Roles of SETD2 in Leukemia—Transcription, DNA-Damage, and Beyond


Abstract
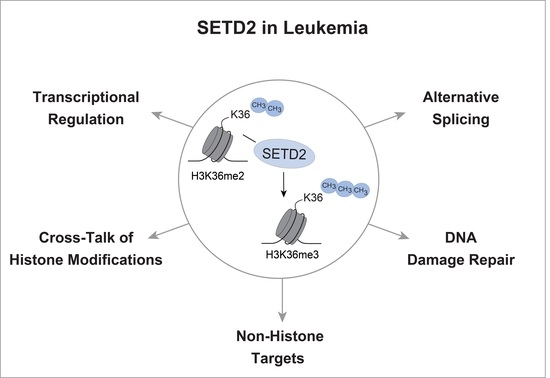
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
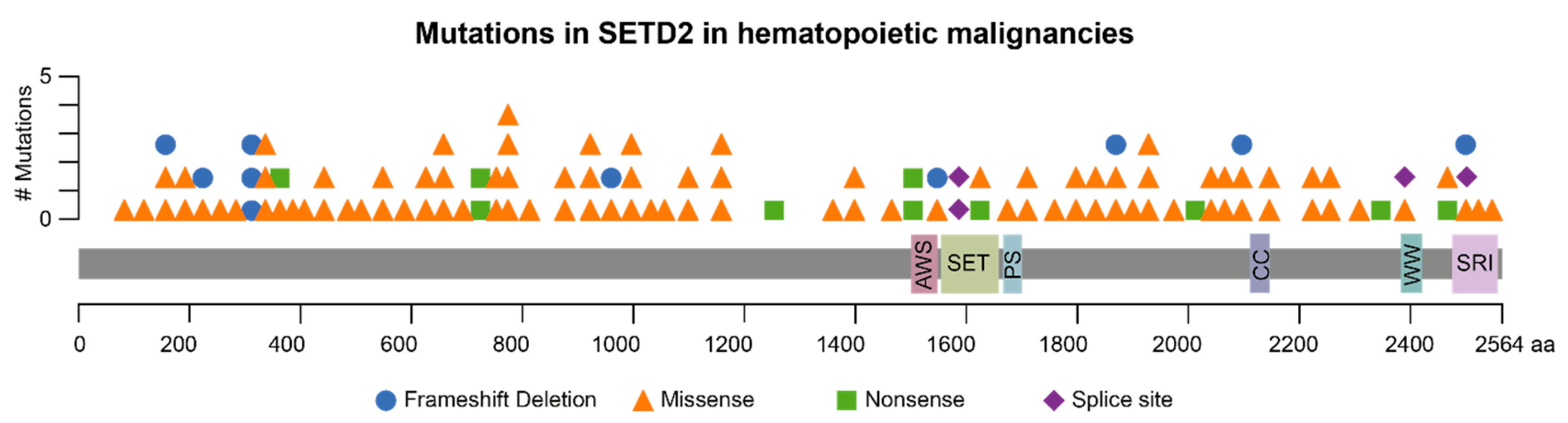
2. SETD2 Mutations in Hematological Malignancies
3. Mechanism of Action of SETD2 in Leukemia and Oncogenesis
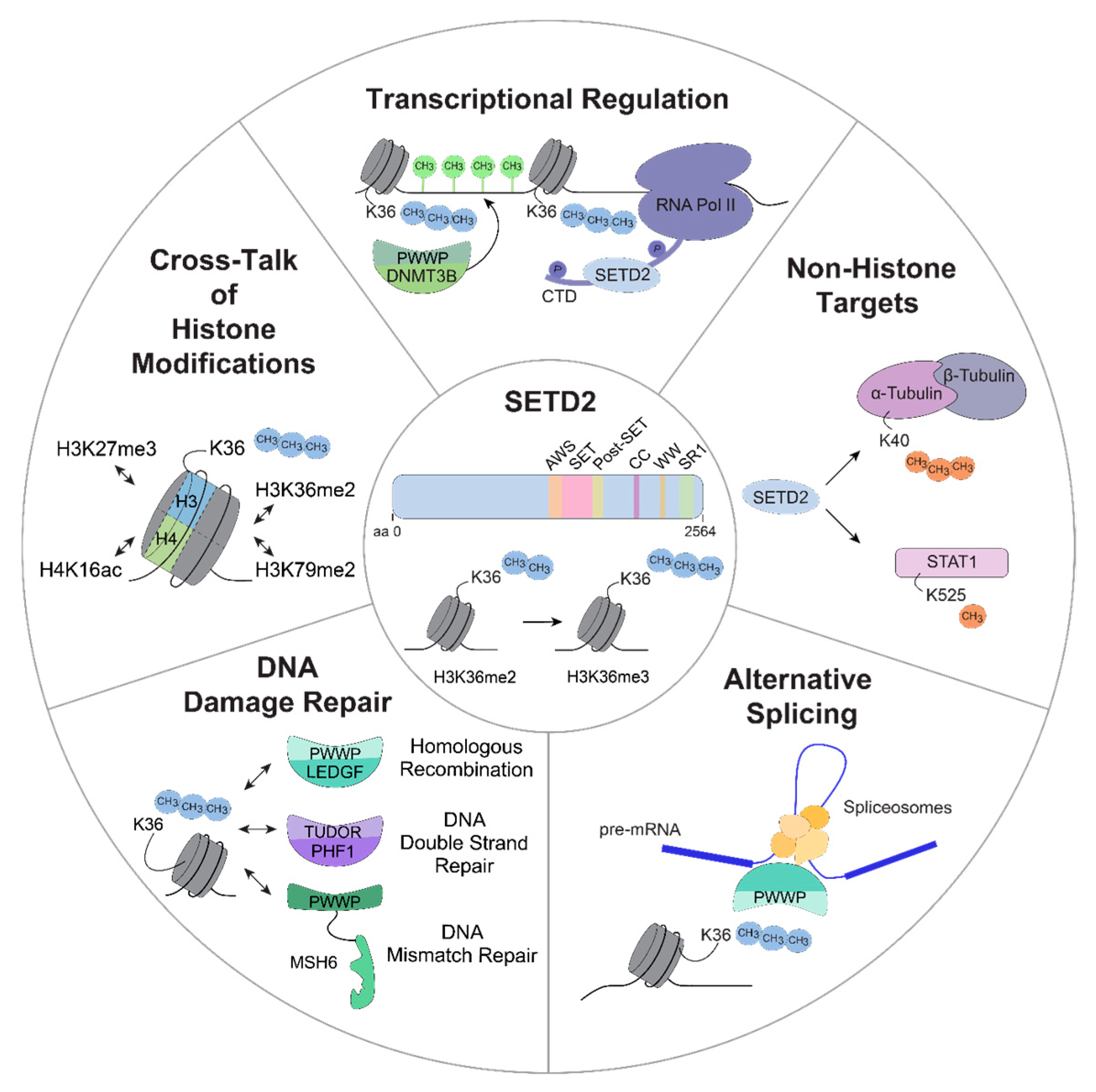
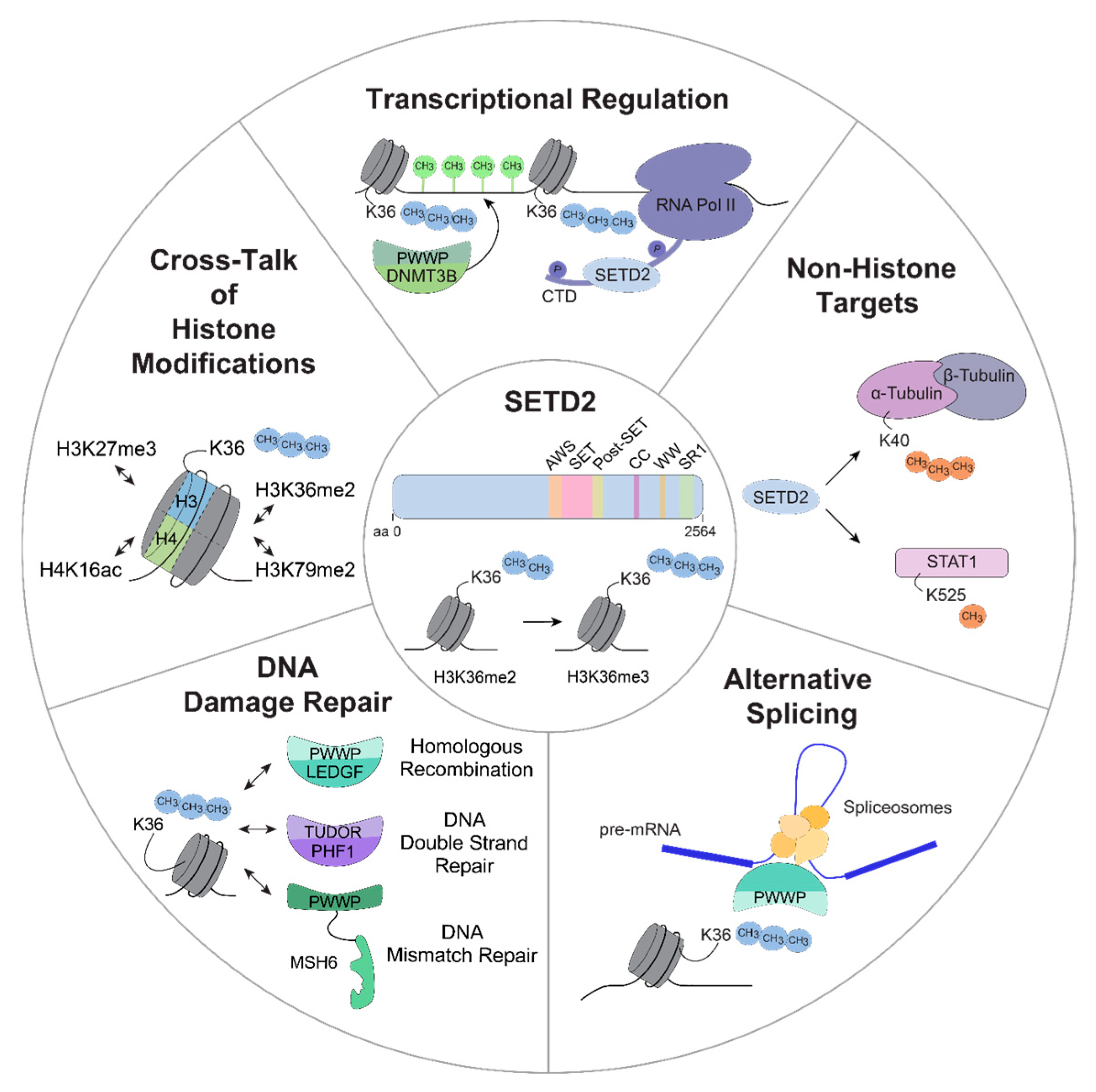
3.1. SETD2-Dependent Regulation of the DNA Damage Response
3.2. Cross-Talk between H3K36me3 and Other Histone Modifications
3.3. SETD2 in Alternative Splicing
3.4. SETD2 in Transcriptional Regulation
3.5. Non-Histone Targets of SETD2
4. Therapeutic Opportunities
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Faber, P.W.; Barnes, G.T.; Srinidhi, J.; Chen, J.; Gusella, J.F.; MacDonald, M.E. Huntingtin interacts with a family of WW domain proteins. Hum. Mol. Genet. 1998, 7, 1463–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, M.; Fu, G.; Wu, J.S.; Zhang, Q.H.; Zhou, J.; Kan, L.X.; Huang, Q.H.; He, K.L.; Gu, B.W.; Han, Z.G.; et al. Identification of genes expressed in human CD34(+) hematopoietic stem/progenitor cells by expressed sequence tags and efficient full-length cDNA cloning. Proc. Natl. Acad. Sci. USA 1998, 95, 8175–8180. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edmunds, J.W.; Mahadevan, L.C.; Clayton, A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2008, 27, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Xie, J.; Long, C.; Erdjument-Bromage, H.; Ding, X.; Zheng, Y.; Tempst, P.; Chen, S.; Zhu, B.; Reinberg, D. Heterogeneous nuclear ribonucleoprotein L is a subunit of human KMT3a/set2 complex required for H3 Lys-36 trimethylation activity in vivo. J. Biol. Chem. 2009, 284, 5701–15707. [Google Scholar] [CrossRef] [PubMed]
- Hacker, K.E.; Fahey, C.C.; Shinsky, S.A.; Chiang, Y.J.; DiFiore, J.V.; Jha, D.K.; Vo, A.H.; Shavit, J.A.; Davis, I.J.; Strahl, B.D.; et al. Structure/function analysis of recurrent mutations in SETD2 protein reveals a critical and conserved role for a SET domain residue in maintaining protein stability and histone H3 Lys-36 trimethylation. J. Biol. Chem. 2016, 291, 21283–21295. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, S.L.; Strahl, B.D. Shaping the cellular landscape with Set2/SETD2 methylation. Cell. Mol. Life Sci. 2017, 74, 3317–3334. [Google Scholar] [CrossRef] [PubMed]
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, C.D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Grant, P.A.; Briggs, S.D.; Sun, Z.W.; Bone, J.R.; Caldwell, J.A.; Mollah, S.; Cook, R.G.; Shabanowitz, J.; Hunt, D.F.; et al. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol. Cell. Biol. 2002, 22, 1298–1306. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Duns, G.; Westers, H.; Sijmons, R.; van den Berg, A.; Kok, K. SETD2: An epigenetic modifier with tumor suppressor functionality. Oncotarget 2016, 7, 50719–50734. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Sun, X.J.; Zhang, Y.L.; Kuang, Y.; Hu, C.Q.; Wu, W.L.; Shen, S.H.; Du, T.T.; Li, H.; He, F.; et al. Histone H3 lysine 36 methyltransferase Hypb/Setd2 is required for embryonic vascular remodeling. Proc. Natl. Acad. Sci. USA 2010, 107, 2956–2961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Yan, X.; Feng, X.; Bu, J.; Dong, Y.; Lin, P.; Hayashi, Y.; Huang, R.; Olsson, A.; Andreassen, P.R.; et al. Setd2 regulates quiescence and differentiation of adult hematopoietic stem cells by restricting RNA polymerase II elongation. Haematologica 2018, 103, 1110–1123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-L.; Sun, J.W.; Xie, Y.Y.; Zhou, Y.; Liu, P.; Song, J.C.; Xu, C.H.; Wang, L.; Liu, D.; Xu, A.N.; et al. Setd2 deficiency impairs hematopoietic stem cell self-renewal and causes malignant transformation. Cell Res. 2018, 28, 476–490. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalgliesh, G.L.; Furge, K.; Greenman, C.; Chen, L.; Bignell, G.; Butler, A.; Davies, H.; Edkins, S.; Hardy, C.; Latimer, C.; et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature 2010, 463, 360–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duns, G.; Hofstra, R.M.; Sietzema, J.G.; Hollema, H.; van Duivenbode, I.; Kuik, A.; Giezen, C.; Jan, O.; Bergsma, J.J.; Bijnen, H.; et al. Targeted exome sequencing in clear cell renal cell carcinoma tumors suggests aberrant chromatin regulation as a crucial step in ccRCC development. Hum. Mutat. 2012, 33, 1059–1062. [Google Scholar] [CrossRef] [PubMed]
- Fontebasso, A.M.; Liu, X.Y.; Sturm, D.; Jabado, N. Chromatin remodeling defects in pediatric and young adult glioblastoma: A tale of a variant histone 3 tail. Brain Pathol. 2013, 23, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Li, N.; Fu, D.; Ren, J.; Hui, J.; Peng, J.; Liu, Y.; Qiu, T.; Jiang, M.; Pan, Q.; et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J. Clin. Investig. 2017, 127, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, G.; Mancini, M.; de Benedittis, C.; Rondoni, M.; Papayannidis, C.; Manfrini, M.; Meggendorfer, M.; Calogero, R.; Guadagnuolo, V.; Fontana, M.C.; et al. SETD2 and histone H3 lysine 36 methylation deficiency in advanced systemic mastocytosis. Leukemia 2018, 32, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, A.B.; Ondrejka, S.L.; McKinney, M.; Rempel, R.E.; Goodlad, J.R.; Teh, C.H.; Leppa, S.; Mannisto, S.; Kovanen, P.E.; Tse, E.; et al. Enteropathy-associated T cell lymphoma subtypes are characterized by loss of function of SETD2. J. Exp. Med. 2017, 214, 1371–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, H.; Strefford, J.C. The mutational signature of chronic lymphocytic leukemia. Biochem J. 2016, 473, 3725–3740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mar, B.G.; Bullinger, L.B.; McLean, K.M.; Grauman, P.V.; Harris, M.H.; Stevenson, K.; Neuberg, D.S.; Sinha, A.U.; Sallan, S.E.; Silverman, L.B.; et al. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat. Commun. 2014, 5, 3469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mar, B.G.; Chu, S.H.; Kahn, J.D.; Krivtsov, A.V.; Koche, R.; Castellano, C.A.; Kotlier, J.L.; Zon, R.L.; McConkey, M.E.; Chabon, J.; et al. SETD2 alterations impair DNA damage recognition and lead to resistance to chemotherapy in leukemia. Blood 2017, 130, 2631–2641. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; He, F.; Zeng, H.; Ling, S.; Chen, A.; Wang, Y.; Yan, X.; Wei, W.; Pang, Y.; Cheng, H.; et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat. Genet. 2014, 46, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef] [PubMed]
- Tzelepis, K.; Koike-Yusa, H.; de Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Post, Y.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [Green Version]
- Shi, J.; Wang, E.; Milazzo, J.P.; Wang, Z.; Kinney, J.B.; Vakoc, C.R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015, 33, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skucha, A.; Ebner, J.; Schmöllerl, J.; Roth, M.; Eder, T.; César-Razquin, A.; Stukalov, A.; Vittori, S.; Muhar, M.; Lu, B.; et al. MLL-fusion-driven leukemia requires SETD2 to safeguard genomic integrity. Nat. Commun. 2018, 9, 1983. [Google Scholar] [CrossRef] [PubMed]
- Musselman, C.A.; Avvakumov, N.; Watanabe, R.; Abraham, C.G.; Lalonde, M.E.; Hong, Z.; Allen, C.; Roy, S.; Nuñez, J.K.; Nickoloff, J.; et al. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat. Struct. Mol. Biol. 2012, 19, 1266–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.M. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, M.; Baude, A.; Fugger, K.; Povlsen, L.K.; Beck, H.; Sørensen, C.S.; Petersen, N.H.; Sorensen, P.H.; Lukas, C.; Bartek, J.; et al. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol. 2012, 19, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.X.; Ahrabi, S.; Zalmas, L.P.; Sarkar, S.; Aymard, F.; Bachrati, C.Z.; Helleday, T.; Legube, G.; La Thangue, N.B.; Porter, A.C.; et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014, 7, 2006–2018. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.; Vítor, A.C.; Sridhara, S.C.; Martins, F.B.; Raposo, A.C.; Desterro, J.M.; Ferreira, J.; de Almeida, S.F. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. elife 2014, 3, e02482. [Google Scholar] [CrossRef] [PubMed]
- Okuda, H.; Kawaguchi, M.; Kanai, A.; Matsui, H.; Kawamura, T.; Inaba, T.; Kitabayashi, I.; Yokoyama, A. MLL fusion proteins link transcriptional coactivators to previously active CpG-rich promoters. Nucleic Acids Res. 2014, 42, 4241–4256. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, Q.; Wong, S.H.; Huang, M.; Klein, B.J.; Shen, J.; Ikenouye, L.; Onishi, M.; Schneidawind, D.; Buechele, C.; et al. ASH1L links histone H3 lysine 36 dimethylation to MLL leukemia. Cancer Discov. 2016, 6, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Bu, J.; Chen, A.; Yan, X.; He, F.; Dong, Y.; Zhou, Y.; He, J.; Zhan, D.; Lin, P.; Hayashi, Y.; et al. SETD2-mediated crosstalk between H3K36me3 and H3K79me2 in MLL-rearranged leukemia. Leukemia 2018, 32, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musselman, C.A.; Lalonde, M.-E.; Côté, J.; Kutateladze, T.G. Perceiving the epigenetic landscape through histone readers. Nat. Struct. Mol. Biol. 2012, 19, 1218–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wang, Y. Cross-talk between the H3K36me3 and H4K16ac histone epigenetic marks in DNA double-strand break repair. J. Biol. Chem. 2017, 292, 11951–11959. [Google Scholar] [CrossRef] [PubMed]
- Kolasinska-Zwierz, P.; Down, T.; Latorre, I.; Liu, T.; Liu, X.S.; Ahringer, J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet. 2009, 41, 376–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Almeida, S.F.; Grosso, A.R.; Koch, F.; Fenouil, R.; Carvalho, S.; Andrade, J.; Levezinho, H.; Gut, M.; Eick, D.; Gut, I.; et al. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat. Struct. Mol. Biol. 2011, 18, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Luco, R.F.; Pan, Q.; Tominaga, K.; Blencowe, B.J.; Pereira-Smith, O.M.; Misteli, T. Regulation of alternative splicing by histone modifications. Science 2010, 327, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Du, J.; Sun, B.; Dong, X.; Xu, G.; Zhou, J.; Huang, Q.; Liu, Q.; Hao, Q.; Ding, J. Structure of human MRG15 chromo domain and its binding to Lys36-methylated histone H3. Nucleic Acids Res. 2006, 34, 6621–6628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Li, Y.; Xi, Y.; Jiang, S.; Stratton, S.; Peng, D.; Tanaka, K.; Ren, Y.; Xia, Z.; Wu, J.; et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 2014, 508, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullah, M.; Pelletier, N.; Xiao, L.; Zhao, S.P.; Wang, K.; Degerny, C.; Tahmasebi, S.; Cayrou, C.; Doyon, Y.; Goh, S.L.; et al. Molecular Architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol. Cell. Biol. 2008, 28, 6828–6843. [Google Scholar] [CrossRef] [PubMed]
- Vezzoli, A.; Bonadies, N.; Allen, M.D.; Freund, S.M.; Santiveri, C.M.; Kvinlaug, B.T.; Huntly, B.J.; Göttgens, B.; Bycroft, M. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat. Struct. Mol. Biol. 2010, 17, 617–619. [Google Scholar] [CrossRef] [PubMed]
- Pradeepa, M.M.; Sutherland, H.G.; Ule, J.; Grimes, G.R.; Bickmore, W.A. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012, 8, e1002717. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Moazed, D.; Gygi, S.P. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J. Biol. Chem. 2002, 277, 49383–49388. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Phatnani, H.P.; Guan, Z.; Sage, H.; Greenleaf, A.L.; Zhou, P. Solution structure of the Set2-Rpb1 interacting domain of human Set2 and its interaction with the hyperphosphorylated C-terminal domain of Rpb1. Proc. Natl. Acad. Sci. USA 2005, 102, 17636–17641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krogan, N.J.; Kim, M.; Tong, A.; Golshani, A.; Cagney, G.; Canadien, V.; Richards, D.P.; Beattie, B.K.; Emili, A.; Boone, C.; et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol. Cell. Biol. 2003, 23, 4207–4218. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S. Structure and mechanism of the RNA polymerase II transcription machinery. Nat. Struct. Mol. Biol. 2004, 11, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, S.; Raposo, A.C.; Martins, F.B.; Grosso, A.R.; Sridhara, S.C.; Rino, J.; Carmo-Fonseca, M.; de Almeida, S.F. Histone methyltransferase SETD2 coordinates FACT recruitment with nucleosome dynamics during transcription. Nucleic Acids Res. 2013, 41, 2881–2893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baubec, T.; Colombo, D.F.; Wirbelauer, C.; Schmidt, J.; Burger, L.; Krebs, A.R.; Akalin, A.; Schübeler, D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015, 520, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 543, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Pfister, S.X.; Markkanen, E.; Jiang, Y.; Sarkar, S.; Woodcock, M.; Orlando, G.; Mavrommati, I.; Pai, C.-C.; Zalmas, L.-P.; Drobnitzky, N.; et al. Inhibiting WEE1 selectively kills histone H3K36me3-deficient cancers by dNTP starvation. Cancer Cell 2015, 28, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Kanu, N.; Grönroos, E.; Martinez, P.; Burrell, R.A.; Yi Goh, X.; Bartkova, J.; Maya-Mendoza, A.; Mistrík, M.; Rowan, A.J.; Patel, H.; et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene 2015, 34, 5699–5708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.Y.; Powell, R.T.; Tripathi, D.N.; Dere, R.; Ho, T.H.; Blasius, T.L.; Chiang, Y.C.; Davis, I.J.; Fahey, C.C.; Hacker, K.E.; et al. Dual chromatin and cytoskeletal remodeling by SETD2. Cell 2016, 166, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, J.; Liu, S.; Xia, M.; Zhang, X.; Han, D.; Jiang, Y.; Wang, C.; Cao, X. Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell 2017, 170, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Sun, Y.; Ding, G.; Wu, Z.; Jiang, H.; Wang, L.; Ding, Q.; Wen, H. PI3Kβ inhibitor TGX221 selectively inhibits renal cell carcinoma cells with both VHL and SETD2 mutations and links multiple pathways. Sci. Rep. 2015, 5, 9465. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Ibáñez, G.; Wu, H.; Blum, G.; Zeng, H.; Dong, A.; Li, F.; Hajian, T.; Allali-Hassani, A.; Amaya, M.F.; et al. Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J. Am. Chem. Soc. 2012, 134, 18004–18014. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skucha, A.; Ebner, J.; Grebien, F. Roles of SETD2 in Leukemia—Transcription, DNA-Damage, and Beyond. Int. J. Mol. Sci. 2019, 20, 1029. https://doi.org/10.3390/ijms20051029
Skucha A, Ebner J, Grebien F. Roles of SETD2 in Leukemia—Transcription, DNA-Damage, and Beyond. International Journal of Molecular Sciences. 2019; 20(5):1029. https://doi.org/10.3390/ijms20051029
Chicago/Turabian StyleSkucha, Anna, Jessica Ebner, and Florian Grebien. 2019. "Roles of SETD2 in Leukemia—Transcription, DNA-Damage, and Beyond" International Journal of Molecular Sciences 20, no. 5: 1029. https://doi.org/10.3390/ijms20051029
APA StyleSkucha, A., Ebner, J., & Grebien, F. (2019). Roles of SETD2 in Leukemia—Transcription, DNA-Damage, and Beyond. International Journal of Molecular Sciences, 20(5), 1029. https://doi.org/10.3390/ijms20051029