Molecular Research in Chronic Thromboembolic Pulmonary Hypertension


Abstract
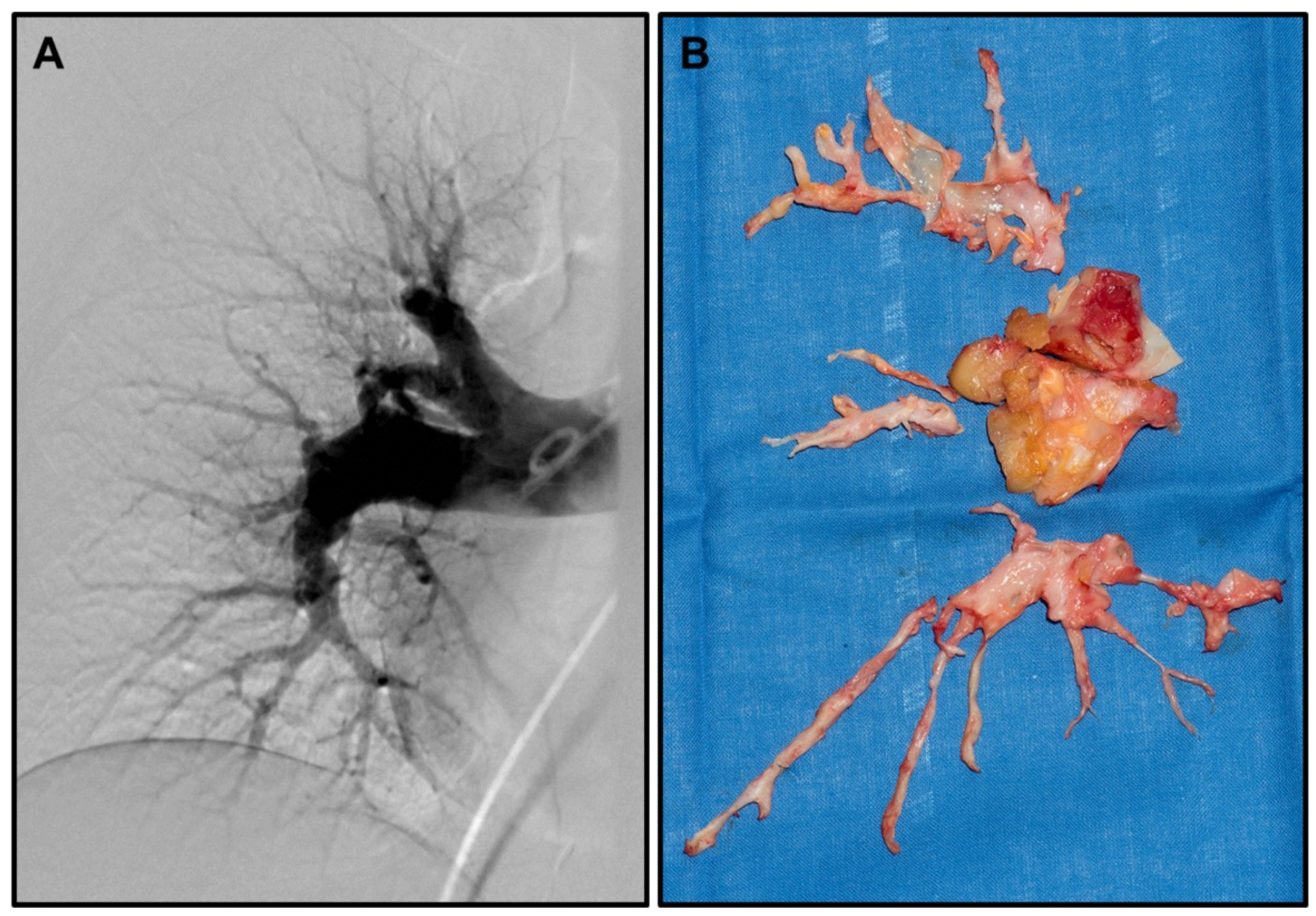
1. Introduction
2. Polymorphisms
3. Mutations
4. Gene Expression
5. MicroRNAs
6. DNA Methylation
7. Transcription Factor FoxO1
8. Summary and Outlook

{kind=link}
| Genetic Alteration | Association with CTEPH | Possible Effect of Alteration | Ref. |
|---|---|---|---|
| Polymorphisms | |||
| Aα Thr312Ala | Frequency ↑ | Resistance to fibrinolysis | [21,24,25,26,27] |
| rs3739817 (ENG) | Frequency ↑ | Unknown | [29] |
| rs55805125 (MAPK10) | Frequency ↑ | MAPK signalling | [29] |
| Mutations | |||
| BMPR2 | Frequency ↑ | Disrupted TGF-β signalling, induction of PASMC proliferation | [29] |
| ACVRL1 | Frequency ↑ | Disrupted TGF-β signalling | [29] |
| SMAD9 | Frequency ↑ | Disrupted TGF-β signalling | [29] |
| CAV1 | Frequency ↑ | Disrupted TGF-β and nitric oxide signalling | [29] |
| KCNK3 | Frequency ↑ | Effect on resting potential of PASMCs | [29] |
| Gene Expression | |||
| Various genes | ↑ and ↓ | Enrichment of dysregulation in cell proliferation, signal transduction, cytokine-related and cancer-related pathways | [50] |
| microRNAs | |||
| miR-759 | Unknown | Degradation of fibrinogen | [65] |
| Let-7d | ↓ | Inhibition of PASMC proliferation | [66] |
| miR-942-5p– circ0002062 axis | ↓ in plasma | Dysregulation of CDK6 signalling | [67] |
| miR-940– circ0022342 axis | ↓ in plasma | Dysregulation of Erb signalling | [67] |
| Let-7b | ↓ in plasma | Regulation of TGBFR1 &ET-1 expression + PAEC and PASMC migration | [69] |
| miR-22 | ↓ in plasma | Unknown | [69] |
| DNA Methylation | |||
| PIC3CA | ↓ | Dysregulation of cancer -related pathways | [73] |
| HFA | ↓ | Dysregulation and actin cytoskeleton regulation of cancer-related pathways | [73] |
| HIC1 | ↑ | Dysregulation and actin cytoskeleton regulation of cancer-related pathways | [73] |
| Transcription Factors | |||
| FoxO1 | ↓ | Modulation of apoptosis in PAECs | [75] |
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart. J. 2016, 37, 67–119. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, M.; Kerr, K.; Fedullo, P. Chronic Thromboembolic Pulmonary Hypertension. Epidemiology and Risk Factors. Ann. Am. Thorac. Soc. 2016, 13, S201–S206. [Google Scholar] [CrossRef] [PubMed]
- Guerin, L.; Couturaud, F.; Parent, F.; Revel, M.P.; Gillaizeau, F.; Planquette, B.; Pontal, D.; Guegan, M.; Simonneau, G.; Meyer, G.; et al. Prevalence of chronic thromboembolic pulmonary hypertension after acute pulmonary embolism. Prevalence of CTEPH after pulmonary embolism. Thromb. Haemost. 2014, 112, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Lang, I. Chronic thromboembolic pulmonary hypertension: A distinct disease entity. Eur. Respir. Rev. 2015, 24, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, S.; Ando, M.; Fukumoto, Y.; Higuchi, Y.; Kaneko, K.; Maeda, K.; Shimokawa, H. Histopathological examination by lung biopsy for the evaluation of operability and postoperative prognosis in patients with chronic thromboembolic pulmonary hypertension. Circ. J. 2014, 78, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Delcroix, M.; Lang, I.; Pepke-Zaba, J.; Jansa, P.; D’Armini, A.M.; Snijder, R.; Bresser, P.; Torbicki, A.; Mellemkjaer, S.; Lewczuk, J.; et al. Long-Term Outcome of Patients With Chronic Thromboembolic Pulmonary Hypertension: Results From an International Prospective Registry. Circulation 2016, 133, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.; Madani, M.; Fadel, E.; D’Armini, A.M.; Mayer, E. Pulmonary endarterectomy in the management of chronic thromboembolic pulmonary hypertension. Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Opitz, I.; Ulrich, S. Chronic thromboembolic pulmonary hypertension. Swiss Med. Wkly. 2018, 148, w14702. [Google Scholar] [CrossRef]
- Mahmud, E.; Behnamfar, O.; Ang, L.; Patel, M.P.; Poch, D.; Kim, N.H. Balloon pulmonary angioplasty for chronic thromboembolic pulmonary hypertension. Interv. Cardiol. Clin. 2018, 7, 103–117. [Google Scholar]
- Pepke-Zaba, J.; Ghofrani, H.A.; Hoeper, M.M. Medical management of chronic thromboembolic pulmonary hypertension. Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; D’Armini, A.M.; Ghofrani, H.A.; Grimminger, F.; Hoeper, M.M.; Jansa, P.; Kim, N.H.; Wang, C.; Wilkins, M.R.; Fritsch, A.; et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension: A long-term extension study (CHEST-2). Eur. Respir. J. 2015, 45, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Bochenek, M.L.; Rosinus, N.S.; Lankeit, M.; Hobohm, L.; Bremmer, F.; Schutz, E.; Klok, F.A.; Horke, S.; Wiedenroth, C.B.; Munzel, T.; et al. From thrombosis to fibrosis in chronic thromboembolic pulmonary hypertension. Thromb. Haemost. 2017, 117, 769–783. [Google Scholar] [CrossRef] [PubMed]
- Lang, I.M.; Dorfmuller, P.; Vonk Noordegraaf, A. The Pathobiology of Chronic Thromboembolic Pulmonary Hypertension. Ann. Am. Thorac. Soc. 2016, 13 (Suppl. 3), S215–S221. [Google Scholar] [CrossRef]
- Sharma, S.; Mathew, A.B.; Chugh, J. miRNAs: Nanomachines That Micromanage the Pathophysiology of Diabetes Mellitus. Adv. Clin. Chem. 2017, 82, 199–264. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Torbicki, A.; Dorfmuller, P.; Kim, N. The pathophysiology of chronic thromboembolic pulmonary hypertension. Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M.; Catto, A.J.; Kohler, H.P.; Ariens, R.A.; Stickland, M.H.; Grant, P.J. alpha-fibrinogen Thr312Ala polymorphism and venous thromboembolism. Blood 2000, 96, 1177–1179. [Google Scholar] [PubMed]
- Francis, C.W. Plasminogen activator inhibitor-1 levels and polymorphisms. Arch. Pathol. Lab. Med. 2002, 126, 1401–1404. [Google Scholar] [CrossRef]
- Francis, C.W. Factor XIII polymorphisms and venous thromboembolism. Arch. Pathol. Lab. Med. 2002, 126, 1391–1393. [Google Scholar] [CrossRef]
- Mansilha, A.; Araujo, F.; Severo, M.; Sampaio, S.M.; Toledo, T.; Albuquerque, R. Genetic polymorphisms and risk of recurrent deep venous thrombosis in young people: Prospective cohort study. Eur. J. Vasc. Endovasc. Surg. 2005, 30, 545–549. [Google Scholar] [CrossRef]
- Carter, A.M.; Catto, A.J.; Grant, P.J. Association of the alpha-fibrinogen Thr312Ala polymorphism with poststroke mortality in subjects with atrial fibrillation. Circulation 1999, 99, 2423–2426. [Google Scholar] [CrossRef]
- Suntharalingam, J.; Goldsmith, K.; van Marion, V.; Long, L.; Treacy, C.M.; Dudbridge, F.; Toshner, M.R.; Pepke-Zaba, J.; Eikenboom, J.C.; Morrell, N.W. Fibrinogen Aalpha Thr312Ala polymorphism is associated with chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2008, 31, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Standeven, K.F.; Grant, P.J.; Carter, A.M.; Scheiner, T.; Weisel, J.W.; Ariens, R.A. Functional analysis of the fibrinogen Aalpha Thr312Ala polymorphism: Effects on fibrin structure and function. Circulation 2003, 107, 2326–2330. [Google Scholar] [CrossRef] [PubMed]
- Curran, J.M.; Fatah-Ardalani, K.; Tornvall, P.; Humphries, S.E.; Green, F.R. A hypothesis to explain the reported association of the alpha-fibrinogen A312 allele with thromboembolic disease. Thromb. Haemost. 2001, 85, 1122–1123. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.A.; Marsh, J.J.; Chiles, P.G.; Magana, M.M.; Liang, N.C.; Soler, X.; Desantis, D.J.; Ngo, D.; Woods, V.L., Jr. High prevalence of dysfibrinogenemia among patients with chronic thromboembolic pulmonary hypertension. Blood 2009, 114, 1929–1936. [Google Scholar] [CrossRef] [PubMed]
- Hennigs, J.K.; Baumann, H.J.; Luneburg, N.; Quast, G.; Harbaum, L.; Heyckendorf, J.; Sydow, K.; Schulte-Hubbert, B.; Halank, M.; Klose, H. Fibrinogen plasma concentration is an independent marker of haemodynamic impairment in chronic thromboembolic pulmonary hypertension. Sci. Rep. 2014, 4, 4808. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Sogawa, K.; Umemura, H.; Sakao, S.; Kasahara, Y.; Tanabe, N.; Kodera, Y.; Takiguchi, Y.; Tatsumi, K.; Nomura, F. Serum level of fibrinogen Aalpha chain fragment increases in chronic thromboembolic pulmonary hypertension. Circ. J. 2011, 75, 2675–2682. [Google Scholar] [CrossRef] [PubMed]
- Li, J.F.; Lin, Y.; Yang, Y.H.; Gan, H.L.; Liang, Y.; Liu, J.; Yang, S.Q.; Zhang, W.J.; Cui, N.; Zhao, L.; et al. Fibrinogen Aalpha Thr312Ala polymorphism specifically contributes to chronic thromboembolic pulmonary hypertension by increasing fibrin resistance. PLoS ONE 2013, 8, e69635. [Google Scholar] [CrossRef]
- Ulrich, S.; Szamalek-Hoegel, J.; Hersberger, M.; Fischler, M.; Garcia, J.S.; Huber, L.C.; Grunig, E.; Janssen, B.; Speich, R. Sequence variants in BMPR2 and genes involved in the serotonin and nitric oxide pathways in idiopathic pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension: Relation to clinical parameters and comparison with left heart disease. Respiration 2010, 79, 279–287. [Google Scholar] [CrossRef]
- Xi, Q.; Liu, Z.; Zhao, Z.; Luo, Q.; Huang, Z. High Frequency of Pulmonary Hypertension-Causing Gene Mutation in Chinese Patients with Chronic Thromboembolic Pulmonary Hypertension. PLoS ONE 2016, 11, e0147396. [Google Scholar] [CrossRef]
- Tuder, R.M.; Archer, S.L.; Dorfmuller, P.; Erzurum, S.C.; Guignabert, C.; Michelakis, E.; Rabinovitch, M.; Schermuly, R.; Stenmark, K.R.; Morrell, N.W. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D4–D12. [Google Scholar] [CrossRef]
- Soubrier, F.; Chung, W.K.; Machado, R.; Grunig, E.; Aldred, M.; Geraci, M.; Loyd, J.E.; Elliott, C.G.; Trembath, R.C.; Newman, J.H.; et al. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2013, 62, D13–D21. [Google Scholar] [CrossRef] [PubMed]
- International, P.P.H.C.; Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Koehler, R.; Grunig, E.; Pauciulo, M.W.; Hoeper, M.M.; Olschewski, H.; Wilkens, H.; Halank, M.; Winkler, J.; Ewert, R.; Bremer, H.; et al. Low frequency of BMPR2 mutations in a German cohort of patients with sporadic idiopathic pulmonary arterial hypertension. J. Med. Genet. 2004, 41, e127. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.R.; Machado, R.D.; Pauciulo, M.W.; Morgan, N.V.; Humbert, M.; Elliott, G.C.; Ward, K.; Yacoub, M.; Mikhail, G.; Rogers, P.; et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J. Med. Genet. 2000, 37, 741–745. [Google Scholar] [CrossRef] [PubMed]
- Gilboa, L.; Nohe, A.; Geissendorfer, T.; Sebald, W.; Henis, Y.I.; Knaus, P. Bone morphogenetic protein receptor complexes on the surface of live cells: A new oligomerization mode for serine/threonine kinase receptors. Mol. Biol. Cell 2000, 11, 1023–1035. [Google Scholar] [CrossRef] [PubMed]
- Grijelmo, C.; Rodrigue, C.; Svrcek, M.; Bruyneel, E.; Hendrix, A.; de Wever, O.; Gespach, C. Proinvasive activity of BMP-7 through SMAD4/src-independent and ERK/Rac/JNK-dependent signaling pathways in colon cancer cells. Cell. Signal. 2007, 19, 1722–1732. [Google Scholar] [CrossRef] [PubMed]
- Nohe, A.; Hassel, S.; Ehrlich, M.; Neubauer, F.; Sebald, W.; Henis, Y.I.; Knaus, P. The mode of bone morphogenetic protein (BMP) receptor oligomerization determines different BMP-2 signaling pathways. J Biol. Chem. 2002, 277, 5330–5338. [Google Scholar] [CrossRef]
- Rudarakanchana, N.; Flanagan, J.A.; Chen, H.; Upton, P.D.; Machado, R.; Patel, D.; Trembath, R.C.; Morrell, N.W. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum. Mol. Genet. 2002, 11, 1517–1525. [Google Scholar] [CrossRef]
- Tian, Q.; He, X.C.; Hood, L.; Li, L. Bridging the BMP and Wnt pathways by PI3 kinase/Akt and 14-3-3zeta. Cell Cycle 2005, 4, 215–216. [Google Scholar] [CrossRef]
- Yamanaka, R.; Otsuka, F.; Nakamura, K.; Yamashita, M.; Otani, H.; Takeda, M.; Matsumoto, Y.; Kusano, K.F.; Ito, H.; Makino, H. Involvement of the bone morphogenetic protein system in endothelin- and aldosterone-induced cell proliferation of pulmonary arterial smooth muscle cells isolated from human patients with pulmonary arterial hypertension. Hypertens. Res. 2010, 33, 435–445. [Google Scholar] [CrossRef]
- Jones, G.; Robertson, L.; Harrison, R.; Ridout, C.; Vasudevan, P. Somatic mosaicism in ACVRL1 with transmission to several offspring affected with severe pulmonary arterial hypertension. Am. J. Med. Genet. A. 2014, 164A, 2121–2123. [Google Scholar] [CrossRef] [PubMed]
- Kraehling, J.R.; Chidlow, J.H.; Rajagopal, C.; Sugiyama, M.G.; Fowler, J.W.; Lee, M.Y.; Zhang, X.; Ramirez, C.M.; Park, E.J.; Tao, B.; et al. Genome-wide RNAi screen reveals ALK1 mediates LDL uptake and transcytosis in endothelial cells. Nat. Commun. 2016, 7, 13516. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Rodriguez, F.; Guerrero-Esteo, M.; Botella, L.M.; Banville, D.; Vary, C.P.; Bernabeu, C. Endoglin regulates cytoskeletal organization through binding to ZRP-1, a member of the Lim family of proteins. J. Biol. Chem. 2004, 279, 32858–32868. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, D.; Ihida-Stansbury, K.; Jones, P.L.; Martin, J.F. Defective pulmonary vascular remodeling in Smad8 mutant mice. Hum. Mol. Genet. 2009, 18, 2791–2801. [Google Scholar] [CrossRef]
- Suntharalingam, J.; Machado, R.D.; Sharples, L.D.; Toshner, M.R.; Sheares, K.K.; Hughes, R.J.; Jenkins, D.P.; Trembath, R.C.; Morrell, N.W.; Pepke-Zaba, J. Demographic features, BMPR2 status and outcomes in distal chronic thromboembolic pulmonary hypertension. Thorax 2007, 62, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Aldred, M.A.; James, V.; Harrison, R.E.; Patel, B.; Schwalbe, E.C.; Gruenig, E.; Janssen, B.; Koehler, R.; Seeger, W.; et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum. Mutat. 2006, 27, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.X.; Liu, D.; Sun, M.L.; Jiang, X.; Sun, N.; Mao, Y.M.; Jing, Z.C. BMPR2 germline mutation in chronic thromboembolic pulmonary hypertension. Lung 2014, 192, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Trembath, R.C.; Thomson, J.R.; Machado, R.D.; Morgan, N.V.; Atkinson, C.; Winship, I.; Simonneau, G.; Galie, N.; Loyd, J.E.; Humbert, M.; et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2001, 345, 325–334. [Google Scholar] [CrossRef]
- Gurney, A.M.; Osipenko, O.N.; MacMillan, D.; McFarlane, K.M.; Tate, R.J.; Kempsill, F.E. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ. Res. 2003, 93, 957–964. [Google Scholar] [CrossRef]
- Gu, S.; Su, P.; Yan, J.; Zhang, X.; An, X.; Gao, J.; Xin, R.; Liu, Y. Comparison of gene expression profiles and related pathways in chronic thromboembolic pulmonary hypertension. Int. J. Mol. Med. 2014, 33, 277–300. [Google Scholar] [CrossRef]
- Wei, L.; Liu, Y.; Kaneto, H.; Fanburg, B.L. Jnk regulates serotonin-mediated proliferation and migration of pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L863–L869. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer Ther. 2017, 16, 2351–2363. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Santarpia, L.; Lippman, S.M.; El-Naggar, A.K. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Stratikopoulos, E.E.; Parsons, R.E. Molecular Pathways: Targeting the PI3K Pathway in Cancer-BET Inhibitors to the Rescue. Clin. Cancer Res. 2016, 22, 2605–2610. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Calin, G.A.; Croce, C.M. MicroRNAs in Cancer. Annu. Rev. Med. 2009, 60, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Schulte, C.; Karakas, M.; Zeller, T. microRNAs in cardiovascular disease—clinical application. Clin. Chem. Lab. Med. 2017, 55, 687–704. [Google Scholar] [CrossRef] [PubMed]
- Caruso, P.; MacLean, M.R.; Khanin, R.; McClure, J.; Soon, E.; Southgate, M.; MacDonald, R.A.; Greig, J.A.; Robertson, K.E.; Masson, R.; et al. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Courboulin, A.; Paulin, R.; Giguere, N.J.; Saksouk, N.; Perreault, T.; Meloche, J.; Paquet, E.R.; Biardel, S.; Provencher, S.; Cote, J.; et al. Role for miR-204 in human pulmonary arterial hypertension. J. Exp. Med. 2011, 208, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, J.; Gou, D.; Turaka, P.; Viktorova, E.; Ramchandran, R.; Raj, J.U. MicroRNA-21 plays a role in hypoxia-mediated pulmonary artery smooth muscle cell proliferation and migration. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L861–L871. [Google Scholar] [CrossRef]
- Chen, Z.; Nakajima, T.; Tanabe, N.; Hinohara, K.; Sakao, S.; Kasahara, Y.; Tatsumi, K.; Inoue, Y.; Kimura, A. Susceptibility to chronic thromboembolic pulmonary hypertension may be conferred by miR-759 via its targeted interaction with polymorphic fibrinogen alpha gene. Hum. Genet. 2010, 128, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Guo, L.J.; Liu, J.; Wang, W.; Yuan, J.X.; Zhao, L.; Wang, J.; Wang, C. MicroRNA expression profile of pulmonary artery smooth muscle cells and the effect of let-7d in chronic thromboembolic pulmonary hypertension. Pulm. Circ. 2013, 3, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Miao, R.; Wang, Y.; Wan, J.; Leng, D.; Gong, J.; Li, J.; Liang, Y.; Zhai, Z.; Yang, Y. Microarray expression profile of circular RNAs in chronic thromboembolic pulmonary hypertension. Medicine (Baltimore) 2017, 96, e7354. [Google Scholar] [CrossRef]
- Miao, R.; Wang, Y.; Wan, J.; Leng, D.; Gong, J.; Li, J.; Zhang, Y.; Pang, W.; Zhai, Z.; Yang, Y. Microarray Analysis and Detection of MicroRNAs Associated with Chronic Thromboembolic Pulmonary Hypertension. Biomed. Res. Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Yang, Y.; Liu, J.; Wang, L.; Li, J.; Wang, Y.; Liu, Y.; Gu, S.; Gan, H.; Cai, J.; et al. Differentially expressed plasma microRNAs and the potential regulatory function of Let-7b in chronic thromboembolic pulmonary hypertension. PLoS ONE 2014, 9, e101055. [Google Scholar] [CrossRef]
- Chun, H.J.; Bonnet, S.; Chan, S.Y. Translational advances in the field of pulmonary hypertension. Translating microrna biology in pulmonary hypertension. It will take more than "mir" words. Am. J. Respir. Crit. Care Med. 2017, 195, 167–178. [Google Scholar] [CrossRef]
- Witwer, K.W. Circulating microrna biomarker studies: Pitfalls and potential solutions. Clin. Chem. 2015, 61, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. Microrna therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, X.; Leng, D.; Li, J.; Wang, L.; Liang, Y.; Wang, J.; Miao, R.; Jiang, T. DNA methylation signatures of pulmonary arterial smooth muscle cells in chronic thromboembolic pulmonary hypertension. Physiol. Genomics 2018, 50, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Wu, D.; Yang, M.; Chen, Y.; Wang, C.; Zhong, Z.; Lian, N.; Chen, H.; Wu, S. Expression of tissue factor and forkhead box transcription factor O-1 in a rat model for chronic thromboembolic pulmonary hypertension. J. Thromb. Thrombolysis 2016, 42, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Zhong, Z.; Wu, D.; Chen, Y.; Lian, N.; Ding, H.; Zhang, Q.; Lin, Q.; Wu, S. Role of FoxO1 and apoptosis in pulmonary vascular remolding in a rat model of chronic thromboembolic pulmonary hypertension. Sci. Rep. 2017, 7, 2270. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Provencher, S.; Guignabert, C.; Perros, F.; Boucherat, O.; Schermuly, R.T.; Hassoun, P.M.; Rabinovitch, M.; Nicolls, M.R.; Humbert, M. Translating research into improved patient care in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2017, 195, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Provencher, S.; Archer, S.L.; Ramirez, F.D.; Hibbert, B.; Paulin, R.; Boucherat, O.; Lacasse, Y.; Bonnet, S. Standards and methodological rigor in pulmonary arterial hypertension preclinical and translational research. Circ. Res. 2018, 122, 1021–1032. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Opitz, I.; Kirschner, M.B. Molecular Research in Chronic Thromboembolic Pulmonary Hypertension. Int. J. Mol. Sci. 2019, 20, 784. https://doi.org/10.3390/ijms20030784
Opitz I, Kirschner MB. Molecular Research in Chronic Thromboembolic Pulmonary Hypertension. International Journal of Molecular Sciences. 2019; 20(3):784. https://doi.org/10.3390/ijms20030784
Chicago/Turabian StyleOpitz, Isabelle, and Michaela B. Kirschner. 2019. "Molecular Research in Chronic Thromboembolic Pulmonary Hypertension" International Journal of Molecular Sciences 20, no. 3: 784. https://doi.org/10.3390/ijms20030784
APA StyleOpitz, I., & Kirschner, M. B. (2019). Molecular Research in Chronic Thromboembolic Pulmonary Hypertension. International Journal of Molecular Sciences, 20(3), 784. https://doi.org/10.3390/ijms20030784