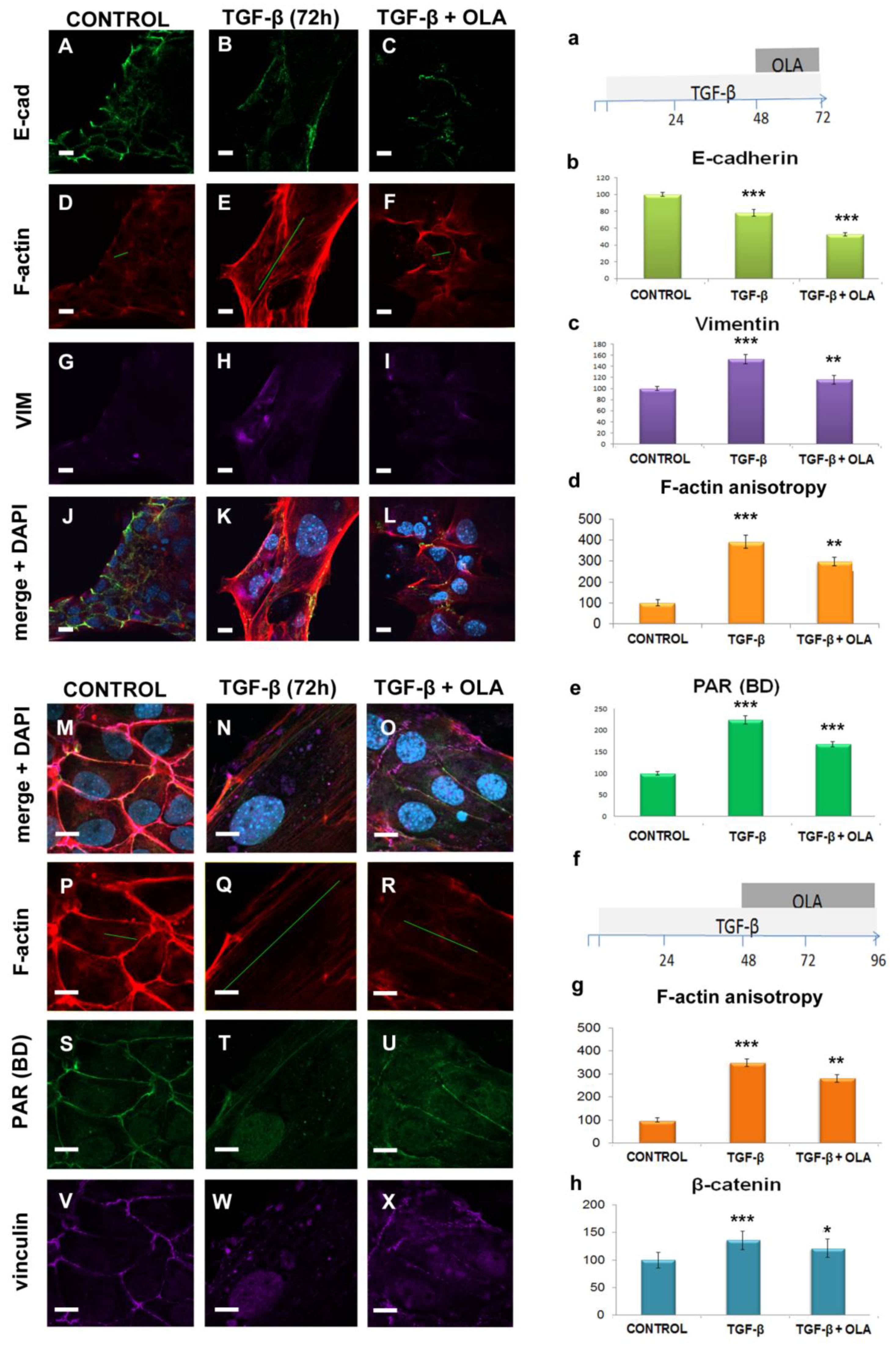
2.1. EMT Induced Total and Nuclear PAR Increase as well as PAR Belt Disassembly
2.2. Anisotropy Measurements of Cell Morphology Changes
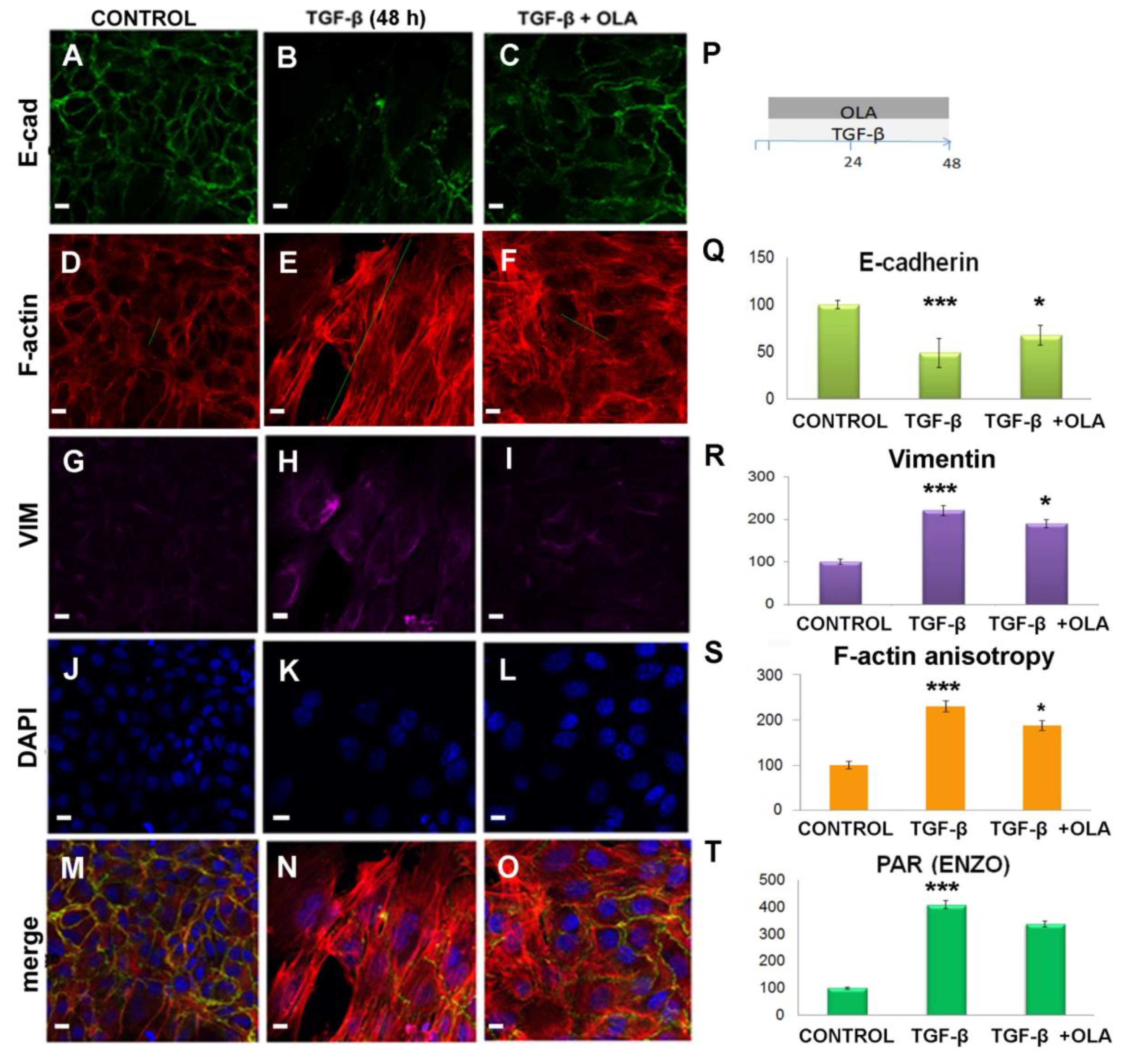
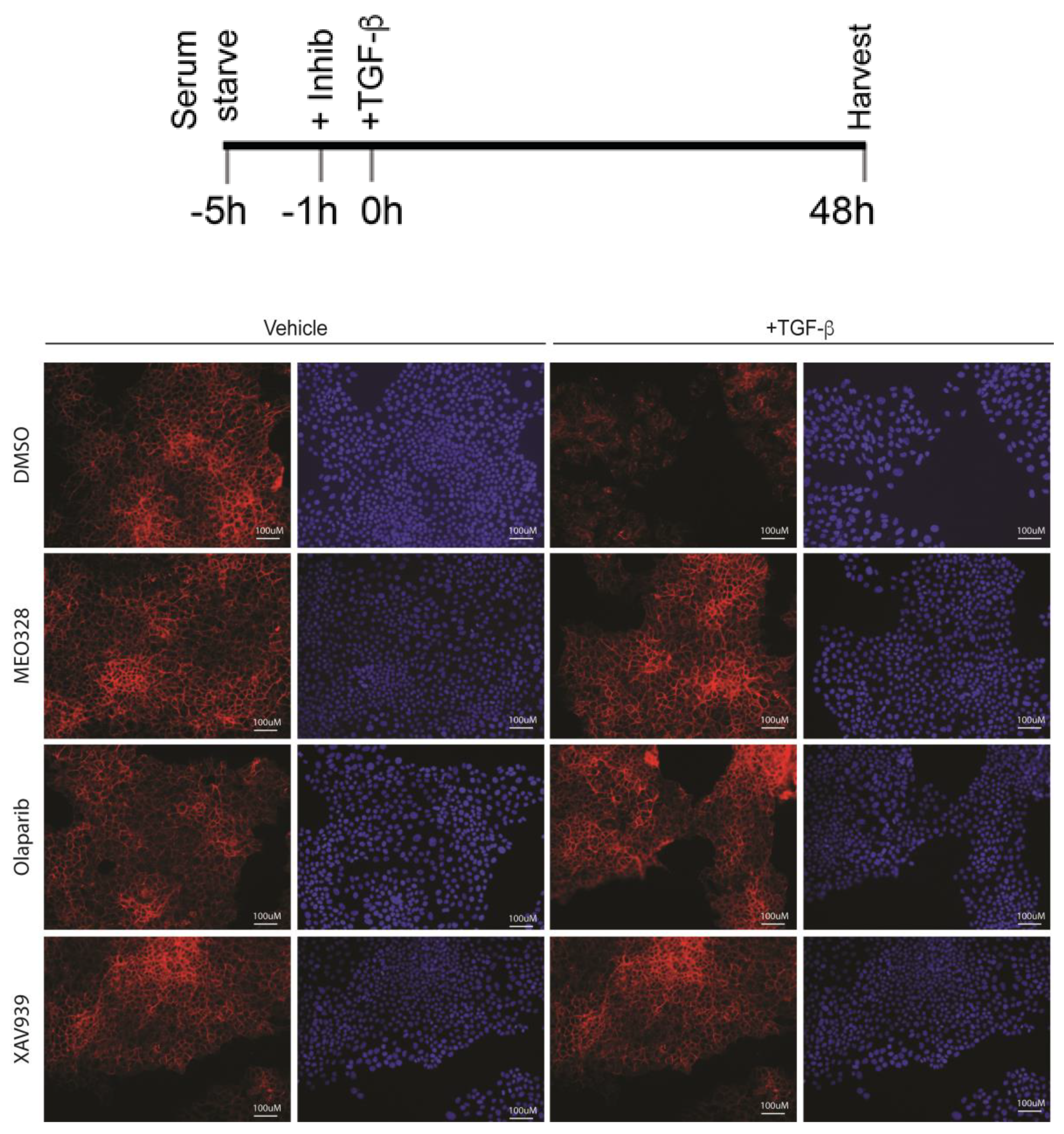
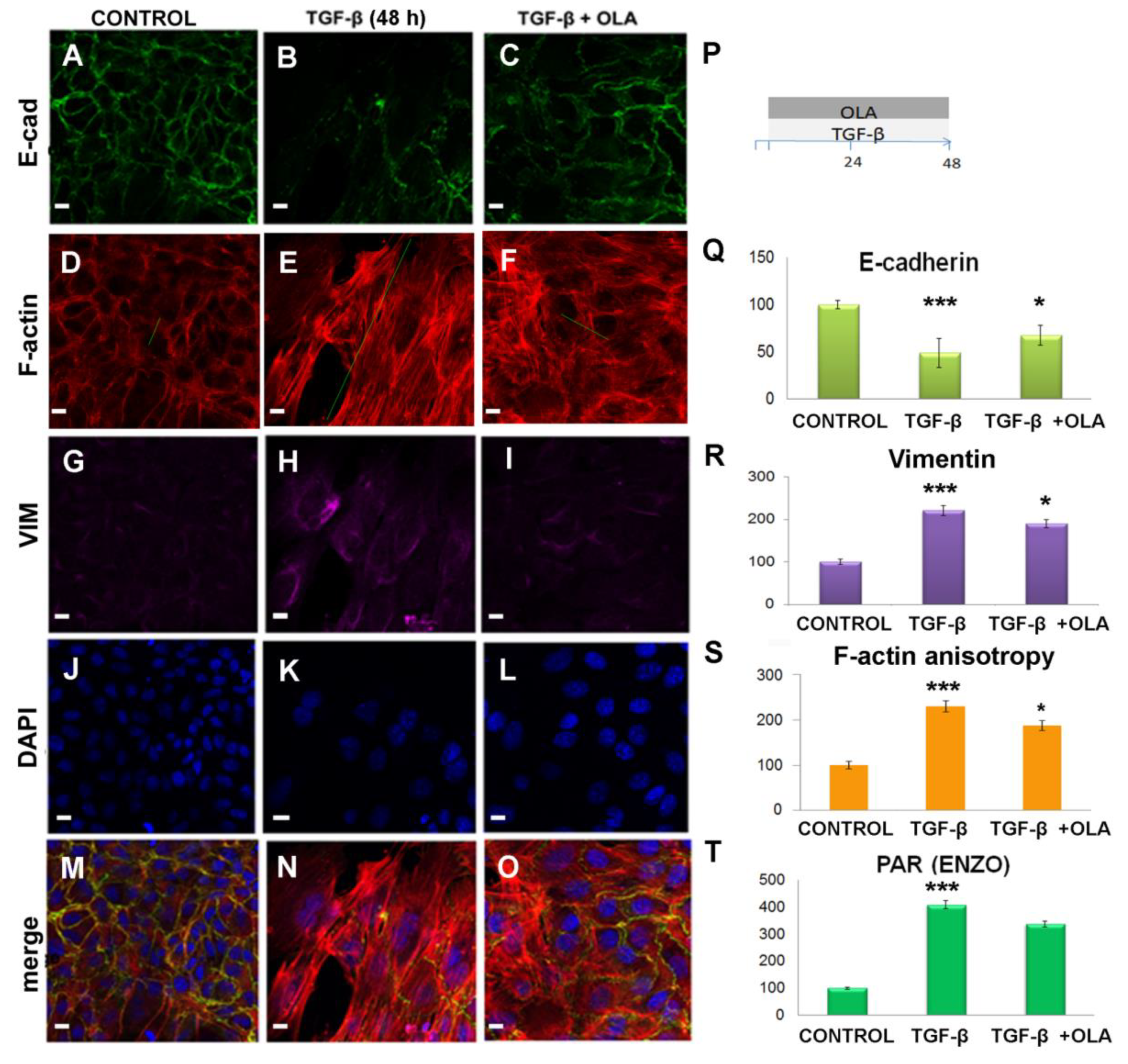
2.3. Treatment with OLA Prevented TGF-β Induced EMT in NMuMG Cells
2.4. EMT Was at Least Partially Reversed by OLA
3. Discussion
- OLA targeted nuclear PARP-1/2 affecting gene expression
- OLA targeted PARP-1 in extracellular vesicles. PARP-1 has been detected in exosomes from colorectal cancers, melanoma, and hepatic carcinoma (EXOCARTA, Reference [53]) and in microvesicles or ectosomes (Vesiclepedia, [54]). It has been indirectly demonstrated that PARP-1 has a role in intercellular communication through exosomes [54,55]. (Vesiclepedia, [54]). Moreover, PAR in extracellular vesicles acts as a paracrine signal [56].
- OLA targeted PARP-3 in addition to PARP-1/2. Although OLA has been well-known in the PARP scientific community for a long time as a specific PARP-1/2 inhibitor, it has recently been demonstrated that it can also inhibit PARP-3 in vitro enzymatic activity [57]. Therefore, a new question arises as to whether OLA can inhibit PARP-3 like MEO, which explains why OLA displayed similar effects to those already who demonstrated for MEO in other systems. While we do not have the answer to this question yet, we do know that PARP-3 does not synthetize PAR. Therefore, neither PAR increase during EMT, nor PAR decrease in the presence of OLA can be explained by invoking only PARP-3. In addition, 5 µM OLA does not significantly diminish PARP-3 activity in the BT-20 breast cell line [58], which suggests that the in vivo effect of OLA on PARP-3 activity is not as evident as in vitro. OLA had non-target effects such as on the ERK (extracellular signal-regulated kinases) pathway ([46,47]) or as a substrate of permeability glycoprotein 1 (Pgp) [48,59], which is a pump that exports toxins or drugs out of cells. Pgp is overexpressed on the surface of many neoplastic cells [60] and in extracellular vesicles that mediate multi-drug resistance in prostate cancer cells [61].
4. Materials and Methods
4.1. Cell Culture and Treatments
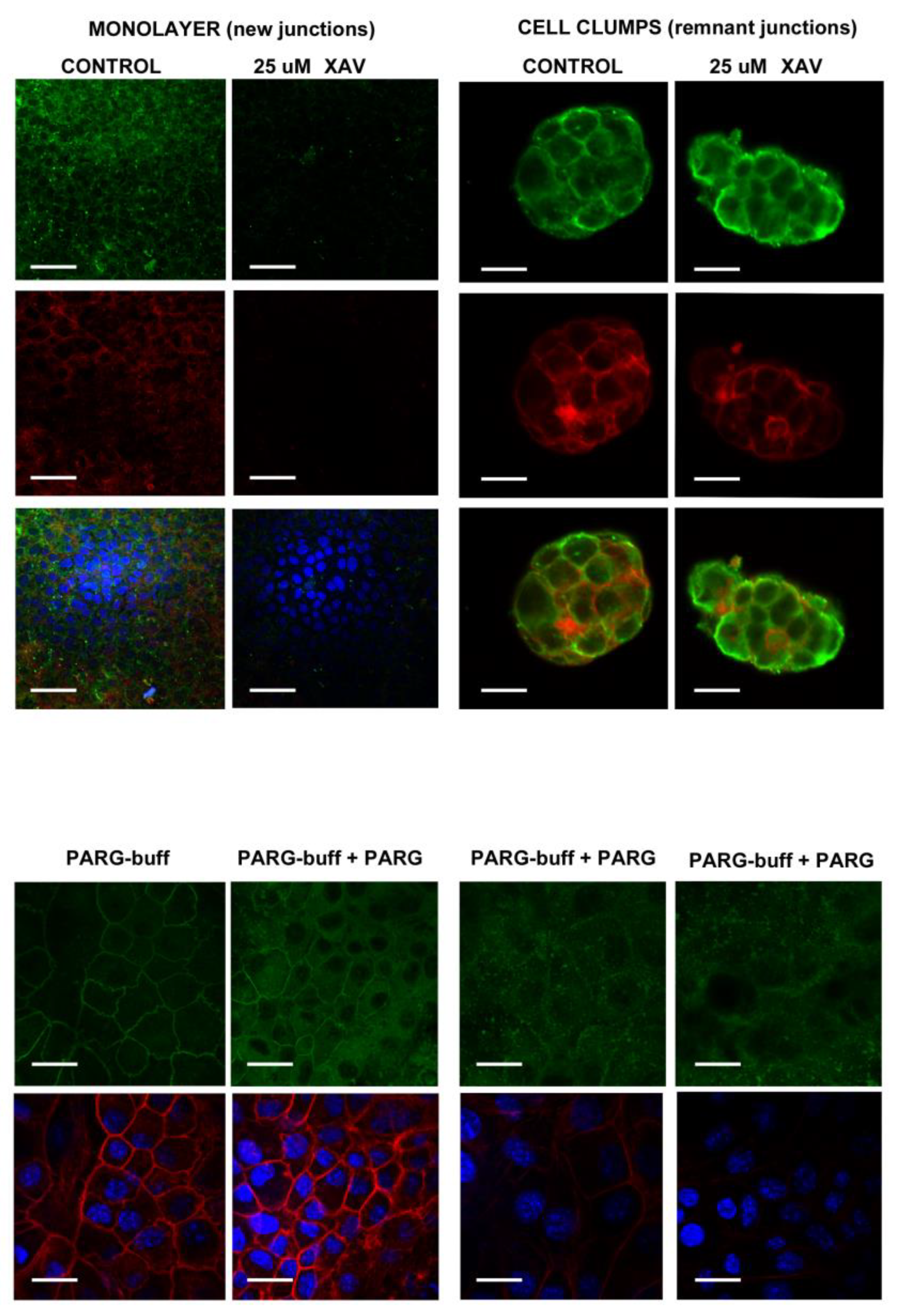
4.2. Specificity of the Anti-PAR Antibodies Signal in NMuMG Cells
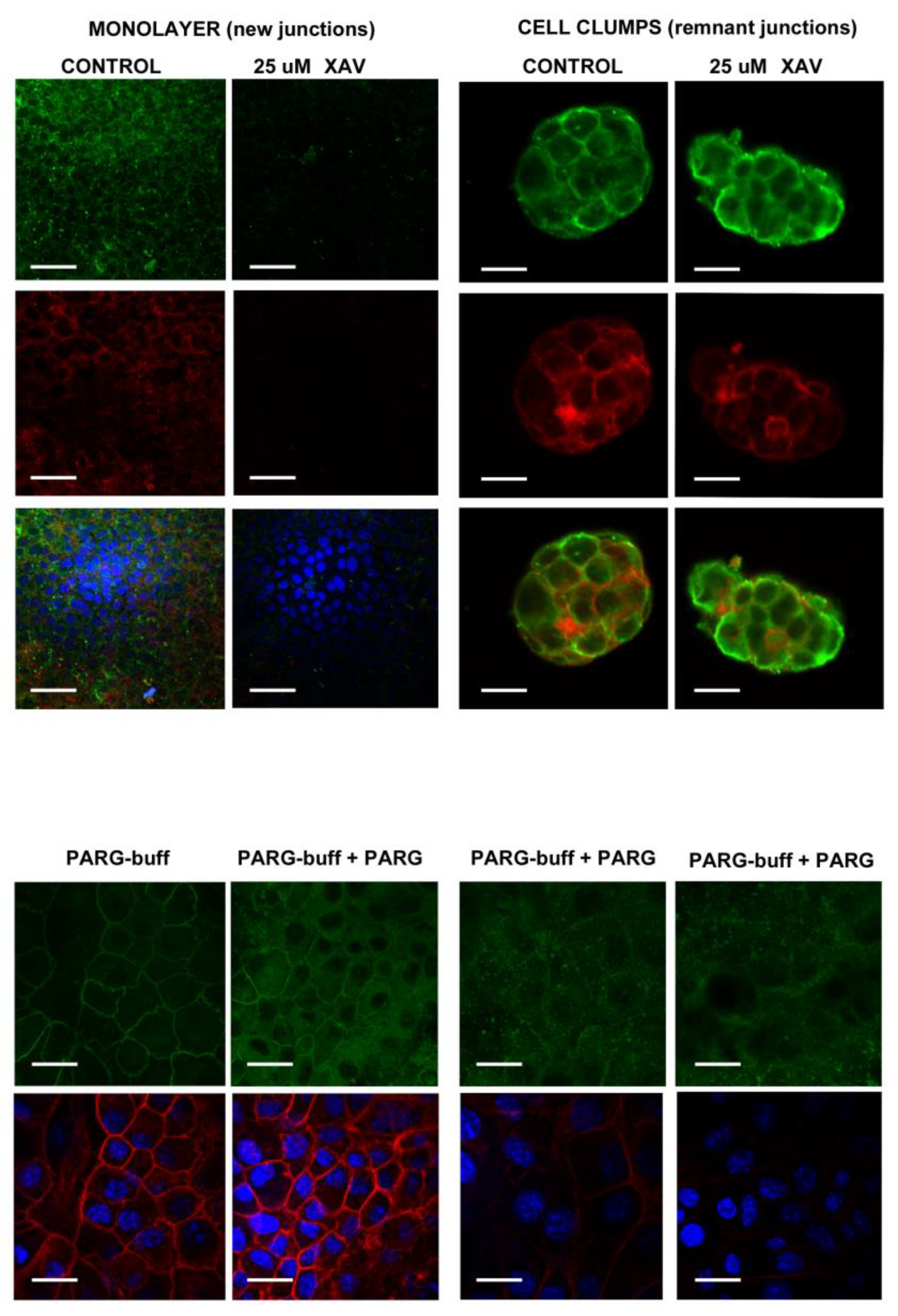
4.2.1. PAR Belt Synthesis Inhibition by XAV
4.2.2. PAR Digestion on Fixed Cells
4.3. Immunocytofluorescence (ICF) and Confocal Microscopy
4.4. Image Analysis
4.4.1. Signal Intensity
4.4.2. Anisotropy
4.4.3. Nuclear Intensity Was Quantified Using the Image J Intensity Ratio Nuclei Cytoplasm Macro
4.5. RNA Isolation and quantitative realtime polymerase chain reaction (qRT-PCR)
4.6. Protein Isolation and Western Blots
4.7. Migration Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DAPI | 4′,6-diamidino-2-phenylindole. Fluorescent DNA marker. |
| DMEM | Dulbecco’s Modified Eagle Medium |
| E-cad | E-cadherin (CDH1) |
| EMT | Epithelial to mesenchymal transition |
| F-actin | Filamentous actin |
| FBS | Fetal bovine serum |
| fFBS | Filtered fetal bovine serum |
| fPBS | Filtered phosphate buffer saline |
| fPFA | Filtered paraformaldehyde |
| ICF | Immuno-cytofluorescence |
| MEO | ME0328, PARP-3 inhibitor (S)-3,4-Dihydro-4-oxo-N-(1-phenylethyl)-2-quinazolinepropanamide |
| MET | Mesenchymal to epithelial transition |
| NAD+ | Nicotinamide adenine dinucleotide |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| OLA | Olaparib (PARP-1/2 inhibitor). Synonym: AZD 2461, AZD-2461 4-[4-Fluoro-3-[(4-methoxypiperidin-1-yl)carbonyl]benzyl]phthalazin-1(2H)-one, 4-[[4-Fluoro-3-[(4-methoxy-1-piperidinyl)carbonyl]phenyl]methyl]-1(2H)-phthalazinone, |
| PAR | Poly(ADP-ribose) |
| PBM | PDZ domain binding motif (PBM) |
| PBZ | PAR-binding zinc-finger domain |
| PARG | Poly(ADP-ribosyl)glycohydrolase |
| PARP | Poly(ADP-ribosyl)polymerase |
| PARPi | PAR inhibitor |
| PARylation | Poly(ADP-ribosylation) |
| RRM | RNA recognition motif domain |
| SB | SB-431542 |
| TBRI | TGF-β receptor type I |
| TBRII | TGF-β receptor type II |
| TBRIII | TGF-β receptor type III |
| TGF-β | Tumor growth factor β1 |
| TNKS | Tankyrase |
| VCL | Vinculin |
| VIM | Vimentin |
| XAV | XAV939 (TNKS inhibitor) 3,5,7,8-Tetrahydro-2-[4-(trifluoromethyl)phenyl]-4H-thiopyrano[4,3-d]pyrimidin-4-one |
| β-cat: | β-catenin |
| Wnt | Wnt is an acronym in the field of genetics that stands for ’Wingless/Integrated’. Wnt proteins are a family of secreted signalling molecules. Wnt pathways begin with Wnt acting on a cell surface receptors and harbor clinical importance in carcinogenesis |
| WWE | protein residue named after three of its conserved residues.W: tryptophans; E:glutamate |
Appendix A. Anti-PAR Antibodies Validation
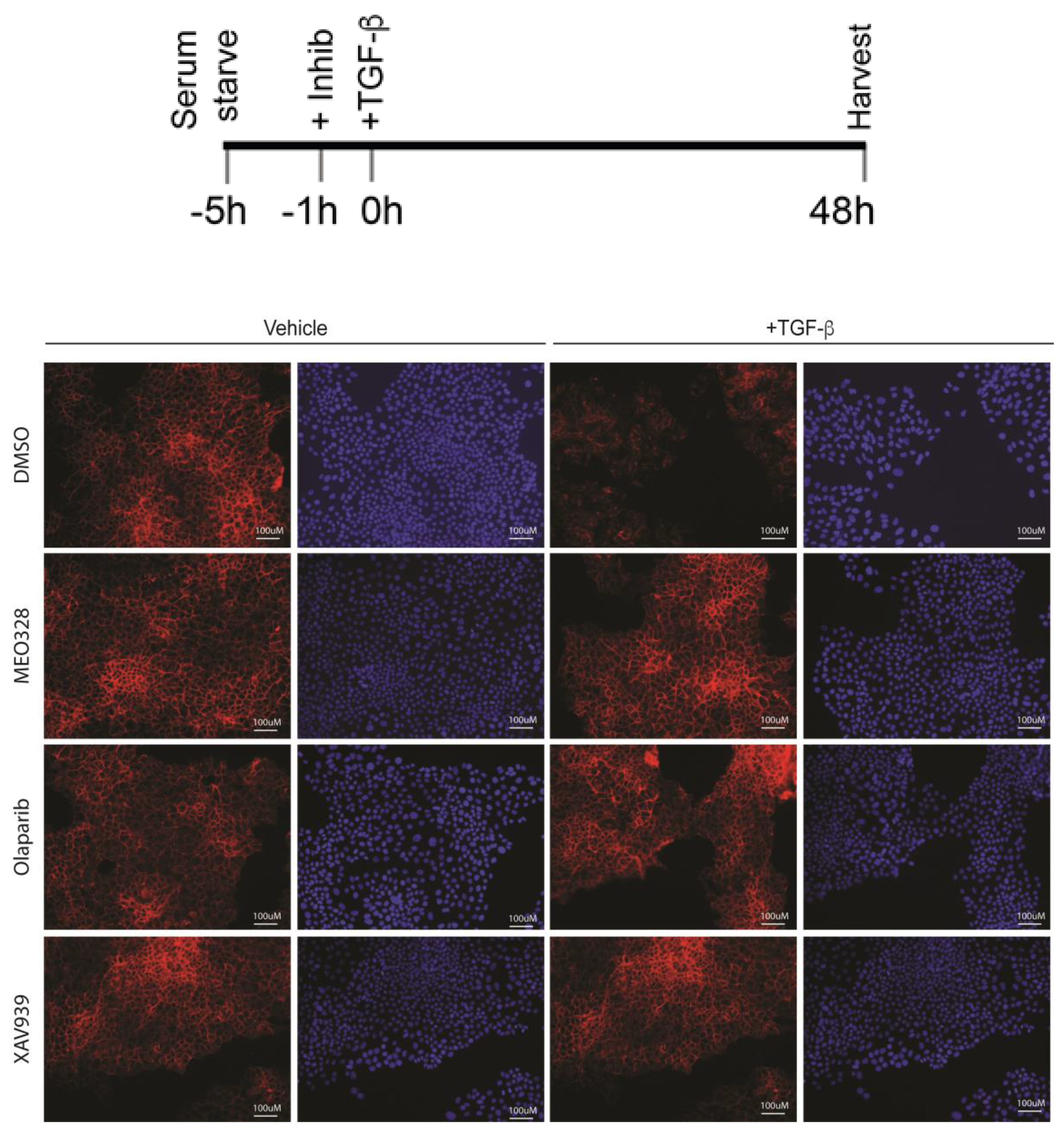
Appendix B. Low-Magnification ICF under the Indicated Schedule. See Figure 3 in the Main Text
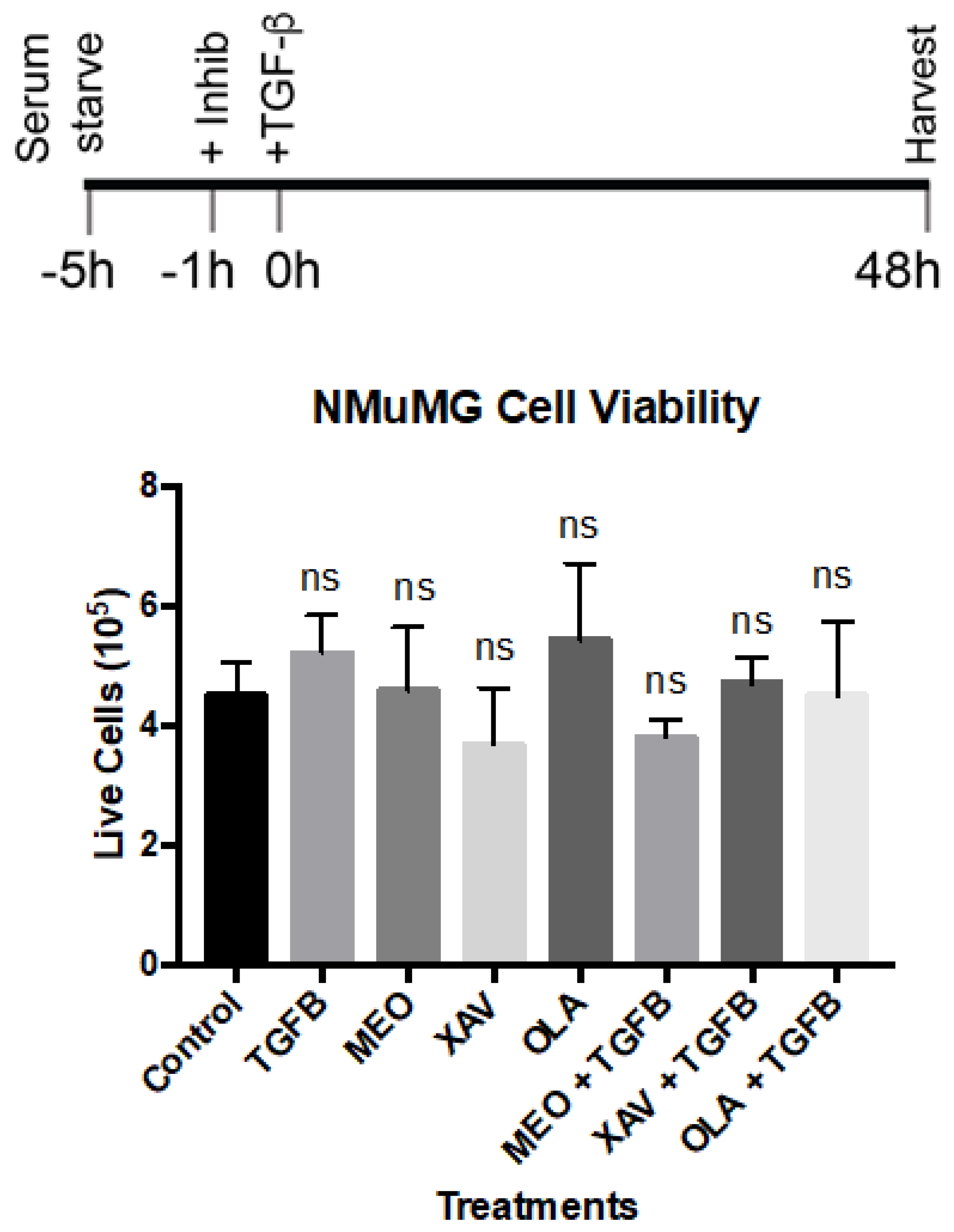
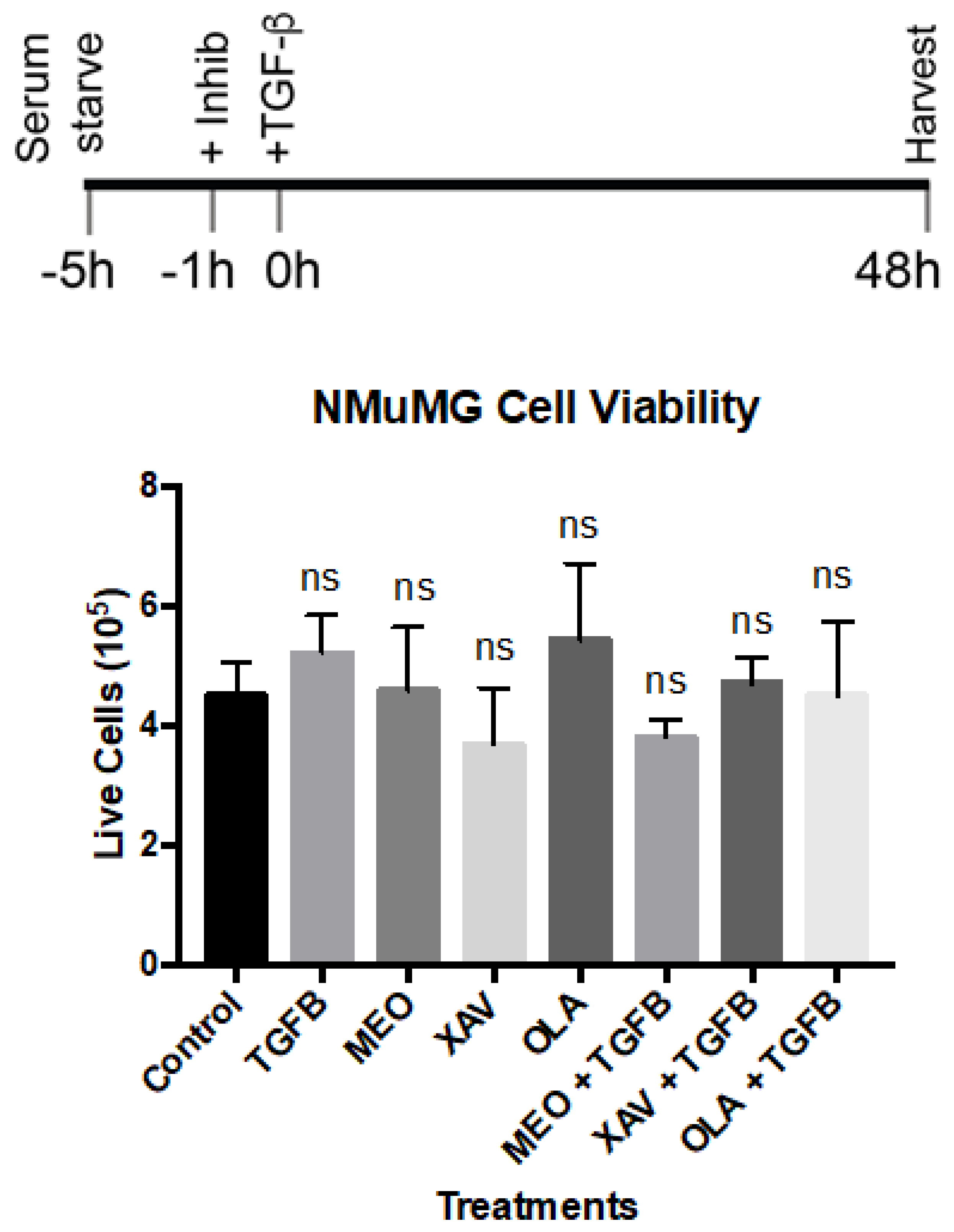
Appendix C. Trypan Blue (Dye Exclusion) Viability Assay. See Figure 3 in the Main Text
References
- Kanai, Y. Overview of poly(ADP-ribose) immuno-biomedicine and future prospects. Proc. Jpn. Acad. 2016, 92, 222–236. [Google Scholar] [PubMed]
- Leung, A.K.L. Poly(ADP-ribose): An organizer of cellular architecture. J. Cell Biol. 2014, 205, 613–619. [Google Scholar] [CrossRef] [Green Version]
- Hottiger, M.O. SnapShot: ADP-Ribosylation Signaling. Mol. Cell 2015, 58, 1134. [Google Scholar] [CrossRef] [PubMed]
- Masutani, M.; Nakagama, H.; Sugimura, T. Poly(ADP-ribosyl)ation in relation to cancer and autoimmune disease. Cell. Mol. Life Sci. 2005, 62, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Lafon-Hughes, L.; Di Tomaso, M.V.; Méndez-Acuña, L.; Martínez-López, W. Chromatin-remodelling mechanisms in cancer. Mutat. Res. Rev. Mutat. Res. 2008, 658, 191–214. [Google Scholar] [CrossRef]
- Cerboni, B.; Di Stefano, A.; Micheli, V.; Morozzi, G.; Pompucci, G.; Sestini, S. PARP activity and NAD concentration in PMC from patients affected by systemic sclerosis and lupus erythematosus. Nucleosides Nucleotides Nucleic Acids 2010, 29, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Virag, L.; Szabo, C. The Therapeutic Potential of Poly(ADP-Ribose) Polymerase Inhibitors. Pharmacol. Rev. 2002, 54, 375–429. [Google Scholar] [PubMed]
- Lafon-Hughes, L.I.; Romeo Cardeillac, C.J.; Cal Castillo, K.B.; Vilchez Larrea, S.C.; Sotelo Sosa, J.R.; Folle Ungo, G.A.; Fernández Villamil, S.H.; Kun González, A.E. Poly(ADP-ribosylation) is present in murine sciatic nerve fibers and is altered in a Charcot-Marie-Tooth-1E neurodegenerative model. PeerJ 2017, 5, e3318. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.; Aslam, H.M.; Anwar, S. PARP inhibitors. Hereditary Cancer Clin. Pract. 2015, 13, 4. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Perina, D.; Mikoč, A.; Ahel, J.; Ćetković, H.; Žaja, R.; Ahel, I. Distribution of protein poly(ADP-ribosyl)ation systems across all domains of life. DNA Repair 2014, 23, 4–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T. Europe PMC Funders Group Family-wide analysis of poly (ADP-ribose) polymerase activity. Nat. Commun. 2014, 1–29. [Google Scholar]
- Sukhanova, M.V.; Abrakhi, S.; Joshi, V.; Pastre, D.; Kutuzov, M.M.; Anarbaev, R.O.; Curmi, P.A.; Hamon, L.; Lavrik, O.I. Single molecule detection of PARP1 and PARP2 interaction with DNA strand breaks and their poly(ADP-ribosyl)ation using high-resolution AFM imaging. Nucleic Acids Res. 2016, 44, e60. [Google Scholar] [CrossRef] [PubMed]
- Bai, P. Review Biology of Poly (ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [PubMed]
- Barkauskaite, E.; Jankevicius, G.; Ahel, I. Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Mol. Cell 2015, 58, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.-H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240. [Google Scholar] [CrossRef] [Green Version]
- Huet, J.; Laval, F. Potentiation of Cell Killing by Inhibitors of Poly (Adenosine Diphosphate-Ribose) Synthesis in Bleomycin-treated Chinese Hamster Ovary Cells. Cancer Res. 1985, 45, 987–991. [Google Scholar] [PubMed]
- Russo, A.L.; Kwon, H.-C.; Burgan, W.E.; Carter, D.; Beam, K.; Weizheng, X.; Zhang, J.; Slusher, B.S.; Chakravarti, A.; Tofilon, P.J.; et al. In vitro and In vivo Radiosensitization of Glioblastoma Cells by the Poly (ADP-Ribose) Polymerase Inhibitor E7016. Clin. Cancer Res. 2009, 15, 607–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vodenicharov, M.D. Mechanism of early biphasic activation of poly(ADP-ribose) polymerase-1 in response to ultraviolet B radiation. J. Cell Sci. 2005, 118, 589–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, W.L. PARPs and ADP-Ribosylation: 50 Years... and Counting. Mol. Cell 2015, 58, 902–910. [Google Scholar] [CrossRef]
- Yeh, T.-Y.J.; Meyer, T.N.; Schwesinger, C.; Tsun, Z.-Y.; Lee, R.M.; Chi, N.-W. Tankyrase recruitment to the lateral membrane in polarized epithelial cells: Regulation by cell–cell contact and protein poly(ADP-ribosyl)ation. Biochem. J. 2006, 399, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Lafon-Hughes, L.; Vilchez Larrea, S.C.; Kun, A.; Fernández Villamil, S.H. VERO cells harbor a poly-ADP-ribose belt partnering their epithelial adhesion belt. PeerJ 2014, 2, e617. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Neilson, E.G. Review series personal perspective Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming Growth Factor-β1 Mediates Epithelial to Mesenchymal Transdifferentiation through a RhoA-dependent Mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhou, B.P. Epithelial-mesenchymal Transition—A Hallmark of Breast Cancer Metastasis. Cancer Hallm. 2013, 1, 38–49. [Google Scholar] [PubMed]
- Cooper, G.; Boston, U. The Development of cancer. In The Cell: A Molecular Approach; Sinauer Associates, Oxford University Press: Boston, MA, USA, 2000; ISBN 10 0-87893-106-6. Available online: https://www.ncbi.nlm.nih.gov/books/NBK9963/ (accessed on 24 January 2019).
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef]
- Tsubakihara, Y.; Moustakas, A. Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β. Int. J. Mol. Sci. 2018, 19, 3672. [Google Scholar] [CrossRef] [PubMed]
- Aroeira, L.S.; Aguilera, A.; Sanchez-Tomero, J.A.; Bajo, M.A.; del Peso, G.; Jimenez-Heffernan, J.A.; Selgas, R.; Lopez-Cabrera, M. Epithelial to Mesenchymal Transition and Peritoneal Membrane Failure in Peritoneal Dialysis Patients: Pathologic Significance and Potential Therapeutic Interventions. J. Am. Soc. Nephrol. 2007, 18, 2004–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Law, B.K.; Aakre, M.E.; Edgerton, M.; Shyr, Y.; Bhowmick, N.A.; Moses, H.L. Transforming growth factor beta-regulated gene expression in a mouse mammary gland epithelial cell line. Breast Cancer Res. 2003, 5, R187. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, M.; Bernabeu, C.; Nez, J.F.S. TGF-β/TGF-β receptor system and its role in physiological and pathological conditions. Clin. Sci. 2011, 251, 233–251. [Google Scholar]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal 2014, 7, re8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb. Perspect. Biol. 2017, 9, a022129. [Google Scholar] [CrossRef] [PubMed]
- Karicheva, O.; Rodriguez-Vargas, J.M.; Wadier, N.; Martin-Hernandez, K.; Vauchelles, R.; Magroun, N.; Tissier, A.; Schreiber, V.; Dantzer, F. PARP3 controls TGFβ and ROS driven epithelial-to-mesenchymal transition and stemness by stimulating a TG2-Snail-E-cadherin axis. Oncotarget 2014, 64109–64123. [Google Scholar]
- Hassa, P.O.; Hottiger, M.O. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of. Cell. Mol. Life Sci. 2002, 59, 1534–1553. [Google Scholar] [CrossRef]
- Huang, D.; Wang, Y.; Wang, L.; Zhang, F.; Deng, S.; Wang, R.; Zhang, Y.; Huang, K. Poly(ADP-ribose) polymerase 1 is indispensable for transforming growth factor-beta Induced Smad3 activation in vascular smooth muscle cell. PLoS ONE 2011, 6, e27123. [Google Scholar]
- Lönn, P.; van der Heide, L.P.; Dahl, M.; Hellman, U.; Heldin, C.H.; Moustakas, A. PARP-1 attenuates smad-mediated transcription. Mol. Cell 2010, 40, 521–532. [Google Scholar] [CrossRef]
- Dahl, M.; Maturi, V.; Lönn, P.; Papoutsoglou, P.; Zieba, A.; Vanlandewijck, M.; van der Heide, L.P.; Watanabe, Y.; Söderberg, O.; Hottiger, M.O.; et al. Fine-Tuning of Smad Protein Function by Poly(ADP-Ribose) Polymerases and Poly(ADP-Ribose) Glycohydrolase during Transforming Growth Factor β Signaling. PLoS ONE 2014, 9, e103651. [Google Scholar] [CrossRef] [PubMed]
- Rajan, J.V.; Wang, M.; Marquis, S.T.; Chodosh, L.A. Brca2 is coordinately regulated with Brca1 during proliferation and differentiation in mammary epithelial cells. Proc. Natl. Acad. Sci. USA 1996, 93, 13078–13083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inman, G.J.; Nicolás, F.J.; Callahan, J.F.; Harling, J.D.; Gaster, L.M.; Reith, A.D.; Laping, N.J.; Hill, C.S. SB-431542 Is a Potent and Specific Inhibitor of Transforming Growth Factor-β Superfamily Type I Activin Receptor-Like Kinase (ALK) Receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 2002, 62, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Boudaoud, A.; Burian, A.; Borowska-Wykręt, D.; Uyttewaal, M.; Wrzalik, R.; Kwiatkowska, D.; Hamant, O. FibrilTool, an ImageJ plug-in to quantify fibrillar structures in raw microscopy images. Nat. Protoc. 2014, 9, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Shen, F.; Yang, L.; Zhang, L.; Guo, W.; Tian, J. Inhibitory effects of XAV939 on the proliferation of small-cell lung cancer H446 cells and Wnt/beta-catenin signaling pathway in vitro. Oncol. Lett. 2018, 16, 1953–1958. [Google Scholar] [PubMed]
- Curtin, N.; Sharma, R. PARP Inhibitors for Cancer Therapy, 1st ed.; Cancer Drug Discovery and Development; Humana Press, Springer: New York, NY, USA, 2015; ISBN 978-3-319-14151-0. [Google Scholar]
- Pu, H.; Horbinski, C.; Hensley, P.J.; Matuszak, E.A.; Atkinson, T.; Kyprianou, N. PARP-1 regulates epithelial-mesenchymal transition (EMT) in prostate tumorigenesis. Carcinogenesis 2014, 35, 2592–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, M.; Johnson, K.R.; Wheelock, M.J. Cadherin switching: Essential for behavioral but not morphological changes during an epithelium-to-mesenchyme transition. J. Cell Sci. 2005, 118, 873–887. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2018. [Google Scholar] [CrossRef]
- Keerthikumar, S.; Chisanga, D.; Ariyaratne, D.; Al Saffar, H.; Anand, S.; Zhao, K.; Samuel, M.; Pathan, M.; Jois, M.; Chilamkurti, N.; et al. ExoCarta: A Web-Based Compendium of Exosomal Cargo. J. Mol. Biol. 2016, 428, 688–692. [Google Scholar] [CrossRef] [Green Version]
- Kalra, H.; Simpson, R.J.; Ji, H.; Aikawa, E.; Altevogt, P.; Askenase, P.; Bond, V.C.; Borras, F.E.; Breakefield, X.; Budnik, V.; et al. Vesiclepedia: A compendium for extracellular vesicles with continuous community annotation. PLoS Biol. 2012, 10, e1001450. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, T.; Sasaki, Y.; Fukuda, I.; Isumi, M.; Nakamoto, K. Next-generation sequencing-based miRNA expression analysis in Parp1-deficient embryonic stem cell-derived exosomes. Biochem. Biophys. Res. Commun. 2018, 499, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Krukenberg, K.A.; Kim, S.; Tan, E.S.; Maliga, Z.; Mitchison, T.J. Extracellular poly(ADP-ribose) is a pro-inflammatory signal for macrophages. Chem. Biol. 2015, 22, 446–452. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, O.L.; Rulten, S.L.; Cranston, A.N.; Odedra, R.; Brown, H.; Jaspers, J.E.; Jones, L.; Knights, C.; Evers, B.; Ting, A.; et al. The PARP Inhibitor AZD2461 Provides Insights into the Role of PARP3 Inhibition for Both Synthetic Lethality and Tolerability with Chemotherapy in Preclinical Models. Cancer Res. 2016, 76, 6084–6094. [Google Scholar] [Green Version]
- Sharif-Askari, B.; Amrein, L.; Aloyz, R.; Panasci, L. PARP3 inhibitors ME0328 and olaparib potentiate vinorelbine sensitization in breast cancer cell lines. Breast Cancer Res. Treat. 2018, 172, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J. PARP inhibitors for cancer therapy. Expert Rev. Mol. Med. 2005, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.L. P-glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 7, DTI.S12519. [Google Scholar] [CrossRef] [PubMed]
- Soekmadji, C.; Nelson, C.C. The Emerging Role of Extracellular Vesicle-Mediated Drug Resistance in Cancers: Implications in Advanced Prostate Cancer. BioMed Res. Int. 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Kumar, J.; Hermanson, K.; Sun, Y.; Qureshi, H.; Perley, D.; Scheidegger, A.; Singh, B.B.; Dhasarathy, A. The calcium channel proteins ORAI3 and STIM1 mediate TGF-β induced Snai1 expression. Oncotarget 2018, 9, 29468–29483. [Google Scholar] [CrossRef]
- Lim, R.; Ahmed, N.; Borregaard, N.; Riley, C.; Wafai, R.; Thompson, E.W.; Quinn, M.A.; Rice, G.E. Neutrophil gelatinase-associated lipocalin (NGAL) an early-screening biomarker for ovarian cancer: NGAL is associated with epidermal growth factor-induced epithelio-mesenchymal transition. Int. J. Cancer 2007, 120, 2426–2434. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.W.; Lim, C.J.; Guo, K.; Ng, C.P.; Lee, I.; Hunziker, W.; Zeng, Q.; Hong, W. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 2008, 68, 2592–2598. [Google Scholar] [CrossRef] [PubMed]
- Baeker, V. ImageJ macro tool sets for biological image analysis. In Proceedings of the ImageJ User and Developer Conference, Luxembourg, 24–26 October 2012. [Google Scholar]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schacke, M.; Kumar, J.; Colwell, N.; Hermanson, K.; Folle, G.A.; Nechaev, S.; Dhasarathy, A.; Lafon-Hughes, L. PARP-1/2 Inhibitor Olaparib Prevents or Partially Reverts EMT Induced by TGF-β in NMuMG Cells. Int. J. Mol. Sci. 2019, 20, 518. https://doi.org/10.3390/ijms20030518
Schacke M, Kumar J, Colwell N, Hermanson K, Folle GA, Nechaev S, Dhasarathy A, Lafon-Hughes L. PARP-1/2 Inhibitor Olaparib Prevents or Partially Reverts EMT Induced by TGF-β in NMuMG Cells. International Journal of Molecular Sciences. 2019; 20(3):518. https://doi.org/10.3390/ijms20030518
Chicago/Turabian StyleSchacke, Michelle, Janani Kumar, Nicholas Colwell, Kole Hermanson, Gustavo A. Folle, Sergei Nechaev, Archana Dhasarathy, and Laura Lafon-Hughes. 2019. "PARP-1/2 Inhibitor Olaparib Prevents or Partially Reverts EMT Induced by TGF-β in NMuMG Cells" International Journal of Molecular Sciences 20, no. 3: 518. https://doi.org/10.3390/ijms20030518
APA StyleSchacke, M., Kumar, J., Colwell, N., Hermanson, K., Folle, G. A., Nechaev, S., Dhasarathy, A., & Lafon-Hughes, L. (2019). PARP-1/2 Inhibitor Olaparib Prevents or Partially Reverts EMT Induced by TGF-β in NMuMG Cells. International Journal of Molecular Sciences, 20(3), 518. https://doi.org/10.3390/ijms20030518