Interleukin-18 in Health and Disease


Abstract
:1. Introduction
2. Production of IL-18
2.1. IL18 Gene Expression
2.1.1. Transcriptional Gene Regulation
2.1.1.1. IL18 Gene Promoter
2.1.1.2. IL18 Gene Repressor
2.1.2. Post-Transcriptional Gene Regulation (miRNA)
2.2. Post-Translational Regulation of IL-18 (Processing of pro-IL-18)
2.2.1. Caspases
2.2.1.1. Caspase-1 (Inflammasome NLRP3, NLRC4, and AIM2)
2.2.1.2. Caspase-8 (upon Fas Ligation)
2.2.2. Other Proteases Involved in the Production of Biologically Active IL-18
2.3. Regulation of Circulating IL-18 by IL-18-Binding Protein (IL-18BP)
3. IL-18 Signaling
3.1. IL-18 Receptor
3.2. IL-18 Signaling Cascade
3.3. IL-18 Binding Protein
4. Physiological Roles of IL-18
4.1. Cytokine and Immune Cell Milieu Determines the Biological Action of IL-18
4.1.1. IFN-γ Production
4.1.2. Innate-Type Basophil and Mast Cell Activation by IL-3 and IL-18
4.1.3. Innate-Type Allergic Inflammation by IL-18
4.1.4. Th1 Cells Produce IFN-γ and IL-13 in Response to IL-18
4.2. IL-18 in Host Defense
4.2.1. IL-18-Mediated Defense Against Extracellular Pathogens
4.2.1.1. Bacteria Infection
4.2.1.2. Helminth Infection
4.2.2. IL-18-Mediated Defense Against Intracellular Pathogens
4.2.2.1 Bacterial Infection
4.2.2.2. Protozoan Infection
4.2.3. IL-18-Mediated Viral Clearance
4.3. IL-18 in Metabolism
4.3.1. IL-18 in Metabolic Homeostasis
4.3.1.1. IL-18 Regulation of Food Intake
4.3.1.2. IL-18 Regulation of Energy Expenditure by Activating Thermogenic Adipose Tissues
4.3.1.3. IL-18 Activation of AMPK and Lipid Oxidation in Skeletal Muscle
4.3.1.4. IL-18 Processed by the NLRP1 Inflammasome Protects Against Metabolic Syndrome.
4.3.2. Detrimental Role of the NLRP3 Inflammasome/IL-1β Axis in the Development of Metabolic Syndrome
4.4. IL-18 in Intestinal Homeostasis
4.4.1. Intestinal Epithelial Cells Constitutively Produce Constituents of the Inflammasome for IL-18 Maturation
4.4.2. Importance of the Gut Microbiome for Homeostatic IL-18 Release
4.4.3. Roles of NLRP6 Inflammasome-Mediated Epithelial IL-18 in Gut Homeostasis
4.4.3.1. The NLRP6 Inflammasome is Indispensable for the Healthy Microbiota
4.4.3.2. Involvement of the NLRP6 Inflammasome in the Formation of the Colonic Mucin Layer
4.4.4. Importance of Pyrin Inflammasome-Mediated Mucosal IL-18 for Tight Junction Formation
4.4.5. Newly Identified NLRP9b-Mediated IL-18 Release is Involved in Rotavirus Clearance
5. IL-18 in Disease
5.1. Endotoxin-Induced Systemic and Tissue Diseases
5.1.1. Induction of Endotoxin Shock in P. acnes-Primed Mice.
5.1.2. LPS-Induced Liver Injury in P. acnes-Primed Mice
5.2. IL-18 in Allergy
5.2.1. Induction of IgE Production by IL-18
5.2.2. Innate-Type Allergic Inflammation Induced by IL-18
5.2.3. The Induction of IFN-γ and IL-13 Producing Super Th1 Cells by IL-2 and IL-18
5.2.4. Bronchial Asthma Induced by the Intranasal Administration of IL-2 and IL-18
5.3. IL-18 in Kidney Diseases
5.3.1. Association between Serum IL-18 Levels and Renal Prognosis in IgA Nephropathy
5.3.2. Urinary IL-18 as A Biomarker of AKI after Cardiac Surgery
5.4. IL-18 in Metabolic Disorders
5.5. IL-18 in Cancer
5.5.1. IL-18 Robustly Expands Human γδT Cells
5.5.2. Mycobiome-Mediated IL-18 Protects Against Colitis-Associated Colorectal Cancer
6. Similarities and Differences between IL-18 and IL-33
7. IL-18 as A Therapeutic Target
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ag | Antigen |
| AIM2 | Absence in melanoma 2 |
| AKI | Acute kidney injury |
| AMPK | Adenosine monophosphate-activated protein kinase |
| AP-1 | Activator protein-1 |
| ASC | Apoptosis-associated speck-like protein containing a C-terminal caspase-recruitment domain |
| BCG | Bacillus Calmette–Guerin |
| Bcl6 | B cell lymphoma 6 |
| Btk | Bruton’s tyrosine kinase |
| CTL | Cytotoxic T lymphocyte |
| ds | Double-stranded |
| DSS | Dextran sulphate sodium |
| FasL | Fas ligand |
| HFD | High-fat diet |
| IBD | Inflammatory bowel disease |
| IFN | Interferon |
| IGIF | IFN-γ inducing factor |
| IL-18BP | IL-18 binding protein |
| IL-18R | IL-18 receptor |
| ILC | Innate lymphoid cell |
| iNOS | Inducible nitric oxide synthase |
| IRAK | IL-1R-associated kinase |
| LDL | Low-density lipoprotein |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinase |
| miRNA | MicroRNA |
| MyD88 | Myeloid differentiation primary response 88 |
| NAIP | NLR family of apoptosis inhibitory protein |
| NF-kB | Nuclear factor (NF)-κB |
| NK | Natural killer |
| NLR | Nucleotide-binding oligomerization domain (NOD)-like receptor |
| NLRC | NLR family CARD domain-containing protein |
| NLRP | Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing |
| NO | Nitric oxide |
| OVA | Ovalbumin |
| PBMC | Peripheral blood mononuclear cell |
| ROS | Reactive oxygen species |
| SNP | Single nucleotide polymorphism |
| SPF | Specific pathogen-free |
| STAT | Signaltransducer and activator of transcription |
| TCR | T cell receptor |
| TIR | TLR/IL-1R |
| TLR | Toll-like receptors |
| TNF | Tumor necrosis factor |
| TRAF6 | TNF receptor-activated factor 6 |
| TRIF | TIR-domain-containing adapter-inducing interferon-β |
References
- Okamura, H.; Tsutsui, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-γ production by T cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Yoshimoto, T.; Tsutsui, H.; Okamura, H. Interleukin-18 regulates both Th1 and Th2 responses. Annu. Rev. Immunol. 2001, 19, 423–474. [Google Scholar] [CrossRef] [PubMed]
- Ushio, S.; Namba, M.; Okura, T.; Hattori, K.; Nukada, Y.; Akita, K.; Tanabe, F.; Konishi, K.; Micallef, M.; Fujii, M.; et al. Cloning of the cDNA for human IFN-g-inducing factor, expression in Escherichia coli, and studies on the biologic activities of the protein. J. Immunol. 1996, 156, 4274–4279. [Google Scholar] [PubMed]
- Gu, Y.; Kuida, K.; Tsutsui, H.; Ku, G.; Hsiao, K.; Fleming, M.A.; Hayashi, N.; Higashino, K.; Okamura, H.; Nakanishi, K.; et al. Activation of interferon-g inducing factor mediated by interleukin-1b converting enzyme. Science 1997, 275, 206–209. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Adachi, O.; Kawai, T.; Takeda, K.; Matsumoto, M.; Tsutsui, H.; Sakagami, M.; Nakanishi, K.; Akira, S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998, 9, 143–150. [Google Scholar] [CrossRef]
- Nakanishi, K. Unique Action of Interleukin-18 on T Cells and Other Immune Cells. Front. Immunol. 2018, 9, 763. [Google Scholar] [CrossRef]
- Bazan, J.F.; Timans, J.C.; Kastelein, R.A. A newly defined interleukin-1? Nature 1996, 379, 591. [Google Scholar] [CrossRef]
- Ghayur, T.; Banerjee, S.; Hugunin, M.; Butler, D.; Herzog, L.; Carter, A.; Quintal, L.; Sekut, L.; Talanian, R.; Paskind, M.; et al. Caspase-1 processes IFN-g-inducing factor and regulates LPS-induced IFN-g production. Nature 1997, 386, 619–623. [Google Scholar] [CrossRef]
- Tone, M.; Thompson, S.A.; Tone, Y.; Fairchild, P.J.; Waldmann, H. Regulation of IL-18 (IFN-g-inducing factor) gene expression. J. Immunol. 1997, 159, 6156–6163. [Google Scholar]
- Kim, Y.-M.; Kang, H.-S.; Paik, S.-G.; Pyun, K.-H.; Anderson, K.L.; Torbett, B.E.; Choi, I. Roles of IFN consensus sequence binding protein and PU.1 in regulating IL-18 gene expression. J. Immunl. 1999, 163, 2000–2007. [Google Scholar]
- Takeuchi, M.; Okura, T.; Mori, T.; Akita, K.; Ohta, T.; Ikeda, M.; Ikegami, H.; Kurimoto, M. Intracellular production of interleukin-18 in human epithelial-like cell lines is enhanced by hyperosmotic stress in vitro. Cell Tissue Res. 1999, 297, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Giedraitis, V.; He, B.; Huang, W.-X.; Hillert, J. Cloning and mutation analysis of the human IL-18 promoter: A possible role of polymorphisms in expression regulation. J. Neuroimmunol. 2001, 112, 146–152. [Google Scholar] [CrossRef]
- Sánchez, E.; Palomino-Morales, R.J.; Ortega-Centeno, N.; Jiménez-Alonso, J.; González-Gay, M.A.; López-Nevot, M.A.; Sánchez-Román, J.; de Ramón, E.; Francisca González-Escribano, M.; Pons-Estel, B.A.; et al. Identification of a new putative functional IL18 gene variant through an association study in systemic lupus erythematosus. Hum. Mol. Genet. 2009, 18, 3739–3748. [Google Scholar] [CrossRef] [PubMed]
- Khripko, O.P.; Sennikova, N.S.; Lopatnikova, J.A.; Khripko, J.I.; Filipenko, M.L.; Khrapov, E.A.; Gelfgat, E.L.; Yakushenko, E.V.; Kozlov, V.A.; Sennikov, S.V. Association of single nucleotide polymorphisms in the IL-18 gene with production of IL-18 protein by mononuclear cells from healthy donors. Mediat. Inflamm. 2008, 2008, 309721. [Google Scholar] [CrossRef]
- Ye, B.H.; Lista, F.; Lo Coco, F.; Knowles, D.M.; Offit, K.; Chaganti, R.S.K.; Dalla-Favera, R. Alternations of a zink finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science 1993, 262, 747–750. [Google Scholar] [CrossRef]
- Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef]
- Takeda, N.; Arima, M.; Tsuruoka, N.; Okada, S.; Hatano, M.; Sakamoto, A.; Kohno, Y.; Tokuhisa, T. Bcl6 is a transcriptional repressor for the IL-18 gene. J. Immunol. 2003, 171, 426–431. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Chekulaeva, M.; Filipowicz, W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr. Opin. Cell Biol. 2009, 21, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Robert Fabian, M.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA Translation and Stability by microRNAs. Annu Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Luiz Marques-Rocha, J.; Samblas, M.; Milagro, F.I.; Bressan, J.; Alfredo Marínez, J.; Marti, A. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J. 2015, 29, 3595–3611. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Sideras, P.; Smith, C.I.; Vorechovsky, I.; Chapman, V.; Paul, W.E. Colocalization of X-Linked Agammaglobulinemia and X-Linked Immunodeficiency Genes. Science 1993, 261, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Alsaleh, G.; Suffert, G.; Semaan, N.; Juncker, T.; Frenzel, L.; Gottenberg, J.E.; Sibilia, J.; Pfeffer, S.; Wachsmann, D. Bruton’s tyrosine kinase is involved in miR-346-related regulation of IL-18 release by lipopolysaccharide-activated rheumatoid fibroblast-like synoviocytes. J. Immunol. 2009, 182, 5088–5097. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, C.; Peng, Z.; Zhao, J.; Gong, G.; Tan, D. miR-197 Expression in Peripheral Blood Mononuclear Cells from Hepatitis B Virus-Infected Patients. Gut Liver 2013, 7, 335–342. [Google Scholar] [CrossRef]
- Alnemri, E.S.; Livingston, D.J.; Nicholson, D.W.; Salvesen, G.; Thornberry, N.A.; Wong, W.W.; Yuan, J. Human ICE/CED-3 protease nomenclature. Cell 1996, 87, 171. [Google Scholar] [CrossRef]
- Lavrik, I.N.; Golks, A.; Krammer, P.H. Caspases: Pharmacological manipulation of cell death. J. Clin. Investig. 2005, 115, 2665–2672. [Google Scholar] [CrossRef]
- Shalini, S.; Dorstyn, L.; Dawer, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Tschopp, J.; Martinon, F.; Burns, K. NALPs: A novel protein family involved in inflammation. Nat. Rev. Mol. Cell Biol. 2003, 4, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermine-mediated programmed necrotic cell death. Trend Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Feng, S.; Fox, D.; Ming Man, S. Mechanisms of Gasdermin familly members in inflammasome signaling and cell death. J. Mol. Biol. 2018, 430, 3068–3080. [Google Scholar] [CrossRef] [PubMed]
- Ming Man, S.; Karki, R.; Kanneganti, T.-D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar]
- Lamkanfi, M.; Vande Walle, L.; Kanneganti, T.-D. Deregulated inflammasome signaling in disease. Immunol. Rev. 2011, 243, 163–173. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef]
- He, Y.; Hara, H.; Núñez, G. Mechanism and regulation of NLRP3 inflammasome activation. Trend Biochem. Sci. 2016, 41, 1012–1020. [Google Scholar] [CrossRef]
- Mathur, A.; Hayward, J.A.; Ming Man, S. Molecular mechanisms of inflammasome signaling. J. Leukoc Biol 2018, 103, 233–257. [Google Scholar] [CrossRef]
- Place, D.E.; Kanneganti, T.-D. Recent advances in infllasome biology. Curr. Opin. Immunol. 2018, 50, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Ozoren, N.; Body-Malapel, M.; Amer, A.; Park, J.H.; Franchi, L.; Whitfield, J.; Barchet, W.; Colonna, M.; Vandenabeele, P.; et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 2006, 440, 233–236. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Yang, Y.; Jin, T.; Jiang, W.; Zhou, R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trend Immunol. 2018, 39, 393–406. [Google Scholar] [CrossRef]
- Hughes, M.M.; O’Neill, L.A.J. Metabolic regulation of NLRP3. Immunol. Rev. 2018, 281, 88–98. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Scjnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Ratner, D.; Orning, M.P.A.; Lien, E. Bacterial secretion systems and regulation of inflammasome activation. J. Leukoc. Biol. 2017, 101, 165–181. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef]
- Duncan, J.A.; Canna, S.W. The NLRC4 inflammasome. Immunol. Rev. 2018, 281, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.-N.; Lu, Q.; Xu, H.; Liou, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Chrrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and fors a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Lugrin, J.; Martinon, F. The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 2018, 281, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Tsutsui, H.; Nakano, H.; Tsuji, N.M.; Hoshino, K.; Adachi, O.; Adachi, K.; Futatsugi, S.; Kuida, K.; Takeuchi, O.; et al. LPS-induced IL-18 secretion from murine Kupffer cells independently of MyD88 that is critically involved in induction of production of IL-12 and IL-1β. J. Immunol. 2001, 166, 2651–2657. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Tsutsui, H.; Yasuda, K.; Uchiyama, R.; Yumikura-Futatsugi, S.; Mitani, K.; Hayashi, S.; Akira, S.; Taniguchi, S.; Van Rooijen, N.; et al. Contribution of TIR domain-containing adapter inducing IFN-β-mediated IL-18 release to LPS-induced liver injury in mice. J. Hepatol. 2009, 51, 333–341. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yaginuma, K.; Tsutsui, H.; Sagara, J.; Guan, X.; Seki, E.; Yasuda, K.; Yamamoto, M.; Akira, S.; Nakanishi, K.; et al. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adapter molecules. Gene Cell 2004, 9, 1055–1067. [Google Scholar] [CrossRef]
- Tsutsui, H.; Imamura, M.; Fujimoto, J.; Nakanishi, K. The TLR4/TRIF-mediated activation of NLRP3 inflammasome underlies endotoxin-induced liver injury in mice. Gastroenterol. Res. Pract. 2010, 2010, 641865. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Österreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-β signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef]
- Tsutsui, H.; Nishiguchi, S. Importance of Kupffer cells in the development of acute liver injuries in mice. Int. J. Mol. Sci. 2014, 15, 7711–7730. [Google Scholar] [CrossRef] [PubMed]
- Miwa, K.; Asano, M.; Horai, R.; Iwakura, Y.; Nagata, S.; Suda, T. Caspase-1-independent IL-1b release and inflammation induced by the apoptosis inducer Fas ligand. Nat. Med. 1998, 4, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kayagaki, N.; Kuida, K.; Nakano, H.; Hayashi, N.; Takeda, K.; Matsui, K.; Kashiwamura, S.-I.; Hada, T.; Akira, S.; et al. Caspase-1-independent, Fas/Fas ligand-mediated IL-18 secretion from macrophages causes acute liver injury in mice. Immunity 1999, 11, 359–367. [Google Scholar] [CrossRef]
- Uchiyama, R.; Yonehara, S.; Tsutsui, H. Fas-mediated inflammatory response in Listeria monocytogenes infection. J. Immunol. 2013, 190, 4245–4254. [Google Scholar] [CrossRef] [PubMed]
- Bassaller, L.; Chiang, P.-I.; Schmidt-Lauber, C.; Ganesan, S.; Kaiser, W.J.; Rathinam, V.A.K.; Mocarski, E.S.; Subramanan, D.; Green, D.R.; Silverman, N.; et al. Fas (CD95) mediates noncanonical IL-1b and IL-18 maturation via caspase-8 in an RIP3-independent manner. J. Immunol. 2012, 189, 5508–5512. [Google Scholar] [CrossRef]
- Uchiyama, R.; Tsutsui, H. Caspases as the key effectors of inflammatory responses against bacterial infection. Arch. Immunol. Ther. Exp. 2015, 63, 1–13. [Google Scholar] [CrossRef]
- Uchiyama, R.; Yonehara, S.; Taniguchi, S.; Ishido, S.; Ishii, K.J.; Tsutsui, H. Inflammasome and Fas-Mediated IL-1beta Contributes to Th17/Th1 Cell Induction in Pathogenic Bacterial Infection In Vivo. J. Immunol. 2017, 199, 1122–1130. [Google Scholar] [CrossRef]
- Kao, R.C.; Wehner, N.G.; Skubitz, K.M.; Gray, B.H.; Hoidal, J.R. Proteinase 3. A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters. J. Clin. Investig. 1988, 1963–1973. [Google Scholar] [CrossRef]
- Sugawara, S.; Uehara, A.; Nochi, T.; Yamaguchi, T.; Ueda, H.; Sugiyama, A.; Hanzawa, K.; Kumagai, K.; Okamura, H.; Takada, H. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J. Immunol. 2001, 167, 6568–6575. [Google Scholar] [CrossRef]
- Omoto, Y.; Tokime, K.; Yamanaka, K.; Habe, K.; Morioka, T.; Kurokawa, I.; Tsutsui, H.; Yamanishi, K.; Nakanishi, K.; Mizutani, H. Human mast cell chymase cleaves pro-IL-18 and generates a novel and biologically active IL-18 fragment. J. Immunol. 2006, 177, 8315–8319. [Google Scholar] [CrossRef]
- Konishi, H.; Tsutsui, H.; Murakami, T.; Yumikura-Futatsugi, S.; Yamanaka, K.; Tanaka, M.; Iwakura, Y.; Suzuki, N.; Takeda, K.; Akira, S.; et al. IL-18 contributes to the spontaneous development of atopic dermatitis-like inflammatory skin lesion independently of IgE/stat6 under specific pathogen-free conditions. Proc. Natl. Acad. Sci. USA 2002, 99, 11340–11345. [Google Scholar] [CrossRef] [PubMed]
- Irani, A.-M.; Bradford, T.R.; Kepley, C.L.; Schechter, N.M.; Schwartz, L.B. Detection of MCT and MCTC types of human mast cells by immunohistochemistry using new monoclonal anti-tryptase and anti-chymase antibodies. J. Histochem. Cytochem. 1989, 37, 1509–1515. [Google Scholar] [CrossRef] [PubMed]
- Catalfamo, M.; Henkart, P.A. Perforin and the granule exocytosis cytotoxicity pathway. Curr. Opin. Immunol. 2003, 15, 522–527. [Google Scholar] [CrossRef]
- Chowdhury, D.; Lieberman, J. Death by a Thousand Cuts: Granzyme Pathways of Programmed Cell Death. Annu. Rev. Immunol. 2008, 26, 389–420. [Google Scholar] [CrossRef] [PubMed]
- Omoto, Y.; Yamanaka, K.; Tokime, K.; Kitano, S.; Kakeda, M.; Akeda, T.; Kurokawa, I.; Gabazza, E.C.; Tsutsui, H.; Katayama, N.; et al. Granzyme B is a novel interleukin-18 converting enzyme. J. Dermatol. Sci. 2010, 59, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Akeda, T.; Yamanaka, K.; Tsuda, K.; Omoto, Y.; Gabazza, E.C.; Mizutani, H. CD8+T cell granzyme B activates keratinocyte endogenous IL-18. Arch. Dermatol. Res. 2014, 306, 125–130. [Google Scholar] [CrossRef]
- Novick, D.; Kim, S.H.; Fantuzzi, G.; Reznikov, L.L.; Dinarello, C.A.; Rubinstein, M. Interleukin-18 binding protein: A novel modulator of the Th1 cytokine response. Immunity 1999, 10, 127–136. [Google Scholar] [CrossRef]
- Aizawa, Y.; Akita, K.; Taniai, M.; Torigoe, K.; Mori, T.; Nishida, Y.; Ushio, S.; Nukada, Y.; Tanimoto, T.; Ikegami, H.; et al. Cloning and expression of interleukin-18 binding protein. FEBS Lett. 1999, 445, 338–342. [Google Scholar] [CrossRef]
- Novick, D.; Rubinstein, M. The tale of soluble receptors and binding proteins: From bench to bedside. Cytokine & Growth Factor Rev. 2007, 18, 525–533. [Google Scholar]
- Kawashima, M.; Yamamura, M.; Taniai, M.; Yamauchi, H.; Tanimoto, T.; Kurimoto, M.; Amano, T.; Takeuchi, T.; Makino, H. Levels of interleukin-18 and its binding inhibitors in the blood circulation of patients with adult-onset Still’s disease. Arthritis Rheum. 2001, 44, 550–560. [Google Scholar] [CrossRef]
- Liao, T.L.; Chen, Y.M.; Hsieh, C.W.; Chen, H.H.; Lee, H.C.; Hung, W.T.; Tang, K.T.; Chen, D.Y. Upregulation of circulating microRNA-134 in adult-onset Still’s disease and its use as potential biomarker. Sci. Rep. 2017, 7, 4214. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Tsutsui, H.; Kawai, T.; Takeda, K.; Nakanishi, K.; Takeda, Y.; Akira, S. Generation of IL-18 receptor-deficient mice: Evidence for IL-1 receptor-related protein as an essential IL-18 binding receptor. J. Immunol. 1999, 162, 5041–5044. [Google Scholar] [PubMed]
- Kim, S.H.; Reznikov, L.L.; Stuyt, R.J.L.; Selzman, C.H.; Fantuzzi, G.; Hoshino, T.; Young, H.A.; Dinarello, C.A. Functional reconstitution and regulation of IL-18 activity by the IL-18Rb chain. J. Immunol. 2001, 166, 148–154. [Google Scholar] [PubMed]
- Wu, C.; Sakorafas, P.; Miller, R.; McCarthy, D.; Scesney, S.; Dixon, R.; Ghayur, T. IL-18 receptor beta-induced changes in the presentation of IL-18 binding sites affect ligand binding and signal transduction. J. Immunol. 2003, 170, 5571–5577. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, N.; Kimura, T.; Arita, K.; Ariyoshi, M.; Ohnishi, H.; Yamamoto, T.; Zuo, X.; Maenaka, K.; Park, E.Y.; Kondo, N.; et al. The structural basis for receptor recognition of human interleukin-18. Nat. Commun. 2014, 5, 5340. [Google Scholar] [CrossRef] [PubMed]
- Tomura, M.; Maruo, S.; Mu, J.; Zhou, X.Y.; Ahn, H.J.; Hamaoka, T.; Okamura, H.; Nakanishi, K.; Clark, S.; Kurimoto, M.; et al. Differential capacities of CD4+, CD8+, and CD4-CD8- T cell subsets to express IL-18 receptor and produce IFN-gamma in response to IL-18. J. Immunol. 1998, 160, 3759–3765. [Google Scholar] [PubMed]
- Nakahira, M.; Tomura, M.; Iwasaki, M.; Ahn, H.-J.; Bian, Y.; Hamaoka, T.; Ohta, T.; Kurimoto, M.; Fujiwara, H. An absolute requirement for STAT4 and a role for IFN-g as an amplifying factor in IL-12 induction fo the functional IL-18 receptor complex. J. Immunol. 2001, 167, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, M.A.; Merlin, T.; Kalis, C.; Chvatchko, Y.; Stübig, H.; Galanos, C. A murine, IL-12-independent pathway of IFN-g induction by gram-negative bacteria based on STAT4 activation by type 1 IFN and IL-18 signaling. J. Immunol. 2002, 169, 1665–1668. [Google Scholar] [CrossRef]
- Matikainen, S.; Paananen, A.; Miettinen, M.; Kurimoto, M.; Timonen, T.; Julkunen, I.; Sareneva, T. IFN-alpha and IL-18 synergistically enhance IFN-gamma production in human NK cells: Differential regulation of Stat4 activation and IFN-gamma gene expression by IFN-alpha and IL-12. Eur. J. Immunol. 2001, 31, 2236–2245. [Google Scholar] [CrossRef]
- Bufler, P.; Azam, T.; Gamboni-Robertson, F.; Reznikov, L.L.; Kumar, S.; Dinarello, C.A.; Kim, S.H. A complex of the IL-1 homologue IL-1F7b and IL-18-binding protein reduces IL-18 activity. Proc. Natl. Acad. Sci. USA 2002, 99, 13723–13728. [Google Scholar] [CrossRef]
- Kumar, S.; Hanning, C.R.; Brigham-Burke, M.R.; Rieman, D.J.; Lehr, R.; Khandekar, S.; Kirkpatrick, R.B.; Scott, G.F.; Lee, J.C.; Lynch, F.J.; et al. Interleukin-1F7B (IL-1H4/IL-1F7) is processed by caspase-1 and mature IL-1F7B binds to the IL-18 receptor but does not induce IFN-gamma production. Cytokine 2002, 18, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Nold-Petry, C.A.; Lo, C.Y.; Rudloff, I.; Elgass, K.D.; Li, S.; Gantier, M.P.; Lotz-Havla, A.S.; Gersting, S.W.; Cho, S.X.; Lao, J.C.; et al. IL-37 requires the receptors IL-18Ralpha and IL-1R8 (SIGIRR) to carry out its multifaceted anti-inflammatory program upon innate signal transduction. Nat. Immunol. 2015, 16, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Allen, J.L.; Tsen, M.; Dubnicoff, T.; Danao, J.; Liao, X.C.; Cao, Z.; Wasserman, S.A. Impaired cytokine signaling in mice lacking the IL-1 receptor-associated kinase. J. Immunol. 1999, 163, 978–984. [Google Scholar] [PubMed]
- Kanakaraj, P.; Ngo, K.; Wu, Y.; Angulo, A.; Ghazal, P.; Harris, C.A.; Siekierka, J.J.; Peterson, P.A.; Fung-Leung, W.-P. Defective interleukin (IL)-18-mediated natural killer and T helper cell type 1 responses in IL-1 receptor-associated kinase (IRAK)-deficient mice. J. Exp. Med. 1999, 189, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Chen, N.-J.; Miller, D.G.; Suzuki, S.; Horacek, T.; Hara, H.; Bouchard, D.; Nakanishi, K.; Penninger, J.M.; Ohashi, P.S.; et al. IRAK-4 is essential for interleukin-18-mediated natural killer and T helper cell type 1 response. J. Immunol. 2003, 170, 4031–4035. [Google Scholar] [CrossRef]
- Robinson, D.; Shibuya, K.; Mui, A.; Zonin, F.; Murphy, E.; Sana, T.; Hartley, S.B.; Menon, S.; Kastelein, R.; Bazan, F.; et al. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NF-kB. Immunity 1997, 7, 571–581. [Google Scholar] [CrossRef]
- Kalina, U.; Kauschat, D.; Koyama, N.; Nuernberger, H.; Ballas, K.; Koschmieder, S.; Bug, G.; Hofmann, W.K.; Hoelzer, D.; Ottmann, O.G. IL-18 activates STAT3 in the natural killer cell line 92, augments cytotoxic activity, and mediates IFN-gamma production by the stress kinase p38 and by the extracellular regulated kinases p44erk-1 and p42erk-21. J. Immunol. 2000, 165, 1307–1313. [Google Scholar] [CrossRef]
- El-Darawish, Y.; Li, W.; Yamanishi, K.; Pencheva, M.; Oka, N.; Yamanishi, H.; Matsuyama, T.; Tanaka, Y.; Minato, N.; Okamura, H. Frontline Science: IL-18 primes murine NK cells for proliferation by promoting protein synthesis, survival, and autophagy. J. Leukoc. Biol. 2018, 104, 253–264. [Google Scholar] [CrossRef]
- Deason, K.; Troutman, T.D.; Jain, A.; Challa, D.K.; Mandraju, R.; Brewer, T.; Ward, E.S.; Pasare, C. BCAP links IL-1R to the PI3K-mTOR pathway and regulates pathogenic Th17 cell differentiation. J. Exp. Med. 2018, 215, 2413–2428. [Google Scholar] [CrossRef]
- Fukao, T.; Tanabe, M.; Terauchi, Y.; Ota, T.; Matsuda, S.; Asano, T.; Kadowaki, T.; Takeuchi, T.; Koyasu, S. PI3K-mediated negative feedback regulation of IL-12 production in DCs. Nat. Immunol. 2002, 3, 875–881. [Google Scholar] [CrossRef]
- Hosotani, Y.; Kashiwamura, S.; Kimura-Shimmyo, A.; Sekiyama, A.; Ueda, H.; Ikeda, T.; Mimura, O.; Okamura, H. Interleukin-18 prevents apoptosis via PI3K/Akt pathway in normal human keratinocytes. J. Dermatol. 2008, 35, 514–524. [Google Scholar] [CrossRef]
- Zhou, J.; Ping, F.F.; Lv, W.T.; Feng, J.Y.; Shang, J. Interleukin-18 directly protects cortical neurons by activating PI3K/AKT/NF-kappaB/CREB pathways. Cytokine 2014, 69, 29–38. [Google Scholar] [CrossRef]
- Kanayama, A.; Seth, R.B.; Sun, L.; Ea, C.K.; Hong, M.; Shaito, A.; Chiu, Y.H.; Deng, L.; Chen, Z.J. TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol. Cell 2004, 15, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Strickson, S. The role of hybrid ubiquitin chains in the MyD88 and other innate immune signalling pathways. Cell Death Differ. 2017, 24, 1153–1159. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Deng, L.; Hong, M.; Akkaraju, G.R.; Inoue, J.; Chen, Z.J. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001, 412, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Horng, T.; Barton, G.M.; Flavell, R.A.; Medzhitov, R. The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 2002, 420, 329–333. [Google Scholar] [CrossRef]
- Ohnishi, H.; Tochio, H.; Kato, Z.; Kawamoto, N.; Kimura, T.; Kubota, K.; Yamamoto, T.; Funasaka, T.; Nakano, H.; Wong, R.W.; et al. TRAM is involved in IL-18 signaling and functions as a sorting adaptor for MyD88. PLoS ONE 2012, 7, e38423. [Google Scholar] [CrossRef]
- Kim, S.H.; Eisenstein, M.; Reznikov, L.; Fantuzzi, G.; Novick, D.; Rubinstein, M.; Dinarello, C.A. Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proc. Natl. Acad. Sci. USA 2000, 97, 1190–1195. [Google Scholar] [CrossRef]
- Faggioni, R.; Cattley, R.C.; Guo, J.; Flores, S.; Brown, H.; Qi, M.; Yin, S.; Hill, D.; Scully, S.; Chen, C.; et al. IL-18-binding protein protects against lipopolysaccharide-induced lethality and prevents the development of Fas/Fas ligand-mediated models of liver disease in mice. J. Immunol. 2001, 167, 5913–5920. [Google Scholar] [CrossRef]
- Banda, N.K.; Vodracek, A.; Kraus, D.; Dinarello, C.A.; Kim, S.-H.; Bendele, A.; Senaldi, G.; Arend, W.P. Mechanisms of inhibition of collagen-induced arthritis by murine IL-18 binding protein. J. Immunol. 2003, 170, 2100–2105. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, B.; Sennello, J.A.; Lehr, H.A.; Senaldi, G.; Dinarello, C.A.; Fantuzzi, G. Frontline: Interferon regulatory factor-1 as a protective gene in intestinal inflammation: Role of TCR gamma delta T cells and interleukin-18-binding protein. Eur. J. Immunol. 2004, 34, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Zaccone, P.; Phillips, J.; Conget, I.; Cooke, A.; Nicoletti, F. IL-18 binding protein fusion construct delays the development of diabetes in adoptive transfer and cyclophosphamide-induced diabetes in NOD mouse. Clin. Immunol. 2005, 115, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Novick, D.; Elbirt, D.; Miller, G.; Dinarello, C.A.; Rubinstein, M.; Sthoeger, Z.M. High circulating levels of free interleukin-18 in patients with active SLE in the presence of elevated levels of interleukin-18 binding protein. J. Autoimmun. 2010, 34, 121–126. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Lu, L.; Altmann, C.; Hoke, T.S.; Ljubanovic, D.; Jani, A.; Dinarello, C.A.; Faubel, S.; Edelstein, C.L. Interleukin-18 binding protein transgenic mice are protected against ischemic acute kidney injury. Am. J. Physiol. Renal Physiol. 2008, 295, F1414–F1421. [Google Scholar] [CrossRef] [PubMed]
- Nowarski, R.; Jackson, R.; Gagliani, N.; de Zoete, M.R.; Palm, N.W.; Bailis, W.; Siong Low, J.; Harman, C.C.D.; Graham, M.; Elinav, E.; et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Immunity 2015, 163, 1444–1456. [Google Scholar] [CrossRef]
- Harms, R.Z.; Creer, A.J.; Lorenzo-Arteaga, K.M.; Ostlund, K.R.; Sarvetnick, N.E. Interleukin (IL)-18 Binding Protein Deficiency Disrupts Natural Killer Cell Maturation and Diminishes Circulating IL-18. Front. Immunol. 2017, 8, 1020. [Google Scholar] [CrossRef]
- Novick, D.; Elbirt, D.; Dinarello, C.A.; Rubinstein, M.; Sthoeger, Z.M. Interleukin-18 binding protein in the sera of patients with Wegener’s granulomatosis. J. Clin. Immunol. 2009, 29, 38–45. [Google Scholar] [CrossRef]
- Mazodier, K.; Marin, V.; Novick, D.; Farnarier, C.; Robitail, S.; Schleinitz, N.; Veit, V.; Paul, P.; Rubinstein, M.; Dinarello, C.A.; et al. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood 2005, 106, 3483–3489. [Google Scholar] [CrossRef]
- Colafrancesco, S.; Priori, R.; Alessandri, C.; Perricone, C.; Pendolino, M.; Picarelli, G.; Valesini, G. IL-18 Serum Level in Adult Onset Still’s Disease: A Marker of Disease Activity. Int. J. Inflamm. 2012, 2012, 156890. [Google Scholar] [CrossRef]
- Gabay, C.; Fautrel, B.; Rech, J.; Spertini, F.; Feist, E.; Kotter, I.; Hachulla, E.; Morel, J.; Schaeverbeke, T.; Hamidou, M.A.; et al. Open-label, multicentre, dose-escalating phase II clinical trial on the safety and efficacy of tadekinig alfa (IL-18BP) in adult-onset Still’s disease. Ann. Rheum. Dis. 2018, 77, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Canna, S.W.; Girard, C.; Malle, L.; de Jesus, A.; Romberg, N.; Kelsen, J.; Surrey, L.F.; Russo, P.; Sleight, A.; Schiffrin, E.; et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J. Allergy Clin. Immunol. 2017, 139, 1698–1701. [Google Scholar] [CrossRef] [PubMed]
- McNamee, E.N.; Masterson, J.C.; Jedlicka, P.; McManus, M.; Grenz, A.; Collins, C.B.; Nold, M.F.; Nold-Petry, C.; Bufler, P.; Dinarello, C.A.; et al. Interleukin 37 expression protects mice from colitis. Proc. Natl. Acad. Sci. USA 2011, 108, 16711–16716. [Google Scholar] [CrossRef] [PubMed]
- Muhl, H.; Kampfer, H.; Bosmann, M.; Frank, S.; Radeke, H.; Pfeilschifter, J. Interferon-g mediates gene expression of IL-18 binding protein in nonleukocytic cells. Biochem. Biophys. Res. Commun. 2000, 267, 960–963. [Google Scholar] [CrossRef] [PubMed]
- Hurgin, V.; Novick, D.; Rubinstein, M. The promoter of IL-18 binding protein: Activation by an IFN-gamma -induced complex of IFN regulatory factor 1 and CCAAT/enhancer binding protein beta. Proc. Natl. Acad. Sci. USA 2002, 99, 16957–16962. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Novick, D.; Rubinstein, M.; Siegmund, B.; Enrich, B.; Koch, R.O.; Vogel, W.; Kim, S.H.; Dinarello, C.A.; Tilg, H. Interferon-alpha induces interleukin-18 binding protein in chronic hepatitis C patients. Clin. Exp. Immunol. 2002, 129, 332–338. [Google Scholar] [CrossRef]
- Ludwiczek, O.; Kaser, A.; Novick, D.; Dinarello, C.A.; Rubinstein, M.; Vogel, W.; Tilg, H. Plasma levels of interleukin-18 and interleukin-18 binding protein are elevated in patients with chronic liver disease. J. Clin. Immunol. 2002, 22, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Nold-Petry, C.A.; Lehrnbecher, T.; Jarisch, A.; Schwabe, D.; Pfeilschifter, J.M.; Muhl, H.; Nold, M.F. Failure of interferon gamma to induce the anti-inflammatory interleukin 18 binding protein in familial hemophagocytosis. PLoS ONE 2010, 5, e8663. [Google Scholar] [CrossRef]
- Wittmann, M.; Doble, R.; Bachmann, M.; Pfeilschifter, J.; Werfel, T.; Muhl, H. IL-27 Regulates IL-18 binding protein in skin resident cells. PLoS ONE 2012, 7, e38751. [Google Scholar] [CrossRef]
- Carbotti, G.; Barisione, G.; Orengo, A.M.; Brizzolara, A.; Airoldi, I.; Bagnoli, M.; Pinciroli, P.; Mezzanzanica, D.; Centurioni, M.G.; Fabbi, M.; et al. The IL-18 antagonist IL-18-binding protein is produced in the human ovarian cancer microenvironment. Clin. Cancer Res. 2013, 19, 4611–4620. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Okamura, H.; Tagawa, Y.; Iwakura, Y.; Nakanishi, K. Interleukin 18 together with interleukin 12 inhibits IgE production by induction of interferon-g production from activated B cells. Proc. Natl. Acad. Sci. USA 1997, 94, 3948–3953. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Takeda, K.; Tanaka, T.; Ohkusu, K.; Kashiwamura, S.; Okamura, H.; Akira, S.; Nakanishi, K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: Synergism with IL-18 for IFN-g production. J. Immunol. 1998, 161, 3400–3407. [Google Scholar] [PubMed]
- Yoshimoto, T.; Tsutsui, H.; Tominaga, K.; Hoshino, K.; Okamura, H.; Akira, S.; Paul, W.E.; Nakanishi, K. IL-18, although anti-allergic when administered with IL-12, stimulates IL-4 and histamine release by basophils. Proc. Natl. Acad. Sci. USA 1999, 96, 13962–13966. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Mizutani, H.; Tsutsui, H.; Noben-Trauth, N.; Yamanaka, K.; Tanaka, M.; Izumi, S.; Okamura, H.; Paul, W.E.; Nakanishi, K. IL-18 induction of IgE: Dependence on CD4+ T cells, IL-4 and STAT6. Nat. Immunol. 2000, 1, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Yoshimoto, T.; Hayashi, N.; Mizutani, H.; Nakanishi, K. Induction of allergic inflammation by interleukin-18 in experimental animal models. Immunol. Rev. 2004, 202, 115–138. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Yoshimoto, T.; Maruyama, H.; Tegoshi, T.; Ohta, N.; Arizono, N.; Nakanishi, K. IL-18 with IL-2 protects against Strongyloides venezuelensis infection by activating mucosal mast cell-dependent type 2 innate immunity. J. Exp. Med. 2005, 202, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, M.; Nakanishi, K. Requirement of GATA-binding protein 3 for II13 gene expression in IL-18-stimulated Th1 cells. Int. Immunol. 2011, 23, 761–772. [Google Scholar] [CrossRef]
- Kinoshita, M.; Miyazaki, H.; Ono, S.; Seki, S. Immunoenhancing therapy with interleukin-18 against bacterial infection in immunocompromised hosts after severe surgical stress. J. Leukoc. Biol. 2013, 93, 689–698. [Google Scholar] [CrossRef]
- Kinoshita, M.; Seki, S.; Ono, S.; Shinomiya, N.; Hiraide, H. Paradoxical effect of IL-18 therapy on the severe and mild Escherichia coli infections in burn-injured mice. Ann. Surg. 2004, 240, 313–320. [Google Scholar] [CrossRef]
- Kinoshita, M.; Kuranaga, N.; Matsumoto, A.; Ono, S.; Shinomiya, N.; Hiraide, H.; Seki, S. Multiple interleukin-18 injections promote both mouse Th1 and Th2 responses after sublethal Escherichia coli infection. Clin. Exp. Immunol. 2006, 143, 41–49. [Google Scholar] [CrossRef]
- Kinoshita, M.; Shinomiya, N.; Ono, S.; Tsujimoto, H.; Kawabata, T.; Matsumoto, A.; Hiraide, H.; Seki, S. Restoration of natural IgM production from liver B cells by exogenous IL-18 improves the survival of burn-injured mice infected with Pseudomonas aeruginosa. J. Immunol. 2006, 177, 4627–4635. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, K.; Yamagata, T.; Nozaki, Y.; Sugiyama, M.; Ikoma, S.; Funauchi, M.; Kanamaru, A. Blockade of IL-18 receptor signaling delays the onset of autoimmune disease in MRL-Faslpr mice. J. Immunol. 2004, 173, 5312–5318. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Miyazaki, H.; Ono, S.; Inatsu, A.; Nakashima, H.; Tsujimoto, H.; Shinomiya, N.; Saitoh, D.; Seki, S. Enhancement of neutrophil function by interleukin-18 therapy protects burn-injured mice from methicillin-resistant Staphylococcus aureus. Infect. Immun. 2011, 79, 2670–2680. [Google Scholar] [CrossRef] [PubMed]
- Finkelman, F.D.; Wynn, T.A.; Donaldson, D.D.; Urban, J.F. The role of IL-13 in helminth-induced inflammation and protective immunity against nematode infections. Curr. Opin. Immunol. 1999, 11, 420–426. [Google Scholar] [CrossRef]
- Grencis, R.K. Th2-mediated host protective immunity to intestinal nematode infections. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1997, 352, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
- Helmby, H.; Takeda, K.; Akira, S.; Grencis, R.K. Interleukin (IL)-18 promotes the development of chronic gastrointestinal helminth infection by downregulating IL-13. J. Exp. Med. 2001, 194, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, M.R.; Hardman, M.J.; Grencis, R.K. The role of sex hormones in the development of Th2 immunity in a gender-biased model of Trichuris muris infection. Eur. J. Immunol. 2010, 40, 406–416. [Google Scholar] [CrossRef]
- Helmby, H.; Grencis, R.K. IL-18 regulates intestinal mastocytosis and Th2 cytokine production independently of IFN-g during Trichinella spiralis infection. J. Immunol. 2002, 169, 2553–2560. [Google Scholar] [CrossRef]
- Kobayashi, K.; Nakata, N.; Kai, M.; Kasama, T.; Hanyuda, Y.; Hatano, Y. Decreased expression of cytokines that induce type 1 helper T cell/interferon-g responses in genetically susceptible mice infected with Mycobacterium avium. Clin. Immunol. Immunopathol. 1997, 85, 112–116. [Google Scholar] [CrossRef]
- Sugawara, I.; Yamada, H.; Kaneko, H.; Mizuno, S.; Takeda, K.; Akira, S. Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-disrupted mice. Infect. Immun. 1999, 67, 2585–2589. [Google Scholar]
- Kinjo, Y.; Kawakami, K.; Uezu, K.; Yara, S.; Miyagi, K.; Koguchi, Y.; Hoshino, T.; Okamoto, M.; Kawase, Y.; Yokota, K.; et al. Contribution of IL-18 to Th1 response and host defense against infection by Mycobacterium tuberculosis: A comparative study with IL-12p40. J. Immunol. 2002, 169, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.E.; Korbel, D.; Hagens, K.; Koch, M.; Raupach, B.; Enders, J.; Kaufmann, S.H.; Mittrucker, H.W.; Schaible, U.E. A role for IL-18 in protective immunity against Mycobacterium tuberculosis. Eur. J. Immunol. 2010, 40, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ouyang, N.; Li, Q.H.; Luo, S.X.; He, Q.; Lei, H.; Liu, Q. The -137G/C single nucleotide polymorphism in IL-18 gene promoter contributes to tuberculosis susceptibility in Chinese Han population. Infect. Genet. Evol. 2015, 36, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, L.; Zhu, D.; An, H.; Yang, Y.; Liang, Y.; Zhao, W.; Ding, W.; Wu, X. Polymorphisms in the interleukin 18 receptor 1 gene and tuberculosis susceptibility among Chinese. PLoS ONE 2014, 9, e110734. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Gao, C.; Chi, X.; Hu, Y.W.; Zheng, L.; Zeng, T.; Wang, Q. IL-37 Expression is Upregulated in Patients with Tuberculosis and Induces Macrophages Towards an M2-like Phenotype. Scand. J. Immunol. 2015, 82, 370–379. [Google Scholar] [CrossRef]
- Liu, H.; Zheng, R.; Wang, P.; Yang, H.; He, X.; Ji, Q.; Bai, W.; Chen, H.; Chen, J.; Peng, W.; et al. IL-37 Confers Protection against Mycobacterial Infection Involving Suppressing Inflammation and Modulating T Cell Activation. PLoS ONE 2017, 12, e0169922. [Google Scholar] [CrossRef] [PubMed]
- Neighbors, M.; Xu, X.; Barrat, F.J.; Ruuls, S.R.; Churakova, T.; Debets, R.; Bazan, J.F.; Kastelein, R.A.; Abrams, J.S.; O’Garra, A. A critical role for interleukin 18 in primary and memory effector responses to Listeria monocytogenes that extends beyond its effects on Interferon gamma production. J. Exp. Med. 2001, 194, 343–354. [Google Scholar] [CrossRef]
- Seki, E.; Tsutsui, H.; Tsuji, N.M.; Hayashi, N.; Adachi, K.; Nakano, H.; Futatsugi-Yumikura, S.; Takeuchi, O.; Hoshino, K.; Akira, S.; et al. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J. Immunol. 2002, 169, 3863–3868. [Google Scholar] [CrossRef]
- Maltez, V.I.; Tubbs, A.L.; Cook, K.D.; Aachoui, Y.; Falcone, E.L.; Holland, S.M.; Whitmire, J.K.; Miao, E.A. Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity 2015, 43, 987–997. [Google Scholar] [CrossRef]
- Lochner, M.; Kastenmuller, K.; Neuenhahn, M.; Weighardt, H.; Busch, D.H.; Reindl, W.; Forster, I. Decreased susceptibility of mice to infection with Listeria monocytogenes in the absence of interleukin-18. Infect. Immun. 2008, 76, 3881–3890. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Hara, H.; Fang, R.; Hernandez-Cuellar, E.; Sakai, S.; Daim, S.; Chen, X.; Dewamitta, S.R.; Qu, H.; Mitsuyama, M.; et al. The adaptor ASC exacerbates lethal Listeria monocytogenes infection by mediating IL-18 production in an inflammasome-dependent and -independent manner. Eur. J. Immunol. 2014, 44, 3696–3707. [Google Scholar] [CrossRef]
- Clark, S.E.; Filak, H.C.; Guthrie, B.S.; Schmidt, R.L.; Jamieson, A.; Merkel, P.; Knight, V.; Cole, C.M.; Raulet, D.H.; Lenz, L.L. Bacterial Manipulation of NK Cell Regulatory Activity Increases Susceptibility to Listeria monocytogenes Infection. PLoS Pathog. 2016, 12, e1005708. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.E.; Schmidt, R.L.; McDermott, D.S.; Lenz, L.L. A Batf3/Nlrp3/IL-18 Axis Promotes Natural Killer Cell IL-10 Production during Listeria monocytogenes Infection. Cell. Rep. 2018, 23, 2582–2594. [Google Scholar] [CrossRef] [PubMed]
- Soudja, S.M.; Ruiz, A.L.; Marie, J.C.; Lauvau, G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity 2012, 37, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Seregin, S.S.; Yang, D.; Fukase, K.; Chamaillard, M.; Alnemri, E.S.; Inohara, N.; Chen, G.Y.; Nunez, G. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 2018, 175, 1651–1664. [Google Scholar] [CrossRef] [PubMed]
- Sansonetti, P.J.; Phalipon, A.; Arondel, J.; Thirumalai, K.; Banerjee, S.; Akira, S.; Takeda, K.; Zychlinsky, A. Caspase-1 activation of IL-1b and IL-18 are essential for Shigella flexneri-induced inflammation. Immunity 2000, 12, 581–590. [Google Scholar] [CrossRef]
- Mastroeni, P.; Clare, S.; Khan, S.; Harrison, J.A.; Hormaeche, C.E.; Okamura, H.; Kurimoto, M.; Dougan, G. Interleukin 18 contributes to host resistance and gamma interferon production in mice infected with virulent Salmonella typhimurium. Infect. Immun. 1999, 67, 478–483. [Google Scholar]
- Raupach, B.; Peuschel, S.K.; Monack, D.M.; Zychlinsky, A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 2006, 74, 4922–4926. [Google Scholar] [CrossRef]
- Bohn, E.; Sing, A.; Zumbihl, R.; Bielfeldt, C.; Okamura, H.; Kurimoto, M.; Heesemann, J.; Autenrieth, I.B. IL-18 (IFN-g-inducing factor) regulates early cytokine production in, and promotes resolution of, bacterial infection in mice. J. Immunol. 1998, 160, 299–307. [Google Scholar]
- Hein, J.; Sing, A.; Di Genaro, M.S.; Autenrieth, I.B. Interleukin-12 and interleukin-18 are indispensable for protective immunity against enteropathogenic Yersinia. Microb. Pathog. 2001, 31, 195–199. [Google Scholar] [CrossRef]
- Diefenbach, A.; Schindler, H.; Donhause, N.; Lorenz, E.; Laskay, T.; MacMicking, J.; Röllinghoff, M.; Gresser, I.; Bogdan, C. Type 1 interferon (IFNa/b) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity 1998, 8, 77–87. [Google Scholar] [CrossRef]
- Ohkusu, K.; Yoshimoto, T.; Takeda, K.; Ogura, T.; Kashiwamura, S.; Iwakura, Y.; Akira, S.; Okamura, H.; Nakanishi, K. Potentiality of interleukin-18 as a useful reagent for treatment and prevention of Leishmania major infection. Infect. Immun. 2000, 68, 2449–2456. [Google Scholar] [CrossRef]
- Monteforte, G.M.; Takeda, K.; Rodriguez-Sosa, M.; Akira, S.; David, J.R.; Satoskar, A.R. Genetically resistant mice lacking IL-18 gene develop Th1 response and control cutaneous Leishmania major infection. J. Immunol. 2000, 164, 5890–5893. [Google Scholar] [CrossRef] [PubMed]
- Gurung, P.; Karki, R.; Vogel, P.; Watanabe, M.; Bix, M.; Lamkanfi, M.; Kanneganti, T.D. An NLRP3 inflammasome-triggered Th2-biased adaptive immune response promotes leishmaniasis. J. Clin. Investig. 2015, 125, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Basso, B.; Cervetta, L.; Moretti, E.; Carlier, Y.; Truyens, C. Acute Trypanosoma cruzi infection: IL-12, IL-18, TNF, sTNFR and NO in T. rangeli-vaccinated mice. Vaccine 2004, 22, 1868–1872. [Google Scholar] [CrossRef] [PubMed]
- Graefe, S.E.; Jacobs, T.; Gaworski, I.; Klauenberg, U.; Steeg, C.; Fleischer, B. Interleukin-12 but not interleukin-18 is required for immunity to Trypanosoma cruzi in mice. Microbes Infect. 2003, 5, 833–839. [Google Scholar] [CrossRef]
- Rodrigues, A.A.; Saosa, J.S.; da Silva, G.K.; Martins, F.A.; da Silva, A.A.; Souza Neto, C.P.; Horta, C.V.; Zamboni, D.S.; da Silva, J.S.; Ferro, E.A.; et al. IFN-gamma plays a unique role in protection against low virulent Trypanosoma cruzi strain. PLoS Negl. Trop. Dis. 2012, 6, e1598. [Google Scholar] [CrossRef] [PubMed]
- Leon Rodriguez, D.A.; Carmona, F.D.; Echeverria, L.E.; Gonzalez, C.I.; Martin, J. IL18 Gene Variants Influence the Susceptibility to Chagas Disease. PLoS Negl. Trop. Dis. 2016, 10, e0004583. [Google Scholar] [CrossRef]
- Esper, L.; Utsch, L.; Soriani, F.M.; Brant, F.; Esteves Arantes, R.M.; Campos, C.F.; Pinho, V.; Souza, D.G.; Teixeira, M.M.; Tanowitz, H.B.; et al. Regulatory effects of IL-18 on cytokine profiles and development of myocarditis during Trypanosoma cruzi infection. Microbes Infect. 2014, 16, 481–490. [Google Scholar] [CrossRef]
- Nogueira, L.G.; Santos, R.H.; Fiorelli, A.I.; Mairena, E.C.; Benvenuti, L.A.; Bocchi, E.A.; Stolf, N.A.; Kalil, J.; Cunha-Neto, E. Myocardial gene expression of T-bet, GATA-3, Ror-gammat, FoxP3, and hallmark cytokines in chronic Chagas disease cardiomyopathy: An essentially unopposed TH1-type response. Mediat. Inflamm. 2014, 2014, 914326. [Google Scholar] [CrossRef]
- Nogueira, L.G.; Frade, A.F.; Ianni, B.M.; Laugier, L.; Pissetti, C.W.; Cabantous, S.; Baron, M.; Peixoto Gde, L.; Borges Ade, M.; Donadi, E.; et al. Functional IL18 polymorphism and susceptibility to Chronic Chagas Disease. Cytokine 2015, 73, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Chudnovskiy, A.; Mortha, A.; Kana, V.; Kennard, A.; Ramirez, J.D.; Rahman, A.; Remark, R.; Mogno, I.; Ng, R.; Gnjatic, S.; et al. Host-Protozoan Interactions Protect from Mucosal Infections through Activation of the Inflammasome. Cell 2016, 167, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Munoz, M.; Liesenfeld, O.; Heimesaat, M.M. Immunology of Toxoplasma gondii. Immunol. Rev. 2011, 240, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Orellana, M.A.; Schreiber, R.D.; Remington, J.S. Interferon-gamma: The major mediator of resistance against Toxoplasma gondii. Science 1988, 240, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Liesenfeld, O.; Kosek, J.; Remington, J.S.; Suzuki, Y. Association of CD4+ T cell-dependent, interferon-gamma-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J. Exp. Med. 1996, 184, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Kastelein, R.; Hunter, C.A. Interleukin-18 (IL-18) enhances innate IL-12-mediated resistance to Toxoplasma gondii. Infect. Immun. 2000, 68, 6932–6938. [Google Scholar] [CrossRef]
- Vossenkamper, A.; Struck, D.; Alvarado-Esquivel, C.; Went, T.; Takeda, K.; Akira, S.; Pfeffer, K.; Alber, G.; Lochner, M.; Forster, I.; et al. Both IL-12 and IL-18 contribute to small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii, but IL-12 is dominant over IL-18 in parasite control. Eur. J. Immunol. 2004, 34, 3197–3207. [Google Scholar] [CrossRef]
- Schulthess, J.; Meresse, B.; Ramiro-Puig, E.; Montcuquet, N.; Darche, S.; Begue, B.; Ruemmele, F.; Combadiere, C.; Di Santo, J.P.; Buzoni-Gatel, D.; et al. Interleukin-15-dependent NKp46+ innate lymphoid cells control intestinal inflammation by recruiting inflammatory monocytes. Immunity 2012, 37, 108–121. [Google Scholar] [CrossRef]
- Munoz, M.; Eidenschenk, C.; Ota, N.; Wong, K.; Lohmann, U.; Kuhl, A.A.; Wang, X.; Manzanillo, P.; Li, Y.; Rutz, S.; et al. Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity 2015, 42, 321–331. [Google Scholar] [CrossRef]
- Miller, L.H.; Baruch, D.I.; Marsh, K.; Doumbo, O.K. The pathogenic basis of malaria. Nature 2002, 415, 673–679. [Google Scholar] [CrossRef]
- Singh, R.P.; Kashiwamura, S.; Rao, P.; Okamura, H.; Mukherjee, A.; Chauhan, V.S. The role of IL-18 in blood-stage immunity against murine malaria Plasmodium yoelii 265 and Plasmodium berghei ANKA. J. Immunol. 2002, 168, 4674–4681. [Google Scholar] [CrossRef] [PubMed]
- Cramer, J.P.; Lepenies, B.; Kamena, F.; Holscher, C.; Freudenberg, M.A.; Burchard, G.D.; Wagner, H.; Kirschning, C.J.; Liu, X.; Seeberger, P.H.; et al. MyD88/IL-18-dependent pathways rather than TLRs control early parasitaemia in non-lethal Plasmodium yoelii infection. Microbes Infect. 2008, 10, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, K.A.; De Souza, J.B.; Riley, E.M. IL-18-induced expression of high-affinity IL-2R on murine NK cells is essential for NK-cell IFN-gamma production during murine Plasmodium yoelii infection. Eur. J. Immunol. 2015, 45, 3431–3440. [Google Scholar] [CrossRef] [PubMed]
- Torre, D.; Giola, M.; Speranza, F.; Matteelli, A.; Basilico, C.; Biondi, G. Serum levels of interleukin-18 in patients with uncomplicated Plasmodium falciparum malaria. Eur. Cytokine Netw. 2001, 12, 361–364. [Google Scholar] [PubMed]
- Nagamine, Y.; Hayano, M.; Kashiwamura, S.; Okamura, H.; Nakanishi, K.; Krudsod, S.; Wilairatana, P.; Looareesuwan, S.; Kojima, S. Involvement of interleukin-18 in severe Plasmodium falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 2003, 97, 236–241. [Google Scholar] [CrossRef]
- Sareneva, T.; Matikainen, S.; Kurimoto, M.; Julkunen, I. Influenza A virus-induced IFN-alpha/beta and IL-18 synergistically enhance IFN-gamma gene expression in human T cells. J. Immunol. 1998, 160, 6032–6038. [Google Scholar] [PubMed]
- Wang, Y.; Chaudhri, G.; Jackson, R.J.; Karupiah, G. IL-12p40 and IL-18 play pivotal roles in orchestrating the cell-mediated immune response to a poxvirus infection. J. Immunol. 2009, 183, 3324–3331. [Google Scholar] [CrossRef]
- Poli, G.; Fauci, A.S. The effect of cytokines and pharmacologic agents on chronic HIV infection. AIDS Res. Hum. Retrovir. 1992, 8, 191–197. [Google Scholar] [CrossRef]
- Shapiro, L.; Puren, A.J.; Barton, H.A.; Novick, D.; Peskind, R.L.; Shenkar, R.; Gu, Y.; Su, M.S.; Dinarello, C.A. Interleukin 18 stimulates HIV type 1 in monocytic cells. Proc. Natl. Acad. Sci. USA 1998, 95, 12550–12555. [Google Scholar] [CrossRef]
- Ahmad, R.; Sindhu, S.T.; Toma, E.; Morisset, R.; Ahmad, A. Elevated levels of circulating interleukin-18 in human immunodeficiency virus-infected individuals: Role of peripheral blood mononuclear cells and implications for AIDS pathogenesis. J. Virol. 2002, 76, 12448–12456. [Google Scholar] [CrossRef]
- Fujioka, N.; Akazawa, R.; Ohashi, K.; Fujii, M.; Ikeda, M.; Kurimoto, M. Interleukin-18 protects mice against acute herpes simplex virus type 1 infection. J. Virol. 1999, 73, 2401–2409. [Google Scholar] [PubMed]
- Harandi, A.M.; Svennerholm, B.; Holmgren, J.; Eriksson, K. Interleukin-12 (IL-12) and IL-18 are important in innate defense against genital herpes simplex virus type 2 infection in mice but are not required for the development of acquired gamma interferon-mediated protective immunity. J. Virol. 2001, 75, 6705–6709. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Chen, B.; Chew, M.V.; Barra, N.G.; Shenouda, M.M.; Nham, T.; van Rooijen, N.; Jordana, M.; Mossman, K.L.; Schreiber, R.D.; et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J. Exp. Med. 2017, 214, 1153–1167. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chassaing, B.; Shi, Z.; Uchiyama, R.; Zhang, Z.; Denning, T.L.; Crawford, S.E.; Pruijssers, A.J.; Iskarpatyoti, J.A.; Estes, M.K.; et al. Viral infection. Prevention and cure of rotavirus infection via TLR5/NLRC4-mediated production of IL-22 and IL-18. Science 2014, 346, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.; Latz, E.; Moore, K.J.; Golenbock, D.T. The Nalp3 inflammsome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Dunne, A.; Subramanian, S.L.; Hull, R.L.; Tannahill, G.M.; Sharp, F.A.; Becker, C.; Franchi, L.; Yoshihara, E.; Chen, Z.; et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1b in type 2 diabetes. Nat. Immunol. 2010, 11, 897. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.B.; Lewis, E.; Jensen, D.R.; Voshol, P.J.; Jan Kullberg, B.; Tack, C.J.; van Krieken, H.; Kim, S.-H.; Stalenhoef, A.F.; et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat. Med. 2006, 12, 650–656. [Google Scholar] [CrossRef]
- Zorrilla, E.P.; Sanchez-Alavez, M.; Sugama, S.; Brennan, M.; Fernandez, R.; Bartfai, T.; Conti, B. Interleukin-18 controls energy homeostasis by suppressing appetite and feed efficiency. Proc. Natl. Acad. Sci. USA 2007, 104, 11097–11102. [Google Scholar] [CrossRef]
- Zorrilla, E.P.; Conti, B. Interleukin-18 null mutation increases weight and food intake and reduces energy expenditure and lipid substrate utilization in high-fat diet fed mice. Brain Behav. Immun. 2014, 37, 45–53. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscle, exercise and obesity: Skeletal muscle as a secretory organ. Nat. Rev. Endocrinol. 2012, 8, 457–465. [Google Scholar] [CrossRef]
- Wang, W.; Seale, P. Control of brown and beige fat development. Nat. Rev. Mol. Cell Biol. 2015, 17, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Virtue, S.; Vidal-Puig, A. Assessmentofbrownadiposetissuefunction. Front. Physiol. 2013, 4, Article128. [Google Scholar]
- Pazos, P.; Lima, L.; Tovar, S.; Gonzalez-Touceda, D.; Dieguez, C.; Garcia, M.C. Divergent responses to thermogenic stimuli in BAT and subcutaneous adipose tissue from interleukin 18 and interleukin 18 receptor 1-deficient mice. Sci. Rep. 2015, 5, 17977. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.S.N.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Brestoff, J.R.; Kim, B.S.; Saenz, S.A.; Stine, R.R.; Monticelli, L.A.; Sonnenberg, G.F.; Thome, J.J.; Farber, D.L.; Lutfy, K.; Seale, P.; et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 2015, 519, 242–246. [Google Scholar] [CrossRef]
- Lee, M.-W.; Odegaard, J.I.; Mukundan, L.; Qin, Y.; Molofsky, A.B.; Nussbaum, J.C.; Yun, K.; Locksley, R.M.; CHawla, A. Activated type 2 innate lyphoid cells regulate beige fat biogenesis. Cell 2015, 160, 74–87. [Google Scholar] [CrossRef]
- Ricardo-Gonzalez, R.R.; Van Dyken, S.J.; Shcneider, C.; Lee, J.; Nussbaum, J.C.; Liang, H.-E.; Vaka, D.; Eckalbar, W.L.; Molofsky, A.B.; Erle, D.J.; et al. Tissue signals imprint ILC2 identity with anticipatory function. Nat. Immunol. 2018, 19, 1093–1099. [Google Scholar] [CrossRef]
- Kjøbsted, R.; Hingst, J.R.; Fentz, J.; Foretz, M.; Sanz, M.-N.; Pehmøller, C.; Shum, M.; Marette, A.; Mounier, R.; Treebak, J.T.; et al. AMPK in skeletal muscle function and metabolism. FASEB J. 2018, 32, 1741–1777. [Google Scholar] [CrossRef]
- Lindegaard, B.; Matthews, V.B.; Brandt, C.; Hojman, P.; Allen, T.L.; Estevez, E.; Watt, M.J.; Bruce, C.R.; Mortensen, O.H.; Syberg, S.; et al. Interleukin-18 activates skeletal muscle AMPK and reduces weight gain and insulin resistance in mice. Diabetes 2013, 62, 3064–3074. [Google Scholar] [CrossRef]
- Murphy, A.J.; Kraakman, M.J.; Kammoun, H.L.; Dragoljevic, D.; Lee, M.K.; Lawlor, K.E.; Wentworth, J.M.; Vasanthakumar, A.; Gerlic, M.; Whitehead, L.W.; et al. IL-18 Production from the NLRP1 Inflammasome Prevents Obesity and Metabolic Syndrome. Cell Metab. 2016, 23, 155–164. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P.-Y. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef] [PubMed]
- De Nardo, D.; Latz, E. NLRP3 inflammasomes link inflammation and metabolic disease. Trend Immunol. 2011, 32, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, Z.; sepahvand, F.; Rashidi, B.; Sahebkar, A.; Mosoudifar, A.; Mizaei, H. NLRP3 inflammasome: Its regulation and involvment in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Chen, S.; Sun, R.; Zhang, X.; Wang, D. The NLRP3 inflammasome: Role in metabolic disorders and regulation by metabolic pathways. Cancer Lett. 2018, 419, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephenes, J.M.; Deep Dixit, V. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Steinstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, H.; van de Veerdonk, F.L.; Perera, D.; Neeale, G.A.; Hooiveld, G.J.; Hijimans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef] [PubMed]
- Wada, J.; Makino, H. Innate immunity in diabetes and diabetic nephropathy. Nat. Rev. Nephrol. 2016, 12, 13–26. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef]
- Stewart, C.R.; stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.C.; Halle, A.; Ryaner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef]
- Klinkner, A.M.; Robbie Waites, C.; Kerns, W.D.; Bugelski, P.J. Evidence of form cell and cholesterol crystal foration in macrophages incubated with oxidized LDL by fluorescence and electron microscopy. J. Histochem. Cytochem. 1995, 43, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauemfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid–induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3024. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Tze-Han Huang, M.; June Brickey, W.; Ting, J.P.-Y. Fatty acid-induced NLRP3-ASC nflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Koenen, T.B.; Stienstra, R.; can Tits, L.J.; de Graaf, J.; Stalenhoef, A.F.H.; Joosten, L.A.B.; Tack, C.J.; Netea, M.G. Hyperglycemia Activates Caspase-1 and TXNIP-Mediated IL-1b Transcription in Human Adipose Tissue. Diabetes 2011, 60, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef]
- Lee, H.-M.; Kim, J.-J.; Shong, M.; Jeong Ku, B.; Jo, E.-K. Upregulated NLRP3 Inflammasome Activation in Patients With Type 2 Diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef]
- Kursawe, R.; Dixit, V.D.; Scherer, P.E.; santoro, N.; Narayan, D.; Gordillo, R.; Giannini, C.; Lopez, X.; Pierpont, B.; Nouws, J.; et al. A role of the inflammasome in the low strage capacity of the abdominal subcutaneous adipose tissue in obese addolescents. Diabetes 2016, 65, 610–618. [Google Scholar] [CrossRef]
- Steinstra, R.; Joosten, L.A.B.; Koenen, T.B.; van Tits, B.; van Diepen, J.A.; van den Berg, S.; Rensen, P.C.N.; van Rooijen, N.; Wabitsch, M.; Kullber, B.-J.; et al. The Inflammasome-Mediated Caspase-1 Activation Controls Adipocyte Differentiation and Insulin Sensitivity. Cell Metab. 2010, 12, 593–605. [Google Scholar] [CrossRef]
- Netae, M.G.; Joosten, L.A.B. The NLRP1-IL18 Connection: A Stab in the Back of Obesity-Induced Inflammation. Cell Metab. 2016, 23, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Biologic basis for interleukin-1 in disease. Blood 1996, 87, 2095–2147. [Google Scholar] [PubMed]
- Hyodo, Y.; Matsui, K.; Hayashi, N.; Tsutsui, H.; Kashiwamura, S.; Yamauchi, H.; Hiroishi, K.; Takeda, K.; Tagawa, Y.; Iwakura, Y.; et al. IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J. Immunol. 1999, 162, 1662–1668. [Google Scholar]
- Takeda, K.; Tsutsui, H.; Yoshimoto, T.; Adachi, O.; Yoshida, N.; Kishimoto, T.; Okamura, H.; Nakanishi, K.; Akira, S. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 1998, 8, 383–390. [Google Scholar] [CrossRef]
- Castordi, A.; Naffah de Souza, C.; Olsen Saraiva Cåmara, N.; Moraes-Vieira, P.M. The Macrophage Switch in Obesity Development. Front. Immunol. 2016, 6, 637. [Google Scholar]
- Appari, M.; Channon, K.M.; McNeill, E. Metabolic Regulation of Adipose Tissue Macrophage Function in Obesity and Diabetes. Antioxid. Redox Signal. 2018, 29, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Fernandes, H.; Mucida, D. Neuro-Immune Interactions at Barrier Surfaces. Cell 2016, 165, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Tanoue, T.; Atarashi, K.; Honda, K. Development and maintenance of intestinal regulatory T cells. Nat. Rev. Immunol. 2016, 16, 295–309. [Google Scholar] [CrossRef]
- Mowat, A.M.; Agace, W.W. Regional specialization within the intestinal immune system. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.K.; Ka-Ming Chan, F. Inflammasome, Inflammation, and Tissue Homeostasis. Trend Mol. Med. 2018, 24, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Lei-Leston, A.C.; Murphy, A.G.; Maloy, K.J. Epithelial cell inflammasomes in intestinal immunity and inflammation. Front. Immunol. 2017, 8, 1168. [Google Scholar] [CrossRef] [PubMed]
- Opipari, A.; Franchi, L. Role of inflammasomes in intestinal inflammation and Crohn’s disease. Inflamm. Bowel. Dis. 2015, 21, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Dahlhamet, J.M.; Zammitti, E.P.; Ward, B.W.; Wheaton, A.G.; Croft, J.B. Prevalence of Inflammatory Bowel Disease Among Adults Aged ≥18 Years—United States, 2015. MMWR Morb. Mortal. Wkly. Rep. 2015, 65, 1166–1169. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Kitani, A.; Strober, W.; Fuss, I.J. The Role of NLRP3 and IL-1beta in the Pathogenesis of Inflammatory Bowel Disease. Front. Immunol 2018, 9, 2566. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Nishizaki, Y.; Sano, O.; Ohta, T.; Ikeda, M.; Kurimoto, M. Immunohistochemical and immuno-electron-microscopic detection of interferon-g-inducing factor (interelukin-18) in mouse intestinal epithelial cells. Cell Tissue Res. 1997, 289, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Pizarro, T.T.; Michie, M.H.; Bents, M.; Woraratanadharm, J.; Smith, M.F., Jr.; Foley, E.; Moskaluk, C.A.; Bickston, S.J.; Cominelli, F. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: Expression and localization in intestinal mucosal cells. J. Immunol. 1999, 162, 6829–6835. [Google Scholar]
- Kempster, S.; Belteki, G.; Forhead, A.J.; Fowden, A.L.; Catalano, R.D.; Lam, B.Y.; McFarlane, I.; Stephen Charnock-Junes, D.; Smith, G.C. Developmental control of the Nlrp6 inflammasome and a substrate, IL-18, in mammalian intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G253–G263. [Google Scholar] [CrossRef]
- Knodler, L.A.; Crowley, S.M.; Pan Sham, H.; Yang, H.; Wrande, M.; Ma, C.; Emst, R.K.; Steele-Mortimer, O.; Celli, J.; Callance, B.A. Noncanonical Inflammasome Activation of Caspase-4/Caspase-11 Mediates Epithelial Defenses against Enteric Bacterial Pathogens. Cell Host Microbe 2014, 16, 249–256. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Zeevi, D.; Dohnalová, L.; Zilbeman-Schapira, G.; Ali Mahdi, J.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 2015, 163, 1428–1443. [Google Scholar] [CrossRef] [PubMed]
- Song-Zhao, G.X.; Srinivasan, N.; Pott, J.; Baban, D.; Frankel, G.; Maloy, K.J. Nlrp3 activation in the intestinal epithelium protects against a mucosal pathogen. Mucosal Immunol. 2014, 7, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Elinav, E.; Huber, S.; Booth, C.J.; Strowig, T.; Jin, C.; Eisenbarth, S.C.; Flavell, R.A. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl. Acad. Sci. USA 2010, 107, 21635–21640. [Google Scholar] [CrossRef] [PubMed]
- Rauch, I.; Deets, K.A.; Ji, D.X.; von Moltke, J.; Tenthorey, J.L.; Lee, A.Y.; Philip, N.H.; Ayres, J.S.; Brodsky, I.E.; Gronert, K.; et al. NAIP-NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL-18 Release via Activation of Caspase-1 and -8. Immunity 2017, 46, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Nordiander, S.; Pott, J.; Moloy, K.J. NLRC4 expression in intestinal epithelial cells mediates protection against an enteric pathogen. Mucosal Immunol. 2015, 7, 775–785. [Google Scholar] [CrossRef] [PubMed]
- sellin, M.E.; Müller, A.A.; Dolowschiak, T.; Diard, M.; Tardivel, A.; Maslowski, K.M.; Hardt, W.-D. Epithelium-Intrinsic NAIP/NLRC4 Inflammasome Drives Infected Enterocyte Expulsion to Restrict Salmonella Replication in the Intestinal Mucosa. Cell Host Microbe 2014, 16, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Normand, S.; Delanoye-Crespin, A.; Bressenot, A.; Huot, L.; HGrandjean, T.; Peyrin-Biroulet, L.; Lemoine, Y.; Hot, D.; Chamaillard, M. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl. Acad. Sci. USA 2011, 108, 9601–9606. [Google Scholar] [CrossRef]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, C.G.; Santee, C.A.; Lych, S.V.; et al. Induction of Intestinal Th17 Cells by Segmented Filamentous Bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2010, 107, 12204–12209. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kumahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of Colonic Regulatory T Cellsby Indigenous Clostridium Species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Chung, H.; Pamp, S.J.; Hill, J.A.; Surana, N.K.; Edelman, S.M.; Troy, E.B.; Reading, N.C.; Villablanca, E.J.; Wang, S.; Mora, J.R.; et al. Gut Immune Maturation Depends on Colonization with a Host-Specific Microbiota. Cell 2012, 149, 1578–1593. [Google Scholar] [CrossRef] [PubMed]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Staley, D.; Luong, S.; Maruya, M.; McKenzie, C.I.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Núñez, G. A Functional Role for Nlrp6 in Intestinal Inflammation and Tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Shapiro, H.; Thaiss, C.A.; Elinav, E. NLRP6: A Multifaceted Innate Immune Sensor. Trend Immunol. 2017, 38, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Somanna, N.K.; Yariswamy, M.; Garagliano, J.M.; Siebenlist, U.; Mummidi, S.; Valete, A.J.; Chadrasekar, B. Aldosterone-induced cardiomyocyte growth, and fibroblast migration and proliferation are mediated by TRAF3IP2. Cell. Signal. 2015, 27, 1928–1938. [Google Scholar] [CrossRef]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.-P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 Inflammasome Orchestrates the Colonic Host-Microbial Interface by Regulating Goblet Cell Mucus Secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Wang, M.L.; Keilbaugh, S.A.; He, W.; Brenes, M.; Swain, G.P.; Knight, P.A.; Donaldson, D.D.; Lazar, M.A.; Miller, H.R.; et al. RELMbeta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc. Natl. Acad. Sci. USA 2004, 101, 13596–13600. [Google Scholar] [CrossRef]
- Tsuji, S.; Uehori, J.; Matsumoto, M.; Suzuki, Y.; Matsuhisa, A.; Toyoshima, K.; Seya, T. Human intelectin is a novel soluble lectin that recognizes galactofuranose in carbohydrate chains of bacterial cell wall. J. Biol. Chem. 2001, 276, 23456–23463. [Google Scholar] [CrossRef]
- Van der Sluis, M.; De Koning, B.A.E.; De Bruun, A.C.J.M.; Velcich, A.; Meuerink, J.P.P.; Van Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-Deficient Mice Spontaneously Develop Colitis, Indicating That Muc2 Is Critical for Colonic Protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef]
- Pelaseyed, T.; Bergström, J.H.; Gustafsson, J.K.; Ermund, A.; Birchenough, G.M.H.; Schütte, A.; van der Post, S.; Svensson, F.; Rodriguez-Plñlro, A.M.; Nystrøm, E.E.L.; et al. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol. Rev. 2014, 260, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Ushio, H.; Ueno, T.; Kojima, Y.; Komatsu, M.; Tanaka, S.; Yamamoto, A.; Ichimura, Y.; Ezki, J.; Nishida, K.; Komazawa-Sakon, S.; et al. Crucial role for autophagy in degranulation of mast cells. J. Allergy Clin. Immunol. 2011, 127, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- consortium, T.i.F. Ancient Missense Mutations in a New Member of the RoRet Gene Family Are Likely to Cause Familial Mediterranean Fever. Cell 1997, 90, 797–807. [Google Scholar] [CrossRef]
- Consortium, T.F.F. A candidate gene for familial Mediterranean fever. Nat. Genet. 1997, 17, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Cattan, D.; Notarnicola, C.; Molinari, N.; Touitou, I. Inflammatory bowel disease in non-Ashkenazi Jews with familial Mediterranean fever. Lancet 2000, 355, 378–379. [Google Scholar] [CrossRef]
- Giaglis, S.; Mimidis, K.; Papadopoulos, V.; Thomopoulos, K.; Sidiropoulos, P.; Rafail, S.; Nikolopoulos, V.; Fragouli, E.; Kartalis, G.; Tzioufas, A.; et al. Increased Frequency of Mutations in the Gene Responsible for Familial Mediterranean Fever (MEFV) in a Cohort of Patients with Ulcerative Colitis: Evidence for a Potential Disease-Modifying Effect? Dig. Dis. Sci. 2006, 51, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Ming Man, S.; Kanneganti, T.-D. Inflammasomes and Cancer. Cancer Immunol. Res. 2017, 5, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Gavrillin, M.A.; Abdelaziz, D.H.A.; Mostafa, M.; Abdulrahman, B.A.; Grandhi, J.; Akhter, A.; Abu Khweek, A.; Aubert, D.F.; Valvano, M.A.; Wewers, M.D.; et al. Activation of the Pyrin Inflammasome by Intracellular Burkholderia cenocepacia. J. Immunol. 2012, 188, 3469–3477. [Google Scholar] [CrossRef]
- Xu, H.; Yang, J.; Gao, W.; Li, L.; Li, P.; Zhang, L.; Gong, Y.-N.; Peng, X.; Jeff Xi, J.; Chen, S.; et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014, 513, 237–241. [Google Scholar] [CrossRef]
- Sharma, D.; Malik, A.; Gui, C.S.; Karki, R.; Vogel, P.; Kanneganti, T.-D. Pyrin inflammasome regulates tight junction integrity to restrict colitis and tumorigenesis. Gastroenterology 2018, 154, 948–964. [Google Scholar] [CrossRef]
- Hasan Zaki, M.; Vogel, P.; Body-Malapel, M.; Lamkafi, M.; Kanneganti, T.-D. IL-18 Production Downstream of the Nlrp3 Inflammasome Confers Protection against Colorectal Tumor Formation. J. Immunol 2010, 185, 4912–4920. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Ding, S.; Wang, P.; Wei, Z.; Pan, W.; Palm, N.W.; Yang, Y.; Yu, H.; Li, H.-B.; Wang, G.; et al. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 2017, 546, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.; Pelletier, J. The biology of DHX9 and its potential as a therapeutic target. Oncotarget 2016, 7, 42716–42739. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, T.; Nakanishi, K.; Hirose, K.; Hiroishi, K.; Okamura, H.; Takemoto, Y.; Kanamaru, A.; Hada, T.; Tamura, T.; Kakishita, E.; et al. High serum IL-6 level reflects susceptible status of the host to endotoxin and IL-1/tumor necrosis factor. J. Immunol. 1992, 148, 3596–3603. [Google Scholar] [PubMed]
- Tsutsui, H.; Matsui, K.; Okamura, H.; Nakanishi, K. Pathophysiological roles of interleukin-18 for inflammatory liver diseases. Immunol. Rev. 2000, 174, 192–209. [Google Scholar] [CrossRef] [PubMed]
- Kawa, K.; Tsutsui, H.; Uchiyama, R.; Kato, J.; Matsui, K.; Iwakura, Y.; Matsumoto, T.; Nakanishi, K. IFN-g is a master regulator of endotoxin shock syndrome in mice primed with heat-killed Propionibacterium acnes. Int. Immunol. 2010, 22, 157–166. [Google Scholar] [CrossRef]
- Sugimoto, T.; Ishikawa, Y.; Yoshimoto, T.; Hayashi, B.; Fujimoto, J.; Nakanishi, K. IL-18 acts on memory Th1 cells to induce airway inflammatin and hyperresponsiveness in a naive host mouse. J. Exp. Med. 2004, 199, 535–545. [Google Scholar] [CrossRef]
- Hayashi, N.; Yoshimoto, T.; Izuhara, K.; Matsui, K.; Tanaka, T.; Nakanishi, K. T helper 1 cells stimulated with ovalbunmin and IL-18 induce airway hyperresponsiveness and lung fibrosis by IFN-g and IL-13 production. Proc. Natl. Acad. Sci. USA 2007, 104, 14765–14770. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Yoshimoto, T.; Nakanishi, K. Contribution of IL-18-induced innate T cell activation to airway inflammation with mucus hypersecretion and airway hyperresponsiveness. Int. Immunol. 2006, 18, 847–855. [Google Scholar] [CrossRef]
- Kondo, Y.; Yoshimoto, T.; Yasuda, K.; Futatsugi-Yumikura, S.; Morimoto, M.; Hayashi, N.; Hoshino, T.; Fujimoto, J.; Nakanishi, K. Administration of IL-33 induces airway hyperresponsiveness and goblet cell hyperplasia in the lung in the absence of adaptive immune system. Int. Immunol. 2008, 20, 791–800. [Google Scholar] [CrossRef]
- Wu, H.; Craft, M.L.; Wang, P.; Wyburn, K.R.; Chen, G.; Ma, J.; Hambly, B.; Chadban, S.J. IL-18 contributes to renal damage after ischemia-reperfusion. J. Am. Soc. Nephrol. 2008, 19, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Faubel, S.; Ljubanovic, D.; Reznikov, L.; Somerset, H.; Dinarello, C.A.; Edelstein, C.L. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int. 2004, 66, 2202–2213. [Google Scholar] [CrossRef] [PubMed]
- Bani-Hani, A.H.; Leslie, J.A.; Asanuma, H.; Dinarello, C.A.; Campbell, M.T.; Meldrum, D.R.; Zhang, H.; Hile, K.; Meldrum, K.K. IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int. 2009, 76, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Masood, H.; Che, R.; Zhang, A. Inflammasomes in the Pathophysiology of Kidney Diseases. Kidney Dis. (Basel) 2015, 1, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Hutton, H.L.; Ooi, J.D.; Holdsworth, S.R.; Kitching, A.R. The NLRP3 inflammasome in kidney disease and autoimmunity. Nephrology (Carlton) 2016, 21, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Purves, J.T.; Hughes, F.M., Jr. Inflammasomes in the urinary tract: A disease-based review. Am. J. Physiol. Renal Physiol. 2016, 311, F653–F662. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, C.; Tan, L.; Miethke, T.; Anand, P.K. Immunity to uropathogens: The emerging roles of inflammasomes. Nat. Rev. Urol. 2017, 14, 284–295. [Google Scholar] [CrossRef]
- D’Amico, G. Natural history of idiopathic IgA nephropathy: Role of clinical and histological prognostic factors. Am. J. Kidney Dis. 2000, 36, 227–237. [Google Scholar] [CrossRef]
- Koyama, A.; Igarashi, M.; Kobayashi, M. Natural history and risk factors for immunoglobulin A nephropathy in Japan. Research Group on Progressive Renal Diseases. Am. J. Kidney Dis. 1997, 29, 526–532. [Google Scholar] [CrossRef]
- Shi, B.; Ni, Z.; Cao, L.; Zhou, M.; Mou, S.; Wang, Q.; Zhang, M.; Fang, W.; Yan, Y.; Qian, J. Serum IL-18 is closely associated with renal tubulointerstitial injury and predicts renal prognosis in IgA nephropathy. Mediat. Inflamm. 2012, 2012, 728417. [Google Scholar] [CrossRef]
- Liu, D.; Xu, M.; Ding, L.H.; Lv, L.L.; Liu, H.; Ma, K.L.; Zhang, A.H.; Crowley, S.D.; Liu, B.C. Activation of the Nlrp3 inflammasome by mitochondrial reactive oxygen species: A novel mechanism of albumin-induced tubulointerstitial inflammation. Int J. Biochem. Cell Biol. 2014, 57, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, V.Y.; Ecder, T.; Fantuzzi, G.; Siegmund, B.; Lucia, M.S.; Dinarello, C.A.; Schrier, R.W.; Edelstein, C.L. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J. Clin. Investig. 2001, 107, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, V.Y.; Faubel, S.; Siegmund, B.; Lucia, M.S.; Ljubanovic, D.; Edelstein, C.L. Neutrophil-independent mechanisms of caspase-1- and IL-18-mediated ischemic acute tubular necrosis in mice. J. Clin. Investig. 2002, 110, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Jani, A.; Melnikov, V.Y.; Faubel, S.; Edelstein, C.L. Urinary interleukin-18 is a marker of human acute tubular necrosis. Am. J. Kidney Dis. 2004, 43, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Mishra, J.; Thiessen-Philbrook, H.; Dursun, B.; Ma, Q.; Kelly, C.; Dent, C.; Devarajan, P.; Edelstein, C.L. Urinary IL-18 is an early predictive biomarker of acute kidney injury after cardiac surgery. Kidney Int. 2006, 70, 199–203. [Google Scholar] [CrossRef]
- Schrezenmeier, E.V.; Barasch, J.; Budde, K.; Westhoff, T.; Schmidt-Ott, K.M. Biomarkers in acute kidney injury—Pathophysiological basis and clinical performance. Acta Physiol. (Oxf.) 2017, 219, 554–572. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Yusuf, B.; Shlipak, M.G.; Garg, A.X.; Parikh, C.R. Long-term risk of mortality and other adverse outcomes after acute kidney injury: A systematic review and meta-analysis. Am. J. Kidney Dis. 2009, 53, 961–973. [Google Scholar] [CrossRef]
- Li, S.; Krawczeski, C.D.; Zappitelli, M.; Devarajan, P.; Thiessen-Philbrook, H.; Coca, S.G.; Kim, R.W.; Parikh, C.R.; Consortium, T.-A. Incidence, risk factors, and outcomes of acute kidney injury after pediatric cardiac surgery: A prospective multicenter study. Crit. Care Med. 2011, 39, 1493–1499. [Google Scholar] [CrossRef]
- Zappitelli, M.; Greenberg, J.H.; Coca, S.G.; Krawczeski, C.D.; Li, S.; Thiessen-Philbrook, H.R.; Bennett, M.R.; Devarajan, P.; Parikh, C.R.; Translational Research Investigating Biomarker Endpoints in Acute Kidney Injury (TRIBE-AKI) Consortium. Association of definition of acute kidney injury by cystatin C rise with biomarkers and clinical outcomes in children undergoing cardiac surgery. JAMA Pediatr. 2015, 169, 583–591. [Google Scholar] [CrossRef]
- Coca, S.G.; Garg, A.X.; Thiessen-Philbrook, H.; Koyner, J.L.; Patel, U.D.; Krumholz, H.M.; Shlipak, M.G.; Parikh, C.R.; Consortium, T.-A. Urinary biomarkers of AKI and mortality 3 years after cardiac surgery. J. Am. Soc. Nephrol. 2014, 25, 1063–1071. [Google Scholar] [CrossRef]
- Feltes, C.M.; Van Eyk, J.; Rabb, H. Distant-organ changes after acute kidney injury. Nephron Physiol. 2008, 109, p80–84. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.J. Distant effects of experimental renal ischemia/reperfusion injury. J. Am. Soc. Nephrol. 2003, 14, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Puthumana, J.; Shlipak, M.G.; Koyner, J.L.; Thiessen-Philbrook, H.; McArthur, E.; Kerr, K.; Kavsak, P.; Whitlock, R.P.; Garg, A.X.; et al. Relationship of Kidney Injury Biomarkers with Long-Term Cardiovascular Outcomes after Cardiac Surgery. J. Am. Soc. Nephrol. 2017, 28, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Hulthe, J.; McPheat, W.; Samnegård, A.; Tornvall, P.; Hamsten, A.; Eriksson, P. Plasma interleukin (IL)-18 concentrations is elevated in patients with previous myocardial infarction and related to severity of coronary atherosclerosis independently of C-reactive protein and IL-6. Atherosclerosis 2006, 188, 4450–4454. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z.; Corbaz, A.; Scoazec, A.; Besnard, S.; Leséche, G.; Chvatchko, Y.; Tedgui, A. Expression of Interleukin-18 in human atherosclerotic plaque and relation to plaque instability. Circulation 2001, 104, 1598–1603. [Google Scholar] [CrossRef]
- Blankenberg, S.; Tiret, L.; Bickel, C.; Peetz, D.; Cambien, F.; Meyer, J.; Rupprecht, H.J.; AtheroGene, I. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation 2002, 106, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Blankenberg, S.; Luc, G.; Ducimetiére, P.; Arveiler, D.; Ferriéres, J.; Amouyel, P.; Evans, A.; Cambien, F.; Tiret, L. Interleukin-18 and the risk of coronary heart disease in european men: The prospective epidemiological study of myocardial infarction (PRIME). Circulation 2003, 108, 2453–2459. [Google Scholar] [CrossRef]
- Jefferis, B.J.M.H.; Papacosta, O.; Owen, C.G.; Toya Wannamethee, S.; Humphries, S.E.; Woodward, M.; Lennon, L.T.; Thomson, A.; Welsh, P.; Rumley, A.; et al. Interleukin 18 and coronary heart disease: Prospective study and systematic review. Atherosclerosis 2011, 217, 227–233. [Google Scholar] [CrossRef]
- Dasu, M.R.; Devaray, S.; Zhao, L.; Hwang, D.H.; Jialal, I. High Glucose Induces Toll-Like Receptor Expression in Human Monocytes. Diabetes 2008, 57, 3090–3098. [Google Scholar] [CrossRef]
- Okamura, H.; Kashiwamura, S.; Tsutsui, H.; Yoshimoto, T.; Nakanishi, K. Regulation of interferon-g production by IL-12 and IL-18. Curr. Opin. Immunol. 1998, 10, 259–264. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Granzin, M.; Wagner, J.; Kohl, U.; Cerwenka, A.; Huppert, V.; Ullrich, E. Shaping of Natural Killer Cell Antitumor Activity by Ex Vivo Cultivation. Front. Immunol. 2017, 8, 458. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Cheon, S.; Cho, D. The Dual Effects of Interleukin-18 in Tumor Progression. Cell. Mol. Immunol 2007, 4, 329–335. [Google Scholar] [PubMed]
- Fabbi, M.; Carbotti, G.; Ferrini, S. Context-dependent role of IL-18 in cancer biology and counter-regulation by IL-18BP. J. Leukoc. Biol. 2015, 97, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Bonneville, M.; O’Brien, R.L.; Born, W.K. γδ T cell effector functions: A blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 2010, 10, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.; Anderson, J. Engineering Approaches in Human Gamma Delta T Cells for Cancer Immunotherapy. Front. Immunol. 2018, 9, 1409. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, F.; Merli, P.; Rutella, S. At the Bedside: Innate immunity as an immunotherapy tool for hematological malignancies. J. Leukoc. Biol. 2013, 94, 1141–1157. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Kubo, S.; Okuda, A.; Yamamoto, H.; Ueda, H.; Tanaka, T.; Nakamura, H.; Yamanishi, H.; Terada, N.; Okamura, H. Effect of IL-18 on Expansion of gd T Cells Stimulated by Zoledronate and IL-2. J. Immunother. 2010, 33, 287–296. [Google Scholar] [CrossRef]
- Tsuda, J.; Li, W.; Yamanishi, H.; Yamamoto, H.; Okuda, A.; Kubo, S.; Ma, Z.; Terada, N.; Tanaka, Y.; Okamura, H. Involvement of CD56brightCD11c+ Cells in IL-18–Mediated Expansion of Human gd T Cells. J. Immunol. 2011, 186, 2003–2012. [Google Scholar] [CrossRef]
- Li, W.; Okuda, A.; Yamamoto, H.; Yamanishi, K.; Terada, N.; Yamanishi, H.; Tanaka, Y.; Okamura, H. Regulation of Development of CD56brightCD11c+ NK-like Cells with Helper Function by IL-18. PLoS ONE 2013, 8, e82586. [Google Scholar] [CrossRef]
- Sugie, T.; Murata-Hirai, K.; Iwasaki, M.; Morita, C.T.; Li, W.; Okamura, H.; Minato, N.; Toi, M.; Tanaka, Y. Zoledronic acid-induced expansion of gammadelta T cells from early-stage breast cancer patients: Effect of IL-18 on helper NK cells. Cancer Immunol. Immunother. 2013, 62, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Gober, H.-J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; De Libero, G. Human T Cell Receptor gamma delta Cells Recognize Endogenous Mevalonate Metabolites in Tumor Cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, A.J.; Jauhiainen, M.; Monkkonen, H.; Rogers, M.J.; Monkkonen, J.; Thompson, K. Peripheral blood monocytes are responsible for gammadelta T cell activation induced by zoledronic acid through accumulation of IPP/DMAPP. Br. J. Haematol. 2009, 144, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Guimont-Desrochers, F.; Boucher, G.; Dong, Z.; Dupuis, M.; Veillette, A.; Lesage, S. Redefining interferon-producing killer dendritic cells as a novel intermediate in NK-cell differentiation. Blood 2012, 119, 4349–4357. [Google Scholar] [CrossRef] [PubMed]
- Guimont-Desrochers, F.; Lesage, S. Revisiting the prominent anti-tumoral potential of pre-mNK cells. Front. Immunol. 2013, 4, Article–446. [Google Scholar] [CrossRef]
- Ma, Z.; Li, W.; Yoshiya, S.; Xu, Y.; Hata, M.; El-Darawish, Y.; Markova, T.; Yamanishi, K.; Yamanishi, H.; Tahara, H.; et al. Augmentation of Immune Checkpoint Cancer Immunotherapy with IL18. Clin. Cancer Res. 2016, 22, 2969–2980. [Google Scholar] [CrossRef]
- Malik, A.; Sharma, D.; Malireddi, R.K.S.; Guy, C.S.; Chang, T.-C.; Olsen, S.R.; Neale, G.A.; Vogel, P.; Kanneganti, T.-D. SYK-CARD9 Signaling Axis Promotes Gut Fungi-Mediated Inflammasome Activation to Restrict Colitis and Colon Cancer. Immunity 2018, 49, 515–530. [Google Scholar] [CrossRef]
- Sartor, R.B.; Wu, G.D. Roles for intestinal bacteria, viruses, and fungi in pathogenesis of inflammatory bowel diseases and therapeutic approaches. Gastroenterology 2017, 152, 327–339. [Google Scholar] [CrossRef]
- Yamasaki, S.; Matsumoto, M.; Takeuchi, O.; Matsuzawa, T.; Ishikawa, E.; Sakuma, M.; Tateno, M.; Uno, J.; Hirabayashi, J.; Mikami, Y.; et al. C-type lectin Mincle is an activating receptor for pathogenic fungus, Malassezia. Proc. Natl. Acad. Sci. USA 2009, 106, 1897–1902. [Google Scholar] [CrossRef]
- Saijo, S.; Fujikado, N.; Furuta, T.; Chung, S.-h.; Kotaki, H.; seki, K.; Sudo, K.; Akira, S.; Adachi, Y.; Ohno, N.; et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat. Immunol. 2007, 8, 39–46. [Google Scholar] [CrossRef]
- Saijo, S.; Ikeda, S.; Yamabe, K.; Kakuta, S.; Ishigame, H.; Akitsu, A.; Fujikado, N.; Kusaka, T.; Kubo, S.; Chung, S.-h.; et al. Dectin-2 Recognition of a-Mannans and Induction of Th17 Cell Differentiation Is Essential for Host Defense against Candida albicans. Immunity 2010, 32, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.-L.; Zhao, X.-Q.; Jiang, C.; You, Y.; Chen, X.-P.; Jiang, Y.-Y.; Jia, X.-M.; Lin, X. C-Type Lectin Receptors Dectin-3 and Dectin-2 Form a Heterodimeric Pattern-Recognition Receptor for Host Defense against Fungal Infection. Immunity 2013, 39, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Drummond, R.A.; Franco, L.M.; Lionakis, M.S. Human CARD9: A Critical Molecule of Fungal Immune Surveillance. Front. Immunol. 2018, 9, 1836. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, Z.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Takatori, H.; Makita, S.; Ito, T.; Matsuki, A.; Nakajima, H. Regulatory Mechanisms of IL-33-ST2-Mediated Allergic Inflammation. Front. Immunol. 2018, 9, 2004. [Google Scholar] [CrossRef]
- Kakkar, R.; Hei, H.; Dobner, S.; Lee, R.T. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J. Biol. Chem. 2012, 287, 6941–6948. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef]
- Zhao, W.; Hu, Z. The enigmatic processing and secretion of interleukin-33. Cell. Mol. Immunol. 2010, 7, 260–262. [Google Scholar] [CrossRef]
- Lefrancais, E.; Roga, S.; Gautier, V.; Gonzalez-de-Peredo, A.; Monsarrat, B.; Girard, J.P.; Cayrol, C. IL-33 is processed into mature bioactive forms by neutrophil elastase and cathepsin G. Proc. Natl. Acad. Sci. USA 2012, 109, 1673–1678. [Google Scholar] [CrossRef]
- Lefrancais, E.; Duval, A.; Mirey, E.; Roga, S.; Espinosa, E.; Cayrol, C.; Girard, J.P. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2014, 111, 15502–15507. [Google Scholar] [CrossRef]
- Imai, Y.; Yasuda, K.; Sakaguchi, Y.; Haneda, T.; Mizutani, H.; Yoshimoto, T.; Nakanishi, K.; Yamanishi, K. Skin-specific expression of IL-33 activates group 2 innate lymphoid cells and elicits atopic dermatitis-like inflammation in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 13921–13926. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Muto, T.; Kawagoe, T.; Matsumoto, M.; Sasaki, Y.; Matsushita, K.; Taki, Y.; Futatsugi-Yumikura, S.; Tsutsui, H.; Ishii, K.J.; et al. Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc. Natl. Acad. Sci. USA 2012, 109, 3451–3456. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, K.; Adachi, T.; Koida, A.; Nakanishi, K. Nematode-Infected Mice Acquire Resistance to Subsequent Infection With Unrelated Nematode by Inducing Highly Responsive Group 2 Innate Lymphoid Cells in the Lung. Front. Immunol. 2018, 9, 2132. [Google Scholar] [CrossRef] [PubMed]
- Simoni, Y.; Fehlings, M.; Kloverpris, H.N.; McGovern, N.; Koo, S.L.; Loh, C.Y.; Lim, S.; Kurioka, A.; Fergusson, J.R.; Tang, C.L.; et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity 2018, 48, 1060. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Bacchi, M.; Bertolino, M. Pharmacokinetics of IL-18 binding protein in healthy volunteers and subjects with rheumatoid arthritis or plaque psoriasis. Eur. J. Drug Metab. Pharmacokinet. 2006, 31, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Mier, J.W.; Logan, T.; Atkins, M.; Koon, H.; Koch, K.M.; Kathman, S.; Pandite, L.N.; Oei, C.; Kirby, L.C.; et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin. Cancer Res. 2006, 12, 4265–4273. [Google Scholar] [CrossRef]
- Robertson, M.J.; Kirkwood, J.M.; Logan, T.F.; Koch, K.M.; Kathman, S.; Kirby, L.C.; Bell, W.N.; Thurmond, L.M.; Weisenbach, J.; Dar, M.M. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin Cancer Res. 2008, 14, 3462–3469. [Google Scholar] [CrossRef] [PubMed]
- Robertson, M.J.; Kline, J.; Struemper, H.; Koch, K.M.; Bauman, J.W.; Gardner, O.S.; Murray, S.C.; Germaschewski, F.; Weisenbach, J.; Jonak, Z.; et al. A dose-escalation study of recombinant human interleukin-18 in combination with rituximab in patients with non-Hodgkin lymphoma. J. Immunother. 2013, 36, 331–341. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Kunert, A.; Chmielewski, M.; Wijers, R.; Berrevoets, C.; Abken, H.; Debets, R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology 2017, 7, e1378842. [Google Scholar] [CrossRef] [PubMed]
- Motavaf, M.; Safari, S.; Moayed Alavian, S. Interleukin-18 Gene Promoter Polymorphisms and Susceptibility to Chronic Hepatitis B Infection: A Review Study. Hepat. Mon. 2014, 14, e19879. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, H. Association between interleukin-18 gene promoter (−607C/A and −137G/C) polymorphisms and chronic hepatitis C virus infections: A meta-analysis. Meta Gene 2015, 5, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-G.; Li, J.-J.; Sun, C.-A.; Jin, Y.; Wu, W.-W. Interleukin-18 promoter polymorphisms and plasma levels are associated with increased risk of periodontitis: A meta-analysis. Inflamm. Res. 2014, 63, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, M.K.; de Carvalho, A.C.G.; Alves, E.H.P.; da Silva, F.R.P.; Pessoa, L.D.S.; Vasconcelos, D.F.P. Genetic Factors and the Risk of Periodontitis Development: Findings from a Systematic Review Composed of 13 Studies of Meta-Analysis with 71,531 Participants. Int. J. Dent. 2017, 2017, 1914073. [Google Scholar] [CrossRef]
- Pan, H.-F.; Leng, R.-X.; Ye, D.-Q. Lack of association of interleukin-18 gene promoter -607 A/C polymorphism with susceptibility to autoimmune diseases: A meta-analysis. Lupus 2011, 20, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Song, G.G.; Choi, S.J.; Ji, J.D.; Lee, Y.H. Association between interleukin-18 polymorphisms and systemic lupus erythematosus: A meta-analysis. Mol. Biol. Rep. 2013, 40, 2581–2587. [Google Scholar] [CrossRef]
- Chen, S.; Jiang, F.; Ren, J.; Liu, J.; Meng, W. Association of IL-18 polymorphisms with rheumatoid arthritis and systemic lupus erythematosus in Asian populations: A meta-analysis. BMC Med. Genet. 2012, 13, 107. [Google Scholar] [CrossRef]
- Guo, B.R.; Rong, J.J.; Wei, Y.H.; Zhong, W.Y.; Li, C.S.; Liu, M.; Li, W.; Wang, X.B.; Wang, L.; Zheng, H.F. Ethnicity-stratified analysis of the association between IL-18 polymorphisms and systemic lupus erythematosus in a European population: A meta-analysis. Arch. Dermatol. Res. 2015, 307, 747–755. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, K.; Yang, Z.; Li, F.; Wang, Z.; Xu, F.; He, C. Association of cytokine gene polymorphisms (IL6, IL12B, IL18) with Behcet’s disease: A meta-analysis. Z. Rheumatol. 2016, 75, 932–938. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.-P.; Zhou, L.-J.; Lu, S.-Y.; Liang, Y.-E.; Chen, X.-Y.; Liu, L.; Lin, J. Association of IL-18 promoter gene polymorphisms with rheumatoid arthritis: A meta-analysis. Mol. Biol. Rep. 2014, 41, 8211–8217. [Google Scholar] [CrossRef] [PubMed]
- Khodaeian, M.; Enayati, S.; Tabatabaei-Malazy, O.; Amoli, M.M. Association between Genetic Variants and Diabetes Mellitus in Iranian Populations: A Systematic Review of Observational Studies. J. Daibetes Res. 2015, 2015, 585917. [Google Scholar] [CrossRef] [PubMed]
- Ho Lee, Y.; Kim, J.-H.; Gyu Song, G. Interleukin-18 promoter -607 C/A and -137 G/C polymorphisms and susceptibility to type 1 diabetes: A meta-analysis. Hum. Immunol. 2015, 76, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.-J.; Zhang, L.; Lu, W.J.; Wang, L.; Chen, L.; Zhu, Z.; Zhu, H.-H. Interleukin-18 genetic polymorphisms contribute differentially to the susceptibility to Crohn’s disease. World J. Gastroenterol. 2015, 21, 8711–8722. [Google Scholar] [CrossRef]
- Zhang, M.-J.; Zhou, Y.; Wang, X.; Chen, X.; Pi, Y.; Guo, L.; Gan, C.-Y.; Li, J.-C.; Zhang, L.-L. Interleukin-18 gene promoter 607A polymorphism, but not 137C polymorphism, is a protective factor for ischemic stroke in the Chinese population: A meta-analysis. Meta Gene 2016, 9, 165–172. [Google Scholar] [CrossRef]
- Mi, Y.-Y.; Yu, Q.-Q.; Yu, M.-L.; Xu, B.; Zhang, L.-F.; Cheng, W.; Zhang, W.; Hua, L.-X.; Feng, N.-H. Review and pooled analysis of studies on -607(C/A) and -137(G/C) polymorphisms in IL-18 and cancer risk. Med. Oncol. 2011, 28, 1107–1115. [Google Scholar] [CrossRef]
- Yang, X.; Qiu, M.-T.; Hu, J.-W.; Jiang, F.; Li, M.; Wang, J.; Zhang, Q.; Yin, R.; Xu, L. Association of Interleukin-18 Gene Promoter 2607 C.A and 2137G.C Polymorphisms with Cancer Risk: A Meta-Analysis of 26 Studies. PLoS ONE 2013, 8, e72671. [Google Scholar]
- Wang, M.; Zhu, X.-Y.; Wang, L.; Lin, Y. The-607C/A Polymorphisms in Interleukin-18 Gene Promoter Contributes to Cancer Risk: Evidence from A Meta-Analysis of 22 Case-Control Studies. PLos ONE 2013, 8, e76915. [Google Scholar] [CrossRef]
- Liang, T.J.; Ma, H.; Wang, C.X.; Liu, Y.R.; Wang, X.G. The-137G>C polymorphism in interleukin-18 promoter region and cancer risk: Evidence from a meta-analysis of 21 studies. Tumour Biol. 2013, 34, 3483–3490. [Google Scholar] [CrossRef]
- Guo, X.-Y.; Xia, Y. The Interleukin-18 Promoter-607C>A Polymorphism Contributes to Nasopharyngeal Carcinoma Risk: Evidence from a Meta-analysis Including 1,886 Subjects. Asian Pac. J. Cancer Prev. 2013, 14, 7577–7581. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gao, Z.M.; Huang, H.B.; Sun, L.S.; Sun, A.Q.; Li, K. Association of IL-8 gene promoter -251 A/T and IL-18 gene promoter -137 G/C polymorphisms with head and neck cancer risk: A comprehensive meta-analysis. Cancer Manag. Res. 2018, 10, 2589–2604. [Google Scholar] [CrossRef]
- Zhu, S.-L.; Zhao, Y.; Hu, X.-Y.; Luo, T.; Chen, Z.-S.; Zhang, Y.; Yang, S.-H.; Zhou, L.; Li, L.-Q. Genetic polymorphisms-137 (rs187238) and -607 (rs1946518) in the interleukin-18 promoter may not be associated with development of hepatocellular carcinoma. Sci. Rep. 2016, 6, 39404. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Xu, J.; Bao, X.; Niu, W.; Wang, L.; Du, L.; Zhang, N.; Sun, Y. Association between Genetic Polymorphisms in Interleukin Genes and Recurrent Pregnancy Loss ± A Systematic Review and Meta-Analysis. PLos ONE 2017, 12, e0169891. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Yang, X.; Du, J.; Lu, M. Interleukin-18 gene polymorphisms and risk of recurrent pregnancy loss: A systematic review and meta-analysis. J. Obstet. Gynecol. Res. 2015, 41, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, K.; Yang, Z. Associations between TNF-alpha and interleukin gene polymorphisms with polycystic ovary syndrome risk: A systematic review and meta-analysis. J. Assist. Reprod. Genet. 2015, 32, 625–634. [Google Scholar] [CrossRef] [PubMed]





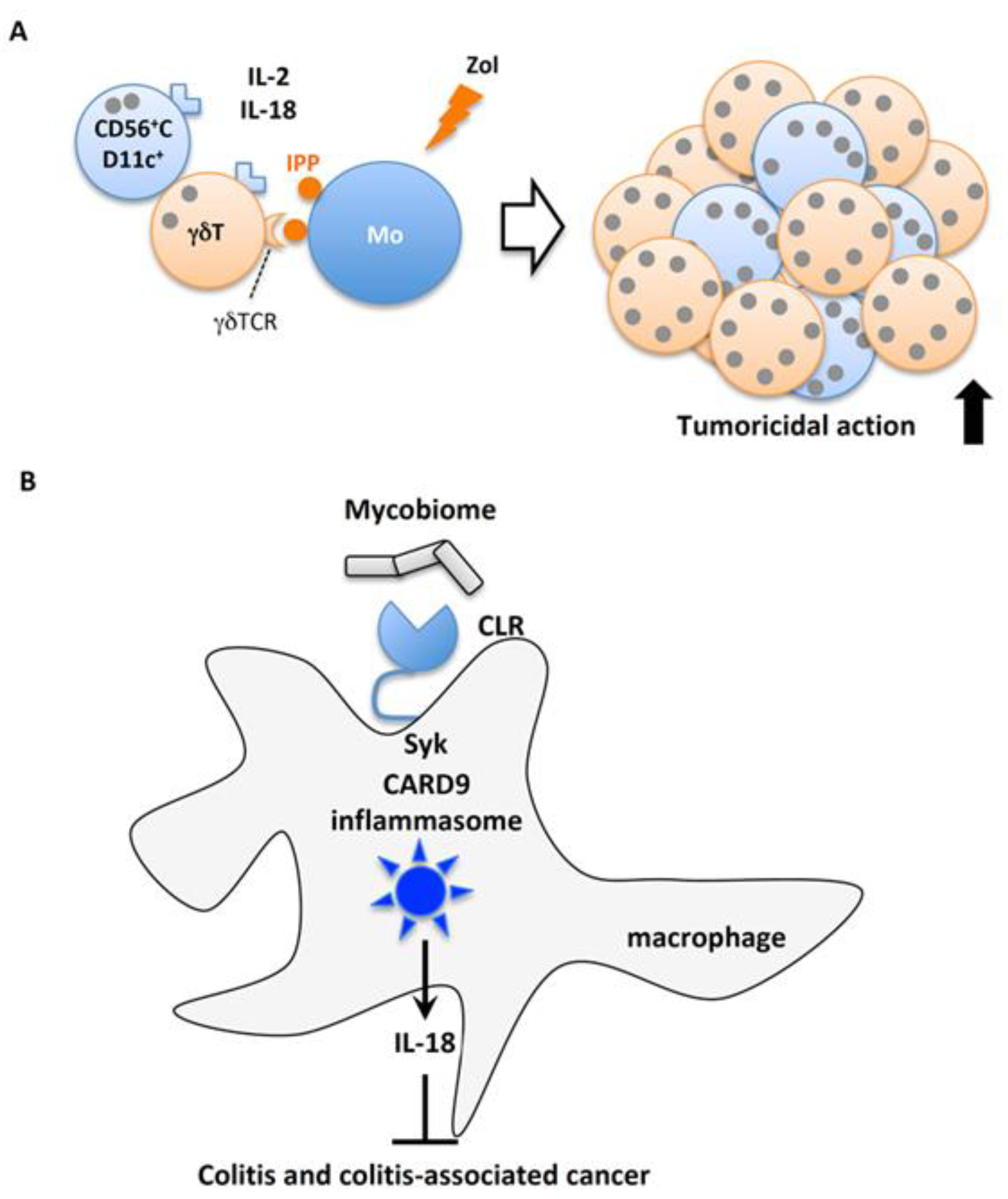

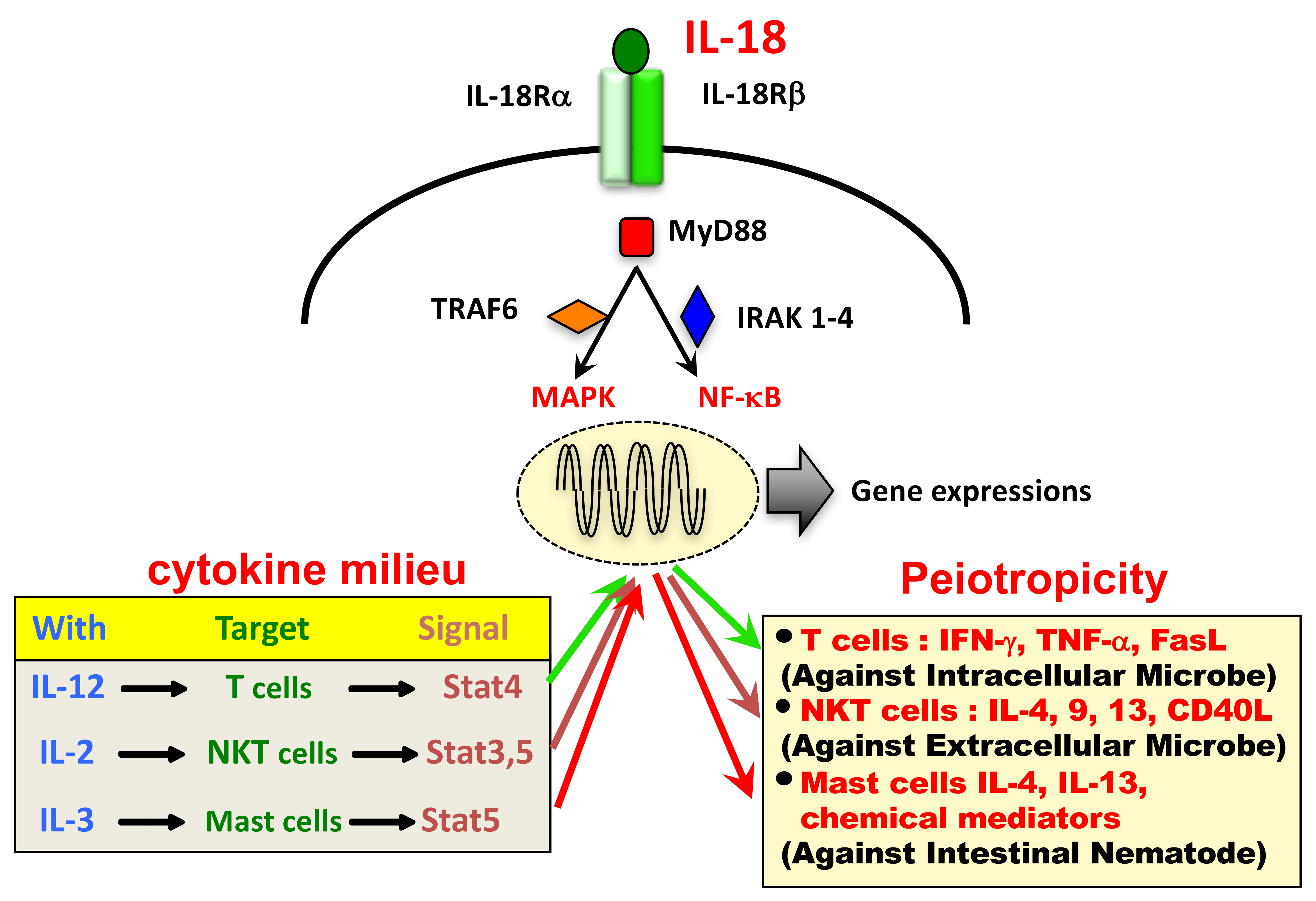{kind=link}
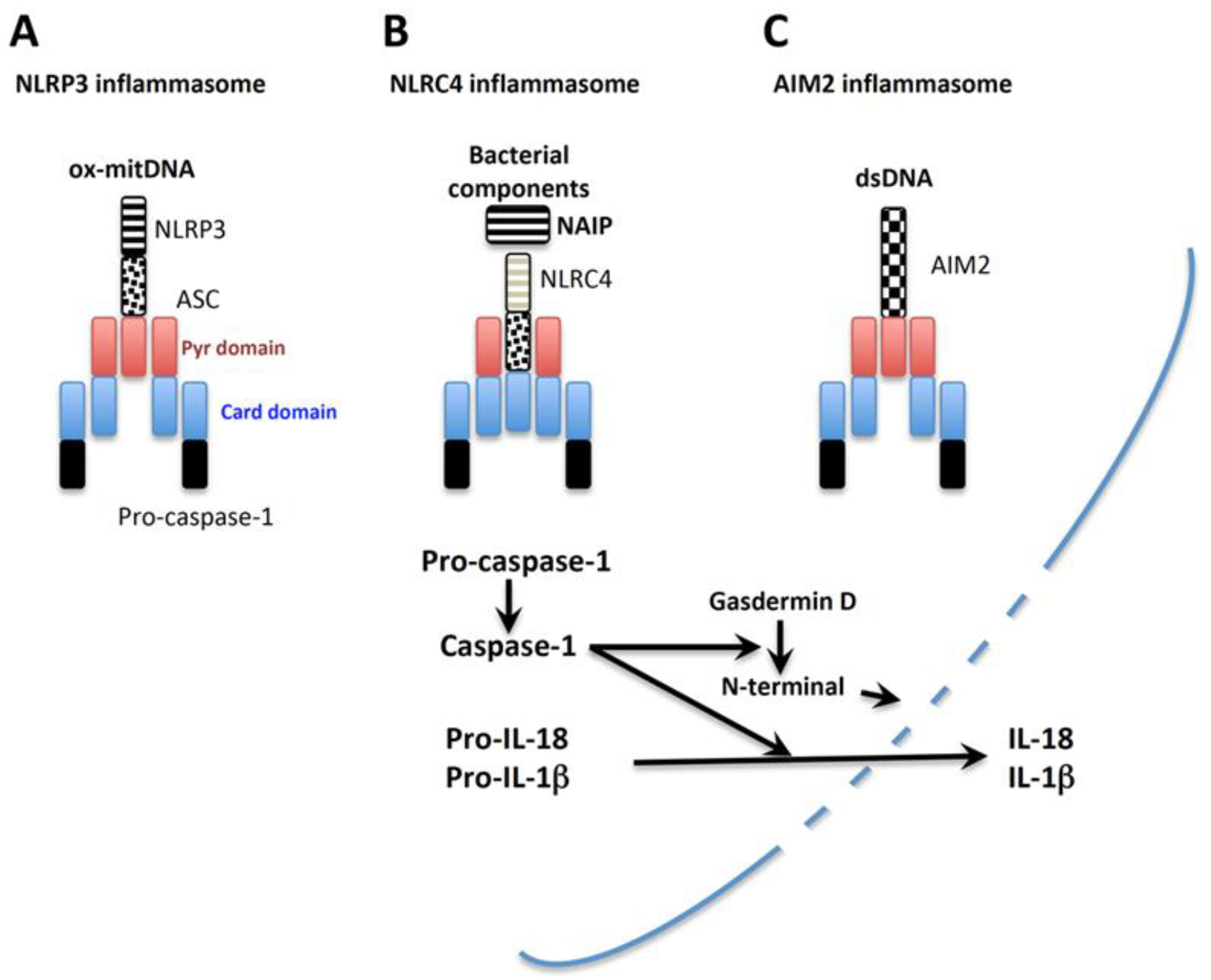{kind=link}
{kind=link}
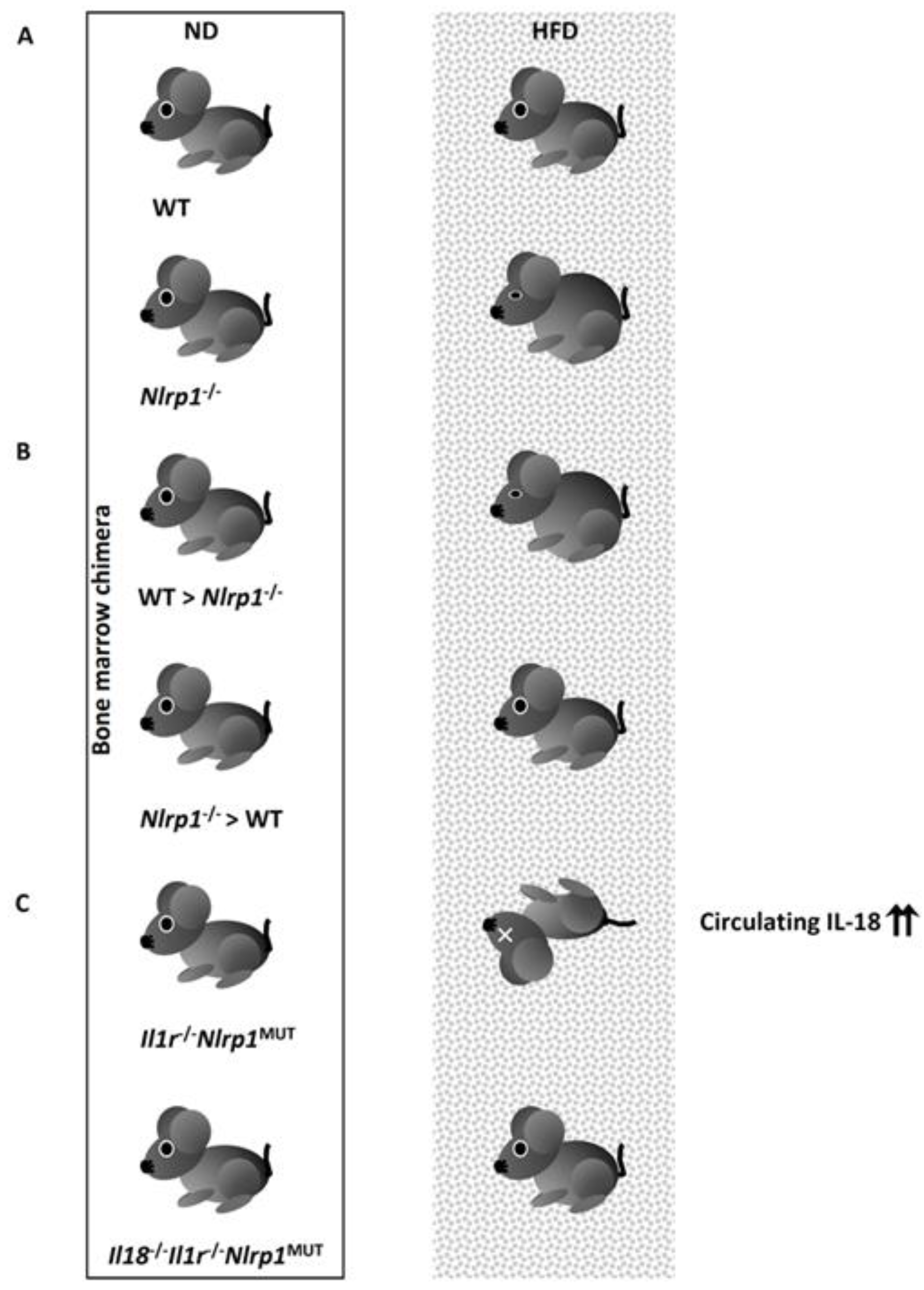{kind=link}
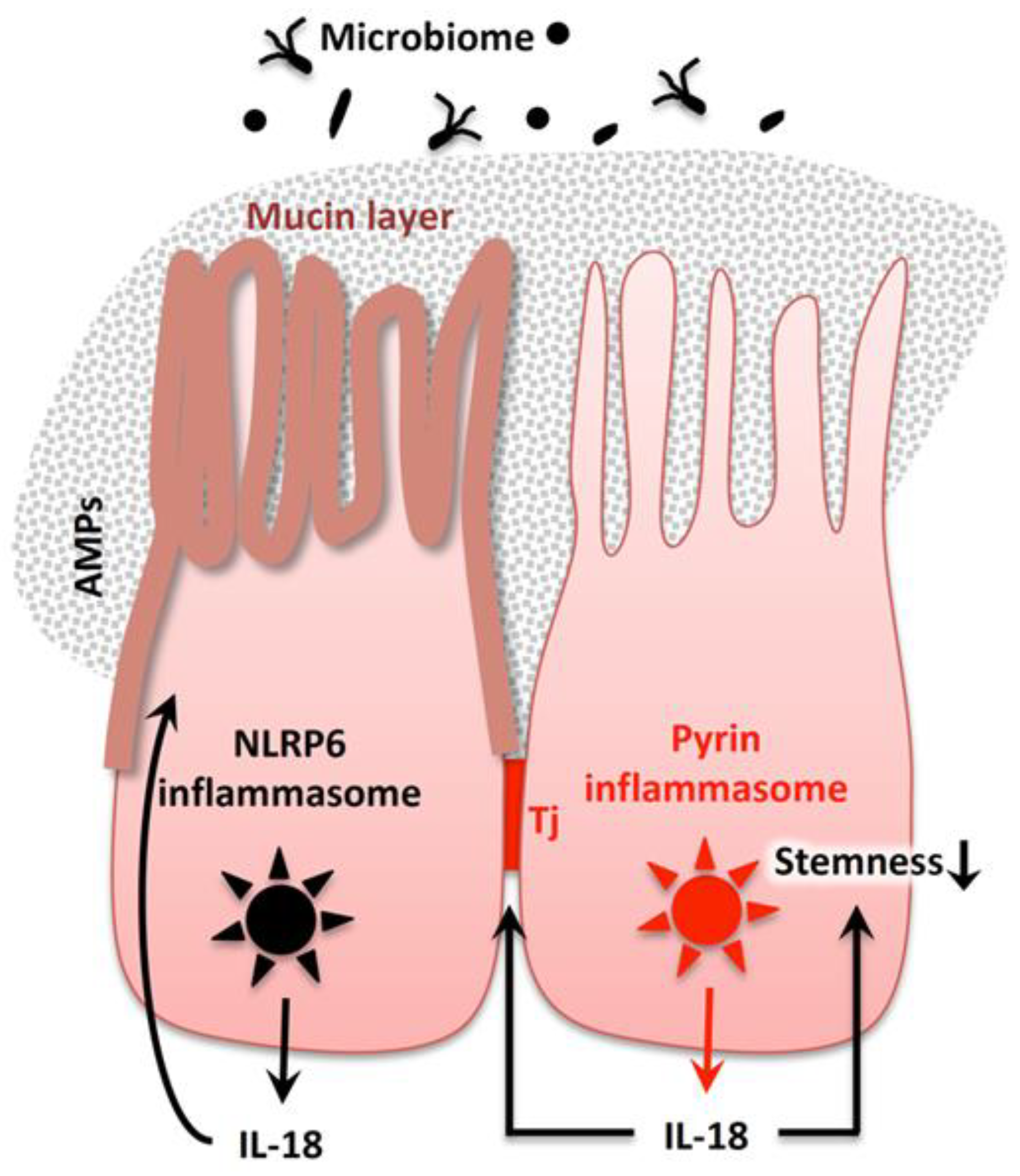{kind=link}
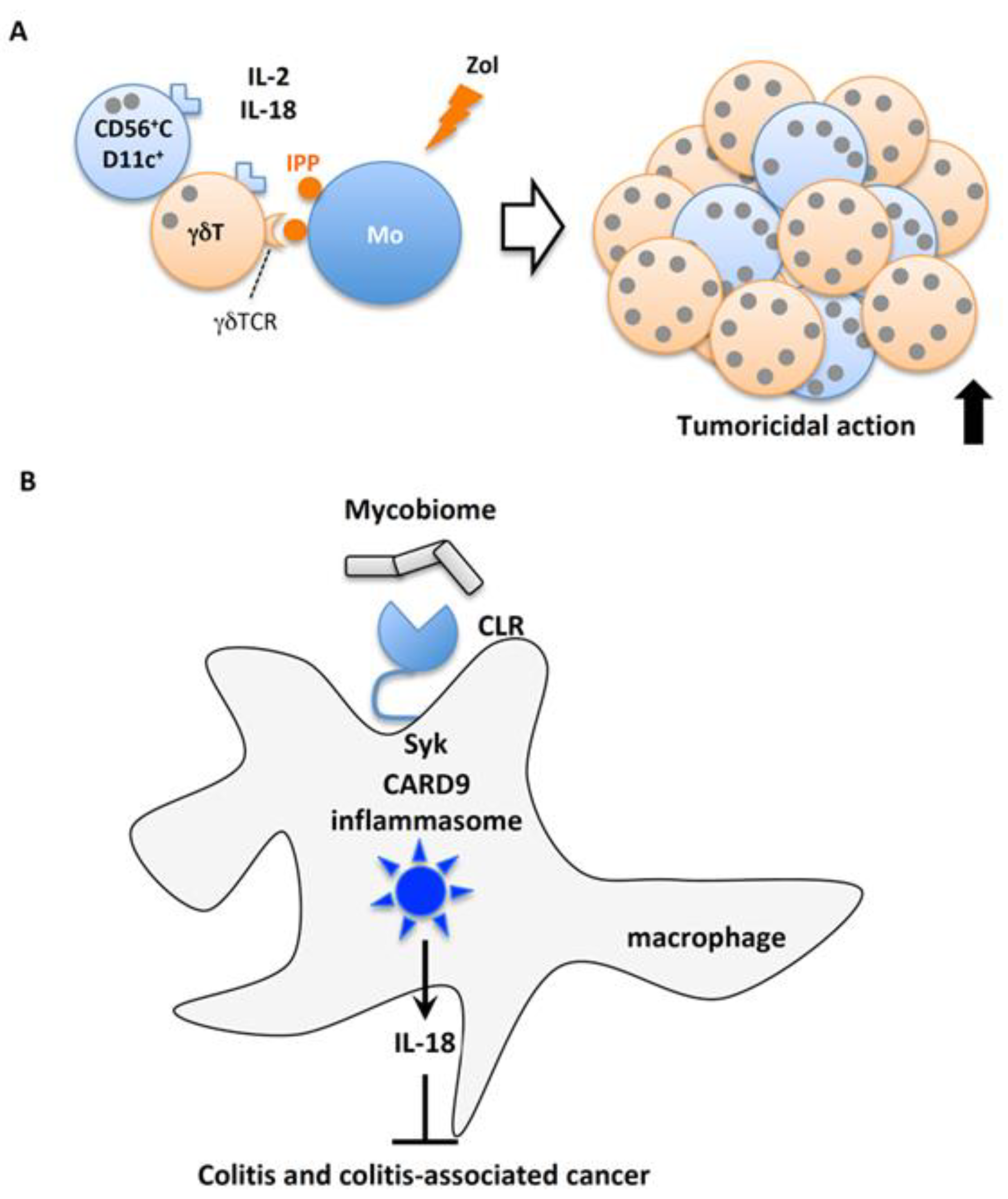{kind=link}
| Disease Association | SNP Polymorphism | Ref | |||||
|---|---|---|---|---|---|---|---|
| Promoters | 5’-UTR | ||||||
| −1297 C/T | −656 G/T | −607 C/A | −137 G/C | +113 T/T | |||
| rs360719 | rs1946519 | rs1946518 | rs187238 | rs360718 | |||
| Chronic viral infection | HBV | ND | ND | + | + | ND | [373] |
| HCV | ND | ND | − | + | ND | [374] | |
| Periodontitis | ND | ND | + | + | ND | [375] | |
| ND | ND | − | − | ND | [376] | ||
| Autoimmune diseases | SLE | ND | ND | − | ND | ND | [377] |
| +a | ND | +a | +b | ND | [378] | ||
| ND | ND | +c | − | ND | [379] | ||
| + | ND | +a | − | ND | [380] | ||
| Behcet’s disease | ND | ND | + | ND | ND | [381] | |
| RA | ND | ND | − | ND | ND | [377] | |
| ND | ND | +c | − | ND | [379] | ||
| ND | ND | +b | − | ND | [382] | ||
| T1D | ND | ND | − | ND | ND | [377] | |
| ND | ND | + | − | ND | [383] | ||
| ND | ND | +b | − | ND | [384] | ||
| CD | ND | − | + | + | + | [385] | |
| ND | ND | − | ND | ND | [377] | ||
| UC | ND | ND | − | ND | ND | [377] | |
| Ischemic stroke | ND | ND | + | − | ND | [386] | |
| Cancer | Total cancer | ND | ND | + | − | ND | [387] |
| ND | ND | − | +b | ND | [388] | ||
| ND | ND | +b | ND | ND | [389] | ||
| ND | ND | ND | +b | ND | [390] | ||
| Nasopharyngeal cancer | ND | ND | + | + | ND | [388] | |
| ND | ND | ND | + | ND | [387] | ||
| ND | ND | ND | + | ND | [390] | ||
| ND | ND | + | ND | ND | [389] | ||
| ND | ND | + | ND | ND | [391] | ||
| Esophageal cancer | ND | ND | + | ND | ND | [389] | |
| Gastric cancer | ND | ND | + | ND | ND | [388] | |
| Head and neck cancer | ND | ND | ND | + | ND | [392] | |
| Hepatocellular carcinoma | ND | ND | − | − | ND | [393] | |
| Recurrent pregnancy loss | ND | ND | + | + | ND | [394] | |
| ND | ND | − | + | ND | [395] | ||
| Polycystic ovary syndrome | ND | ND | − | − | ND | [396] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasuda, K.; Nakanishi, K.; Tsutsui, H. Interleukin-18 in Health and Disease. Int. J. Mol. Sci. 2019, 20, 649. https://doi.org/10.3390/ijms20030649
Yasuda K, Nakanishi K, Tsutsui H. Interleukin-18 in Health and Disease. International Journal of Molecular Sciences. 2019; 20(3):649. https://doi.org/10.3390/ijms20030649
Chicago/Turabian StyleYasuda, Koubun, Kenji Nakanishi, and Hiroko Tsutsui. 2019. "Interleukin-18 in Health and Disease" International Journal of Molecular Sciences 20, no. 3: 649. https://doi.org/10.3390/ijms20030649
APA StyleYasuda, K., Nakanishi, K., & Tsutsui, H. (2019). Interleukin-18 in Health and Disease. International Journal of Molecular Sciences, 20(3), 649. https://doi.org/10.3390/ijms20030649