Kidney Injury by Variants in the COL4A5 Gene Aggravated by Polymorphisms in Slit Diaphragm Genes Causes Focal Segmental Glomerulosclerosis

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Results
2.1. Clinical Presentation
2.2. Genetic Analyses of COL4A3/4/5 GBM Genes and NPHS1/2 Slit Diaphragm Genes
2.3. Slit Diaphragm Gene Polymorphisms Aggravate Glomerular Architecture towards FSGS in Patients with GBM Mutations
3. Discussion
4. Methods
4.1. Ethical Considerations
4.2. Genetic Analyses
4.3. Morphological Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- D’Agati, V.D.; Kaskel, F.J.; Falk, R.J. Focal segmental glomerulosclerosis. N. Engl. J. Med. 2011, 365, 2398–2411. [Google Scholar] [CrossRef] [PubMed]
- Brinkkoetter, P.T.; Ising, C.; Benzing, T. The role of the podocyte in albumin filtration. Nat. Rev. Nephrol. 2013, 9, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.G.; Tryggvason, K.; Sundaramoorthy, M.; Neilson, E.G. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N. Engl. J. Med. 2003, 348, 2543–2556. [Google Scholar] [CrossRef] [PubMed]
- Kruegel, J.; Rubel, D.; Gross, O. Alport syndrome—Insights from basic and clinical research. Nat. Rev. Nephrol. 2013, 9, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Storey, H.; Savige, J.; Sivakumar, V.; Abbs, S.; Flinter, F.A. COL4A3/COL4A4 mutations and features in individuals with autosomal recessive Alport syndrome. J. Am. Soc. Nephrol. 2013, 24, 1945–1954. [Google Scholar] [CrossRef] [PubMed]
- Gubler, M.C. Podocyte differentiation and hereditary proteinuria/nephrotic syndromes. J. Am. Soc. Nephrol. 2003, 14 (Suppl. 1), S22–S26. [Google Scholar] [CrossRef] [PubMed]
- Niaudet, P. Genetic forms of nephrotic syndrome. Pediatr. Nephrol. 2004, 19, 1313–1318. [Google Scholar] [CrossRef]
- Fogo, A.B. Causes and pathogenesis of focal segmental glomerulosclerosis. Nat. Rev. Nephrol. 2015, 11, 76–87. [Google Scholar] [CrossRef]
- Malone, A.F.; Phelan, P.J.; Hall, G.; Cetincelik, U.; Homstad, A.; Alonso, A.S.; Jiang, R.; Lindsey, T.B.; Wu, G.; Sparks, M.A.; et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014, 86, 1253–1259. [Google Scholar] [CrossRef]
- Megged, O.; Stein, J.; Ben-Meir, D.; Shulman, L.M.; Yaniv, I.; Shalit, I.; Levy, I. BK-virus-associated hemorrhagic cystitis in children after hematopoietic stem cell transplantation. J. Pediatr. Hematol. Oncol. 2011, 33, 190–193. [Google Scholar] [CrossRef]
- Kloos, R.Q.; Boelens, J.J.; de Jong, T.P.; Versluys, B.; Bierings, M. Hemorrhagic cystitis in a cohort of pediatric transplantations: Incidence, treatment, outcome, and risk factors. Biol. Blood Marrow Transplant. 2013, 19, 1263–1266. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Licht, C.; Anders, H.J.; Hoppe, B.; Beck, B.; Tönshoff, B.; Höcker, B.; Wygoda, S.; Ehrich, J.H.; Pape, L.; et al. Early angiotensin-converting enzyme inhibition in Alport syndrome delays renal failure and improves life expectancy. Kidney Int. 2012, 81, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Friede, T.; Hilgers, R.; Görlitz, A.; Gavénis, K.; Ahmed, R.; Dürr, U. Safety and Efficacy of the ACE-Inhibitor Ramipril in Alport Syndrome: The Double-Blind, Randomized, Placebo-Controlled, Multicenter Phase III EARLY PRO-TECT Alport Trial in Pediatric Patients. ISRN Pediatr. 2012, 2012, 436046. [Google Scholar] [CrossRef] [PubMed]
- Gross, O.; Netzer, K.O.; Lambrecht, R.; Seibold, S.; Weber, M. Meta-analysis of genotype-phenotype correlation in X-linked Alport syndrome: Impact on clinical counselling. Nephrol. Dial. Transplant. 2002, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.O.; Flinter, F.; et al. X-linked Alport syndrome: Natural history and genotype-phenotype correlations in girls and women belonging to 195 families: A “European Community Alport Syndrome Concerted Action” study. J. Am. Soc. Nephrol. 2003, 14, 2603–2610. [Google Scholar] [CrossRef] [PubMed]
- Knebelmann, B.; Breillat, C.; Forestier, L.; Arrondel, C.; Jacassier, D.; Giatras, I.; Drouot, L.; Deschênes, G.; Grünfeld, J.P.; Broyer, M.; et al. Spectrum of mutations in the COL4A5 collagen gene in X-linked Alport syndrome. Am. J. Hum. Genet. 1996, 59, 1221–1232. [Google Scholar] [PubMed]
- Voskarides, K.; Arsali, M.; Athanasiou, Y.; Elia, A.; Pierides, A.; Deltas, C. Evidence that NPHS2-R229Q predisposes to proteinuria and renal failure in familial hematuria. Pediatr. Nephrol. 2012, 27, 675–679. [Google Scholar] [CrossRef]
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681–691. [Google Scholar] [CrossRef]
- Morinière, V.; Dahan, K.; Hilbert, P.; Lison, M.; Lebbah, S.; Topa, A.; Bole-Feysot, C.; Pruvost, S.; Nitschke, P.; Plaisier, E.; et al. Improving Mutation Screening in Familial Hematuric Nephropathies through Next Generation Sequencing. J. Am. Soc. Nephrol. 2014, 25, 2740–2751. [Google Scholar] [CrossRef]
- Gibson, J.; Gilbert, R.D.; Bunyan, D.J.; Angus, E.M.; Fowler, D.J.; Ennis, S. Exome analysis resolves differential diagnosis of familial kidney disease and uncovers a potential confounding variant. Genet. Res. 2014, 28, 1–9. [Google Scholar] [CrossRef]
- Bullich, G.; Trujillano, D.; Santín, S.; Ballarín, J.; Torra, R.; Estivill, X.; Ars, E. Targeted next-generation sequencing in steroid-resistant nephrotic syndrome: Mutations in multiple glomerular genes may influence disease severity. Eur. J. Hum. Genet. 2014, 23, 1192. [Google Scholar] [CrossRef] [PubMed]
- Cravedi, P.; Kopp, J.B.; Remuzzi, G. Recent progress in the pathophysiology and treatment of FSGS recurrence. Am. J. Transplant. 2013, 13, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Sonnenberg, A. Cell-matrix adhesion of podocytes in physiology and disease. Nat. Rev. Nephrol. 2013, 9, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Ingelfinger, J.R. Blood-pressure control and delay in progression of kidney disease in children. N. Engl. J. Med. 2009, 361, 1701–1703. [Google Scholar] [CrossRef] [PubMed]
- Adam, J.; Connor, T.M.; Wood, K.; Lewis, D.; Naik, R.; Gale, D.P.; Sayer, J.A. Genetic testing can resolve diagnostic confusion in Alport syndrome. Clin. Kidney J. 2014, 7, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Fidler, A.L.; Vanacore, R.M.; Chetyrkin, S.V.; Pedchenko, V.K.; Bhave, G.; Yin, V.P.; Stothers, C.L.; Rose, K.L.; McDonald, W.H.; Clark, T.A.; et al. A unique covalent bond in basement membrane is a primordial innovation for tissue evolution. Proc. Natl. Acad. Sci. USA 2014, 111, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Welsh, G.I.; Saleem, M.A. The podocyte cytoskeleto—Key to a functioning glomerulus in health and disease. Nat. Rev. Nephrol. 2011, 8, 14–21. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Sudhakar, A.; Le, T.C.; Nguyen, T.; Yao, J.; Schwimmer, J.A.; Schachter, A.D.; Poch, E.; Abreu, P.F.; Appel, G.B.; et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J. Clin. Invest. 2002, 110, 1659–1666. [Google Scholar] [CrossRef]
- Ozaltin, F.; Ibsirlioglu, T.; Taskiran, E.Z.; Baydar, D.E.; Kaymaz, F.; Buyukcelik, M.; Kilic, B.D.; Balat, A.; Iatropoulos, P.; Asan, E.; et al. Disruption of PTPRO causes childhood-onset nephrotic syndrome. Am. J. Hum. Genet. 2011, 89, 139–147. [Google Scholar] [CrossRef]
- Tory, K.; Menyhárd, D.K.; Woerner, S.; Nevo, F.; Gribouval, O.; Kerti, A.; Stráner, P.; Arrondel, C.; Huynh Cong, E.; Tulassay, T.; et al. Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome. Nat. Genet. 2014, 46, 299–304. [Google Scholar] [CrossRef]
- Kerti, A.; Csohány, R.; Wagner, L.; Jávorszky, E.; Maka, E.; Tory, K. NPHS2 homozygous p.R229Q variant: Potential modifier instead of causal effect in focal segmental glomerulosclerosis. Pediatr. Nephrol. 2013, 28, 2061–2429. [Google Scholar] [CrossRef] [PubMed]
- Philippe, A.; Weber, S.; Esquivel, E.L.; Houbron, C.; Hamard, G.; Ratelade, J.; Kriz, W.; Schaefer, F.; Gubler, M.C.; Antignac, C. A missense mutation in podocin leads to early and severe renal disease in mice. Kidney Int. 2008, 73, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Tonna, S.; Wang, Y.Y.; Wilson, D.; Rigby, L.; Tabone, T.; Cotton, R.; Savige, J. The R229Q mutation in NPHS2 may predispose to proteinuria in thin-basement-membrane nephropathy. Pediatr. Nephrol. 2008, 23, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Beirowski, B.; Weber, M.; Gross, O. Chronic renal failure and shortened lifespan in COL4A3+/− mice: An animal model for thin basement membrane nephropathy. J. Am. Soc. Nephrol. 2006, 17, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.; Meehan, D.T.; Grunkemeyer, J.A.; Kornak, J.M.; Sayers, R.; Hunter, W.J.; Samuelson, G.C. Collagen COL4A3 knockout: A mouse model for autosomal Alport syndrome. Genes Dev. 1996, 10, 2981–2992. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
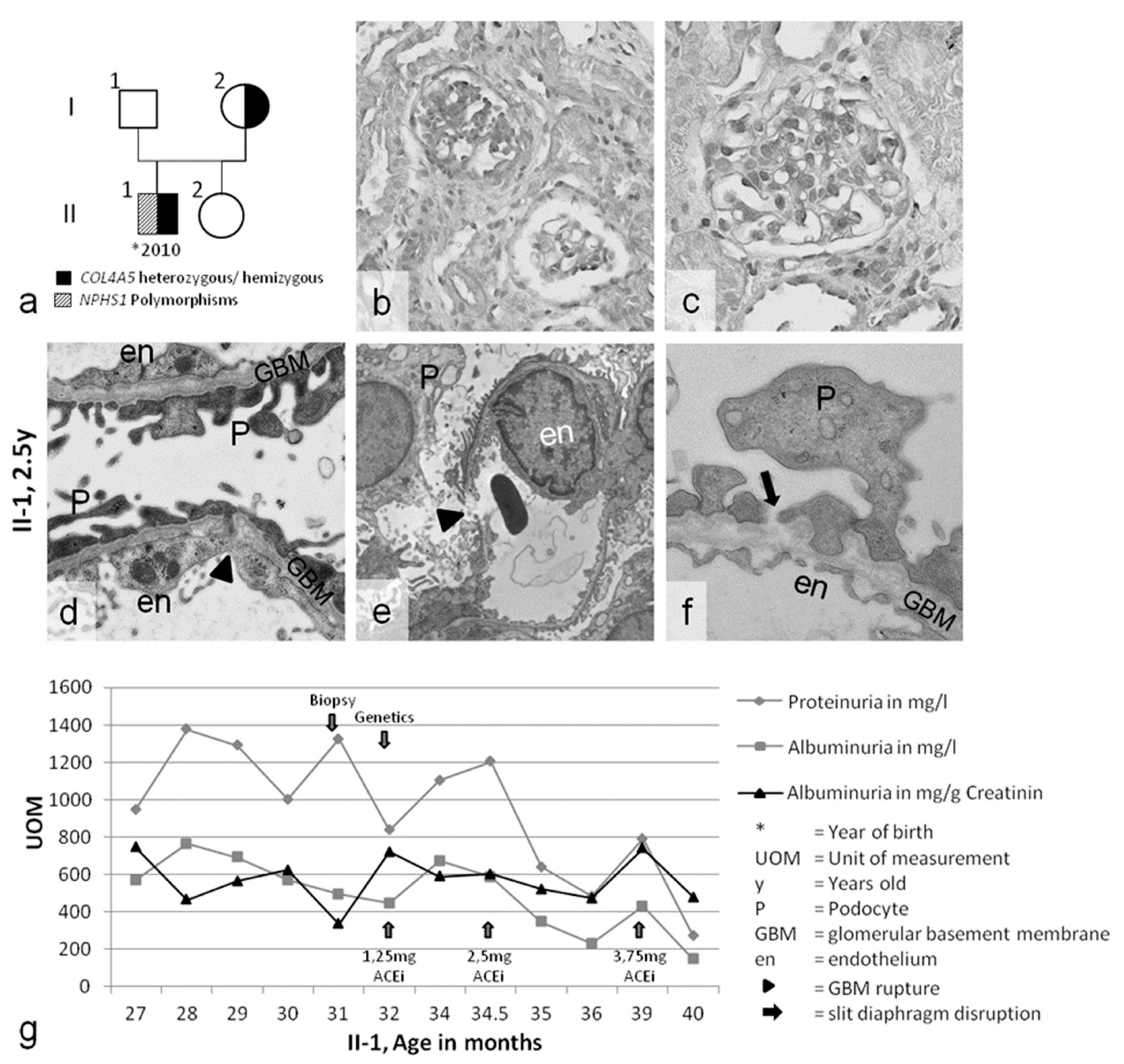
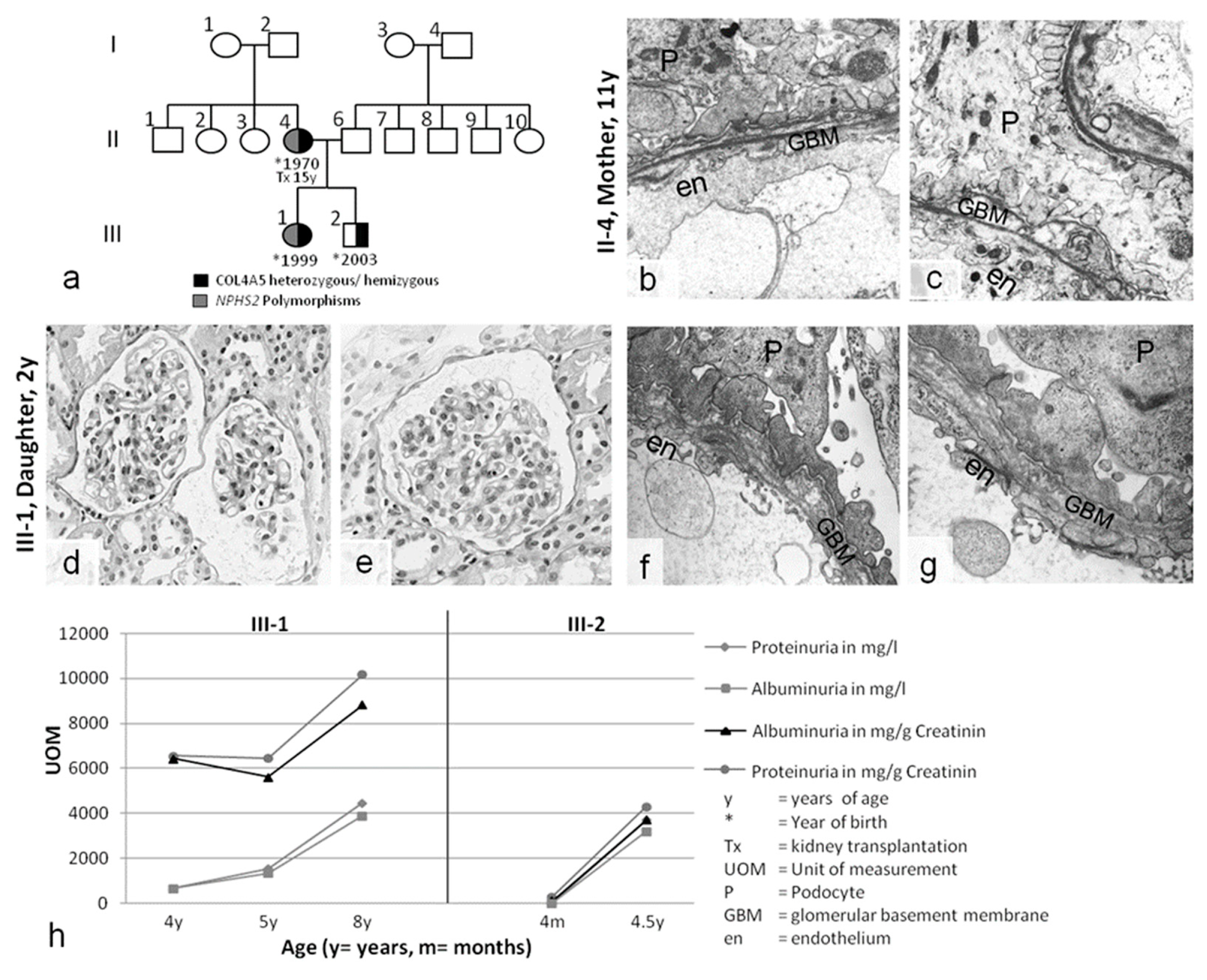
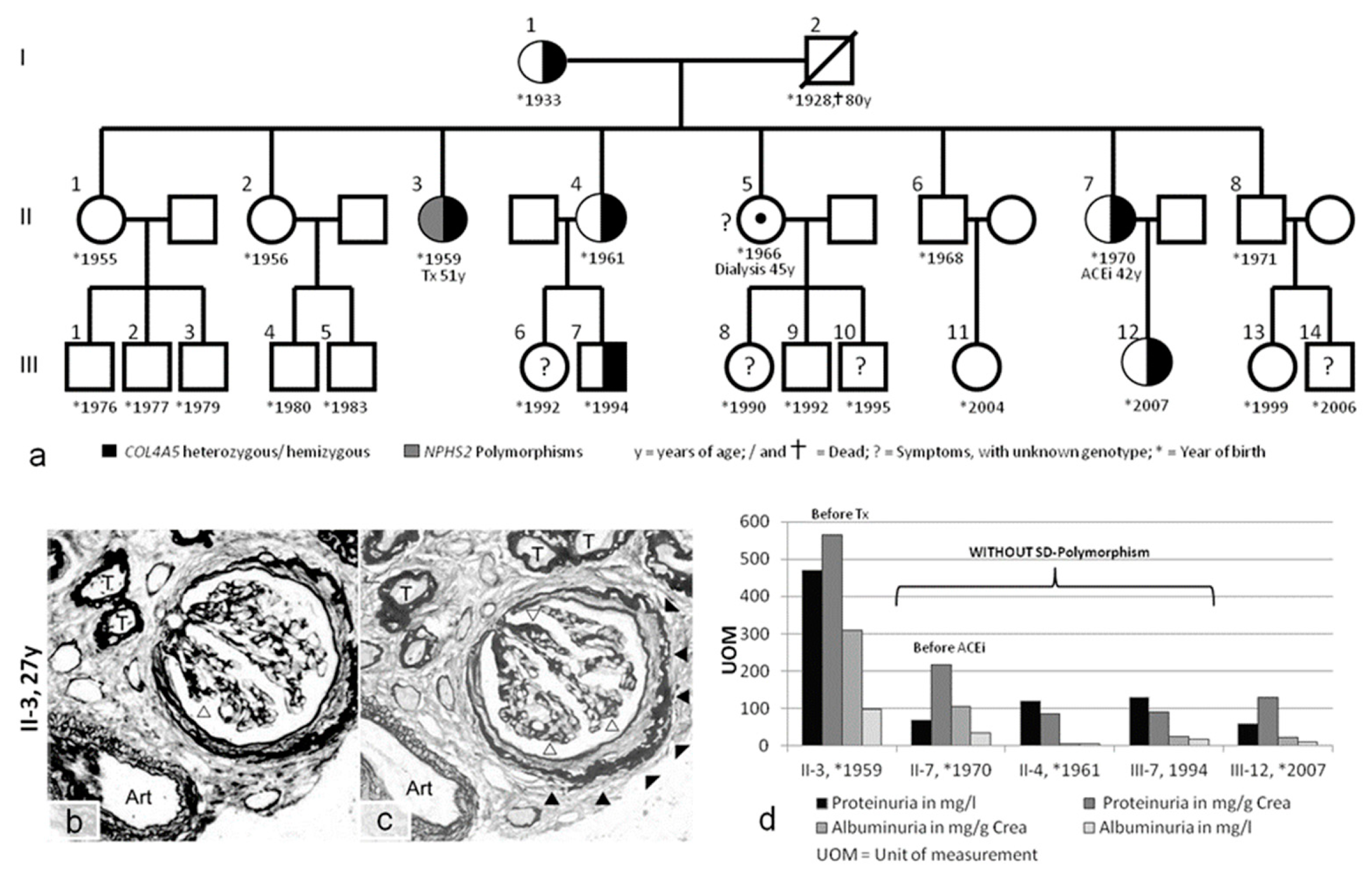
| Patient | Sex | First Clinical Presentation > Symptoms | Genotype COL4A5 | Genotype Nphs-1/-2 | Ear/Eye | Dialysis/Tx | Medication | Affected Family Members |
|---|---|---|---|---|---|---|---|---|
| Case 1 A.N. II-1 | ♂ | 27 months macrohematuria acanthocytes proteinuria urinary tract infection | COL4A5 p.W1538X (TGG>TGA) hemizygous | Nphs1: p.R408Q (CGG>CAG) polymorphism heterozygous Nphs1:p.S1105S(TCG>TCA) polymorphism homozygous Nphs2: p.G34G (GGA>GGG) polymorphism homozygous | no pathological findings | none | 2012–today ACEi | no kidney diseases known mother without symptoms: Nphs1: p.R408Q (CGG>CAG) polymorphism heterozygous |
| Case 2 C.V. II-4 | ♀ | 11 years macrohematuria proteinuria urinary tract infection | COL4A5 Exon 49, Codon 1510, IVS49+3A>G heterozygous | Nphs2: p.R229Q (CGA>CAA) polymorphism heterozygous | no pathological findings | Tx age: 15 | /refused treatment | no other family members affected |
| S.V. III-1 | ♀ | 1 year macrohematuria Proteinuria | COL4A5 Exon 49, Codon 1510, IVS49+3A>G heterozygous | Nphs2: p.R229Q (CGA>CAA) polymorphism heterozygous | no pathological findings | none | /refused treatment | |
| M.V. III-2 | ♂ | 4 months macrohematuria proteinuria | COL4A5 Exon 49, Codon 1510, IVS49+3A>G hemizygous | none | no pathological findings | none | /refuse treatment | |
| Case 3 L.U. II-3 | ♀ | 27 years hematuria proteinuria | COL4A5 p.G624D (GGT>GAT) heterozygous | Nphs2: p.R229Q (CGA>CAA) polymorphism heterozygous Nphs2: p.G34G(GGA>GGG) polymorphism homozygous | minimal high-frequency hearing loss (2013) | Tx age: 51 | Before Tx: ACEi from 33 years | see pedigree of family (Figure 3) |
| W.T. II-7 | ♀ | 40 years: microhematuria 42 years: proteinuria | COL4A5 p.G624D (GGT>GAT) heterozygous | none | retinal detachment | none | 02/2013: ACEi (discontinued due to angioedema) | |
| T.O. II.4 | ♀ | microhematuria | COL4A5 p.G624D (GGT>GAT) heterozygous | none | high-frequency hearing loss (2013) | none | none | |
| S.O. III-7 | ♂ | microhematuria | COL4A5 p.G624D (GGT>GAT) heterozygous | none | no pathological findings (2013) | none | none | |
| O.T. III-12 | ♀ | no symptoms | COL4A5 p.G624D (GGT>GAT) heterozygous | not investigated | not investigated | none | none |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frese, J.; Kettwig, M.; Zappel, H.; Hofer, J.; Gröne, H.-J.; Nagel, M.; Sunder-Plassmann, G.; Kain, R.; Neuweiler, J.; Gross, O. Kidney Injury by Variants in the COL4A5 Gene Aggravated by Polymorphisms in Slit Diaphragm Genes Causes Focal Segmental Glomerulosclerosis. Int. J. Mol. Sci. 2019, 20, 519. https://doi.org/10.3390/ijms20030519
Frese J, Kettwig M, Zappel H, Hofer J, Gröne H-J, Nagel M, Sunder-Plassmann G, Kain R, Neuweiler J, Gross O. Kidney Injury by Variants in the COL4A5 Gene Aggravated by Polymorphisms in Slit Diaphragm Genes Causes Focal Segmental Glomerulosclerosis. International Journal of Molecular Sciences. 2019; 20(3):519. https://doi.org/10.3390/ijms20030519
Chicago/Turabian StyleFrese, Jenny, Matthias Kettwig, Hildegard Zappel, Johannes Hofer, Hermann-Josef Gröne, Mato Nagel, Gere Sunder-Plassmann, Renate Kain, Jörg Neuweiler, and Oliver Gross. 2019. "Kidney Injury by Variants in the COL4A5 Gene Aggravated by Polymorphisms in Slit Diaphragm Genes Causes Focal Segmental Glomerulosclerosis" International Journal of Molecular Sciences 20, no. 3: 519. https://doi.org/10.3390/ijms20030519
APA StyleFrese, J., Kettwig, M., Zappel, H., Hofer, J., Gröne, H.-J., Nagel, M., Sunder-Plassmann, G., Kain, R., Neuweiler, J., & Gross, O. (2019). Kidney Injury by Variants in the COL4A5 Gene Aggravated by Polymorphisms in Slit Diaphragm Genes Causes Focal Segmental Glomerulosclerosis. International Journal of Molecular Sciences, 20(3), 519. https://doi.org/10.3390/ijms20030519