Role of Arginase 2 in Systemic Metabolic Activity and Adipose Tissue Fatty Acid Metabolism in Diet-Induced Obese Mice

 ,
, 

Abstract
1. Introduction
2. Results
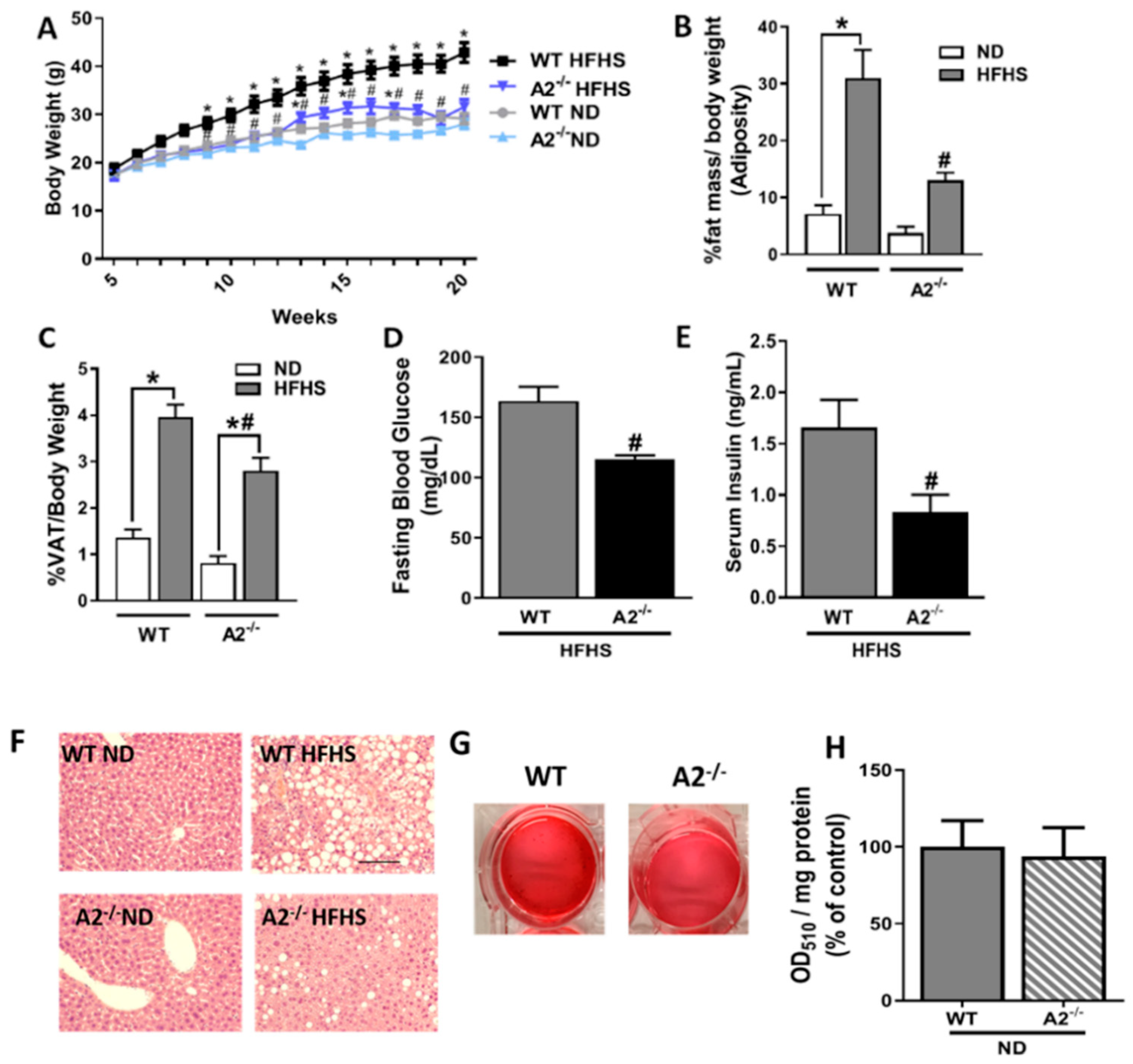
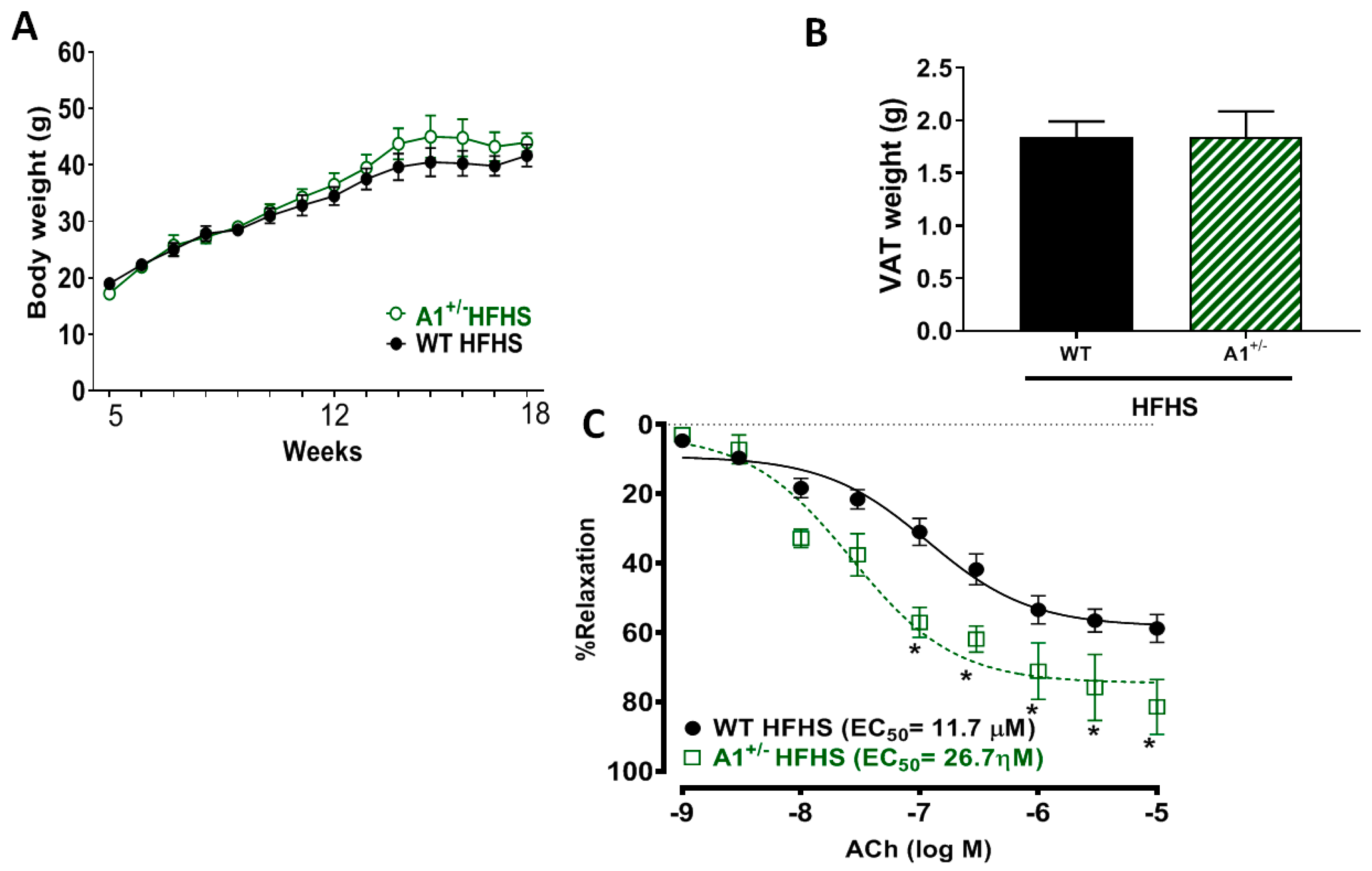
2.1. A2 Deletion Prevents High Fat/High Sucrose (HFHS) Diet-Induced Obesity
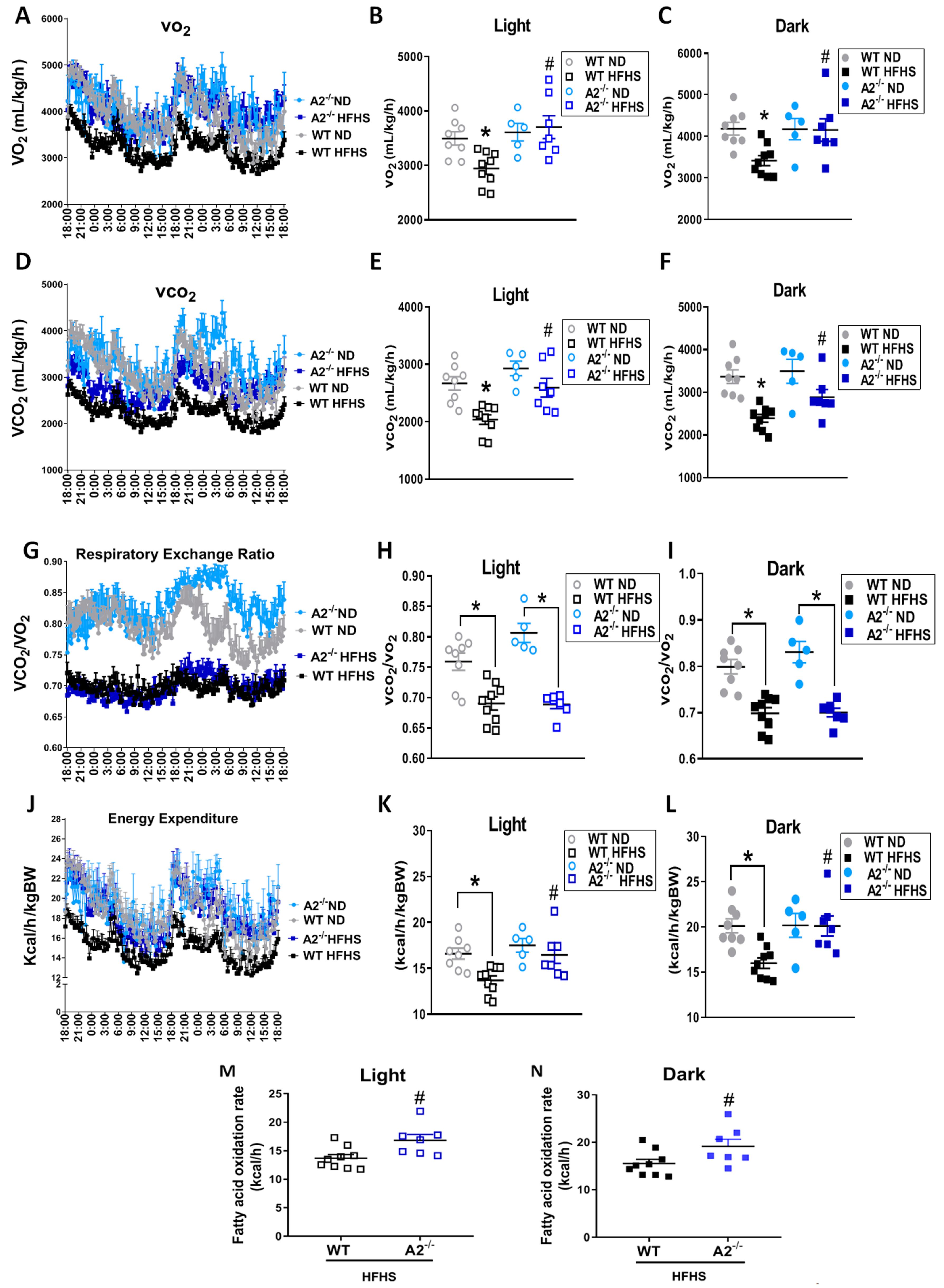
2.2. A2 Deletion Ameliorates the HFHS Diet-Induced Decrease in Metabolic Rate and Energy Expenditure
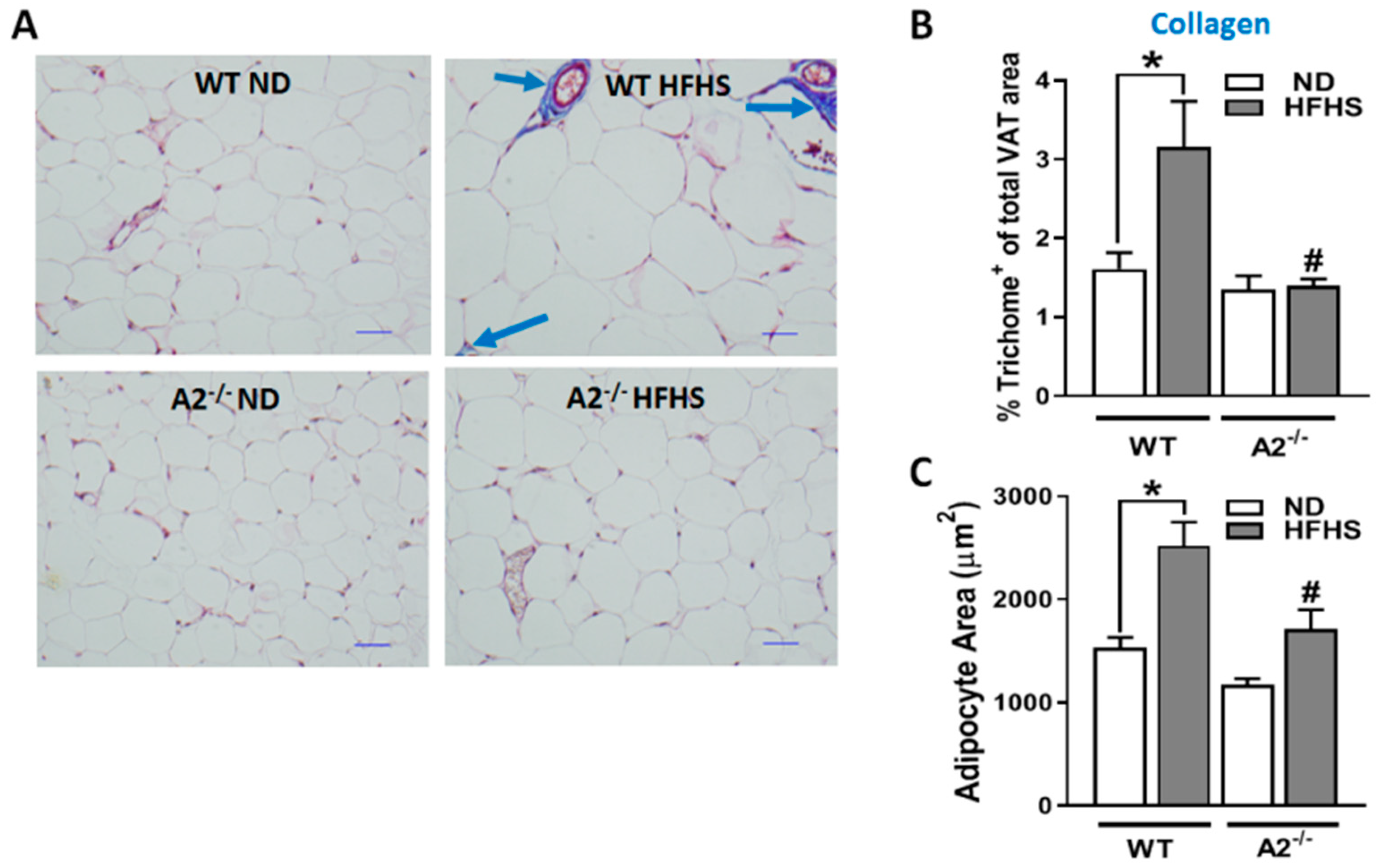
2.3. A2 Deletion Protected against HFHS Diet-Induced Adipocyte Hypertrophy and VAT Fibrosis
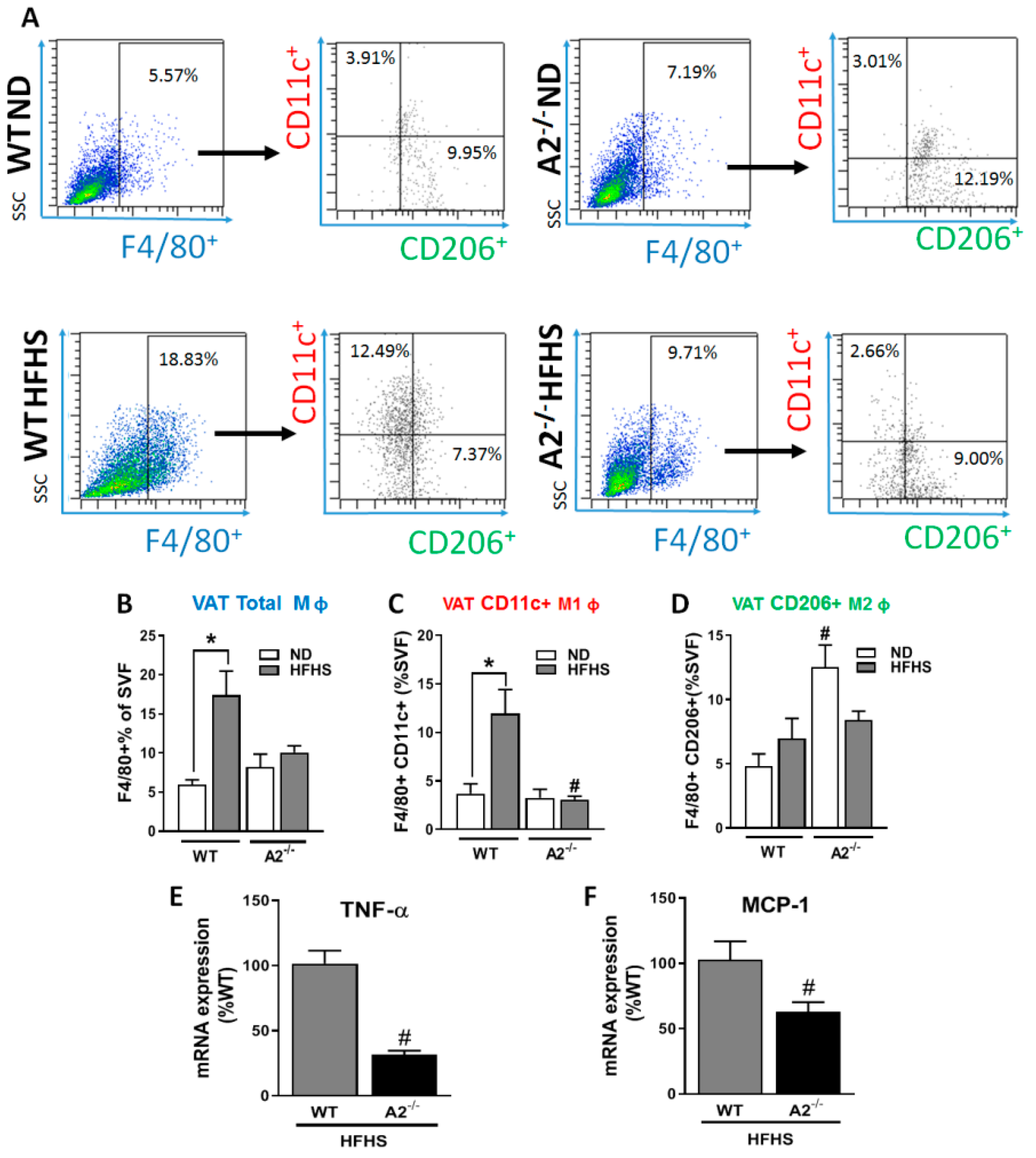
2.4. A2 Deletion Protected against HFHS Diet-Induced Pro-Inflammatory Macrophage Infiltration in the VAT
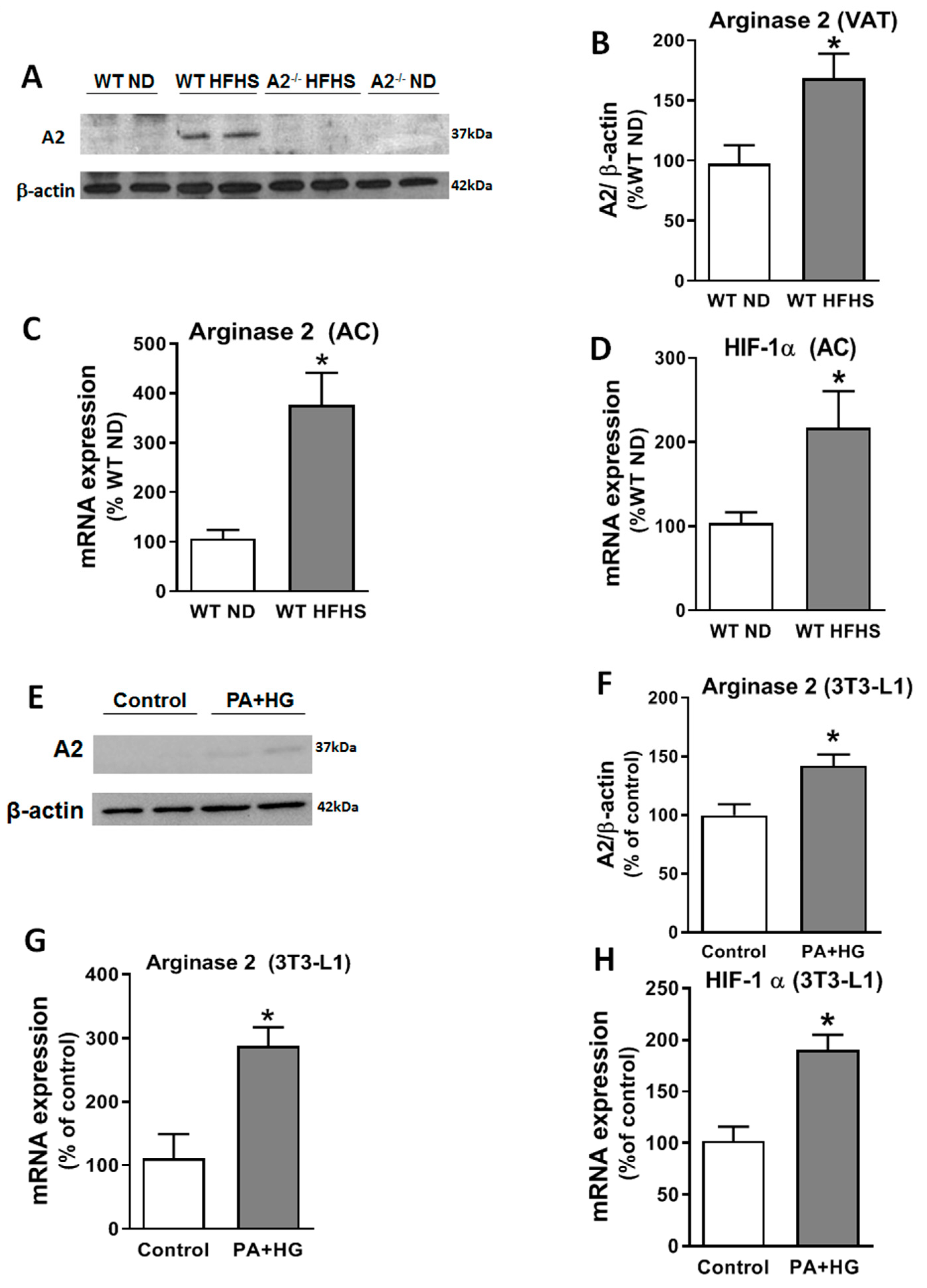
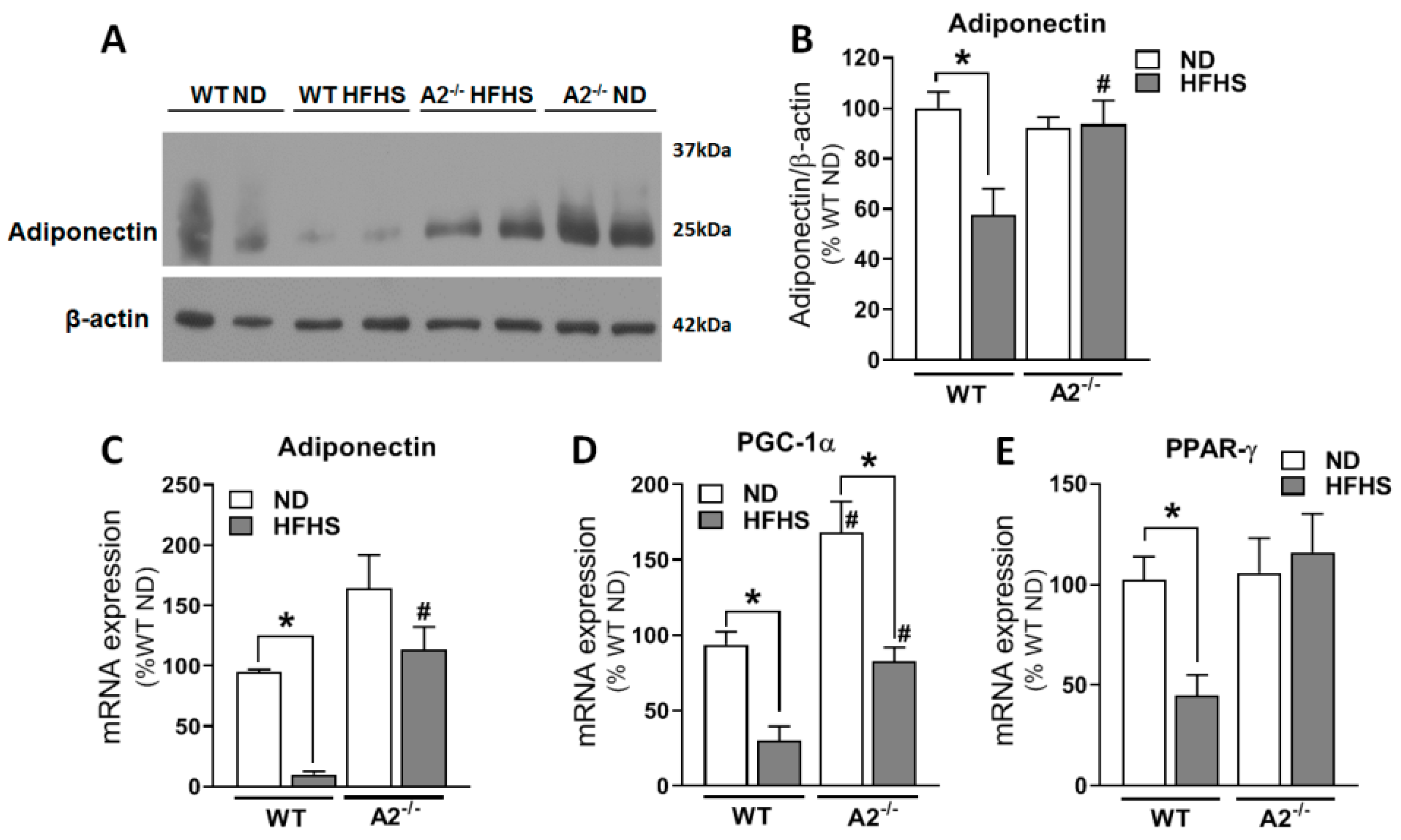
2.5. The HFHS Diet Increased A2 and Hypoxia-Inducible Factor (HIF)-1α Expressions in the VAT
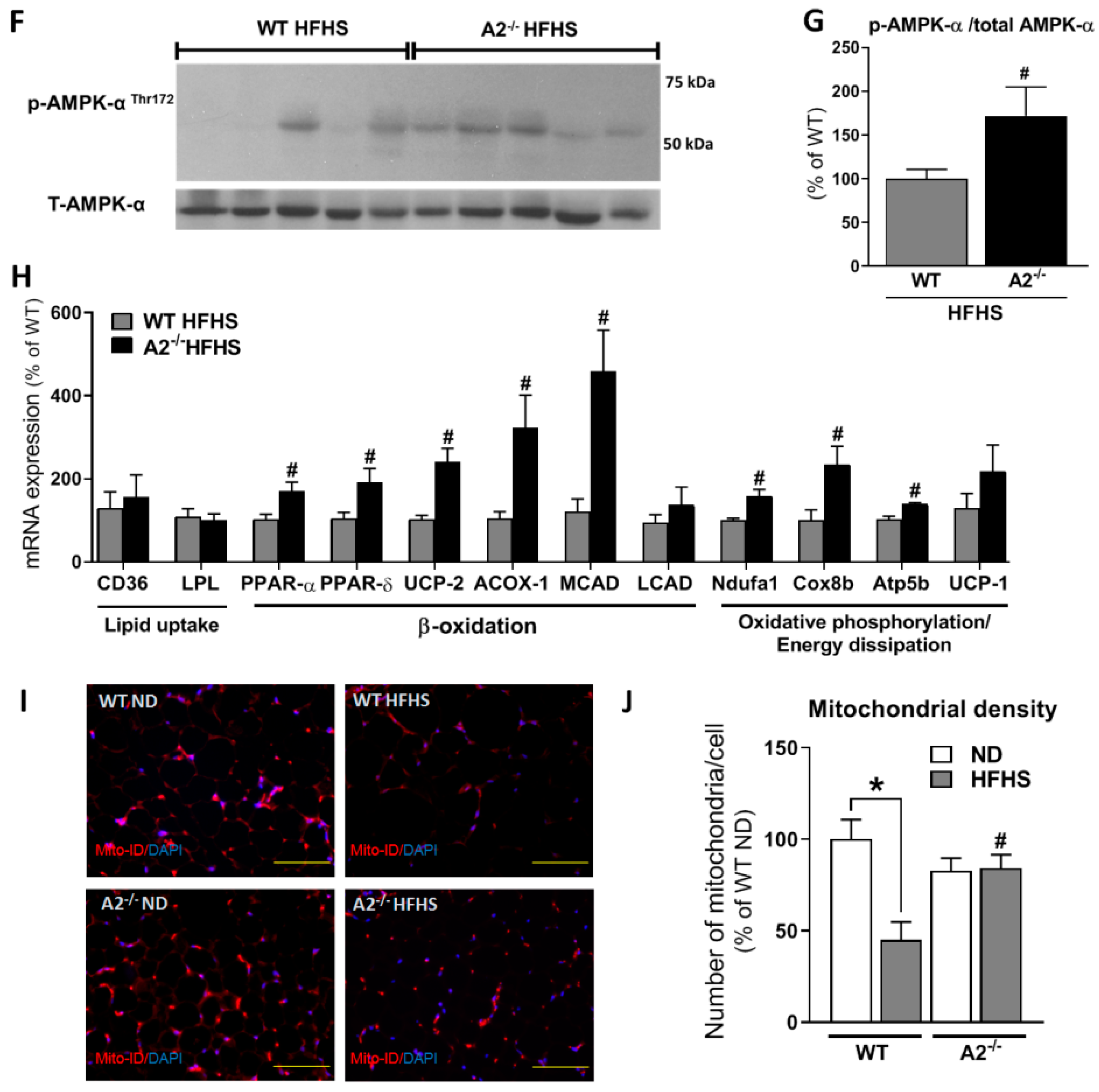
2.6. A2 Deletion Enhanced Expression of Genes Involved in Fatty Acid β-Oxidation and Preserved Mitochondrial Density
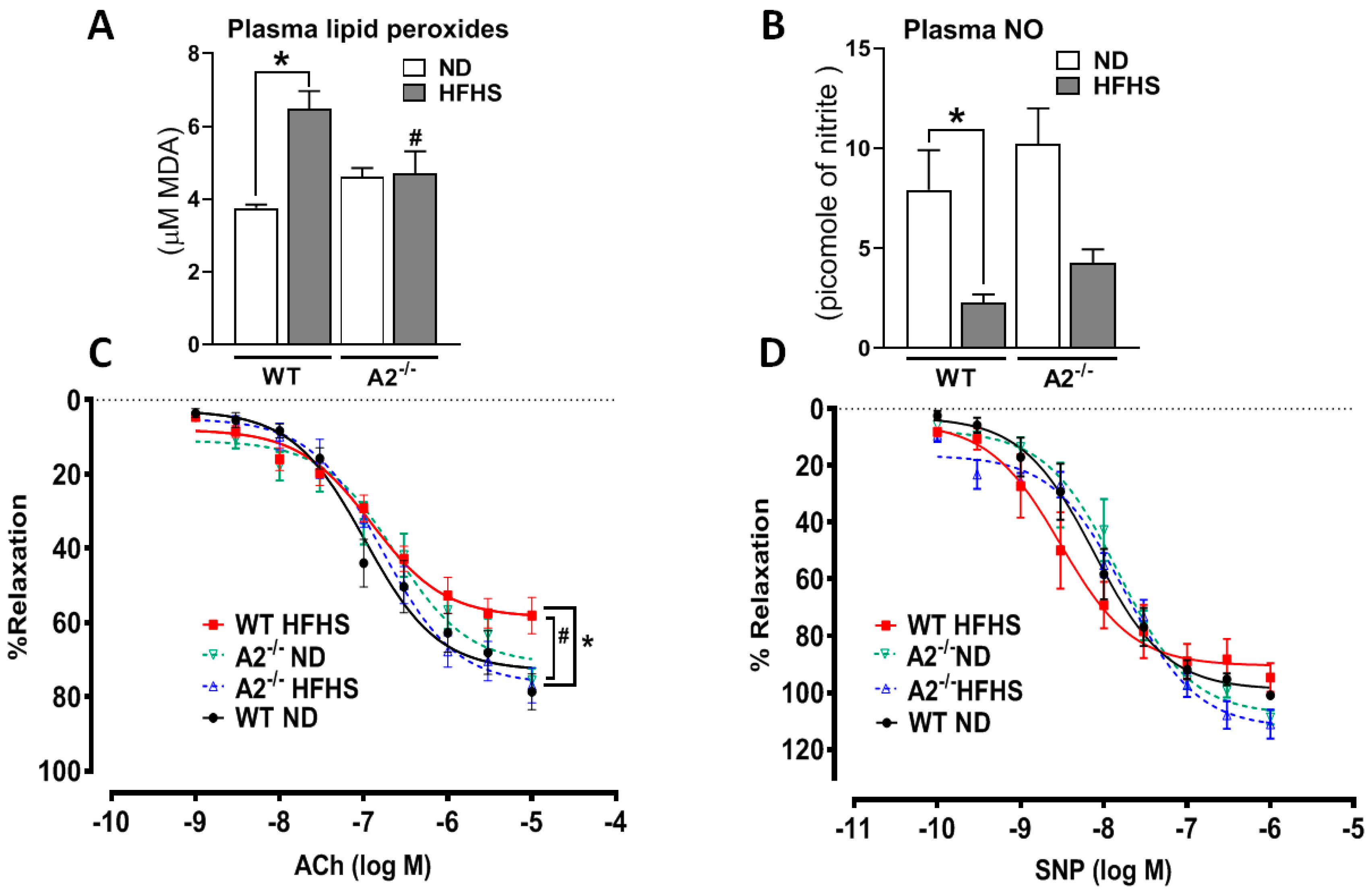
2.7. A2 Deletion Attenuated Circulating Oxidative Species and Preserved NO Levels and Vascular Function
2.8. Arginase 1 Deletion Preserved Vascular Function in HFHS Fed Mice without Affecting the Gain in Body Weight
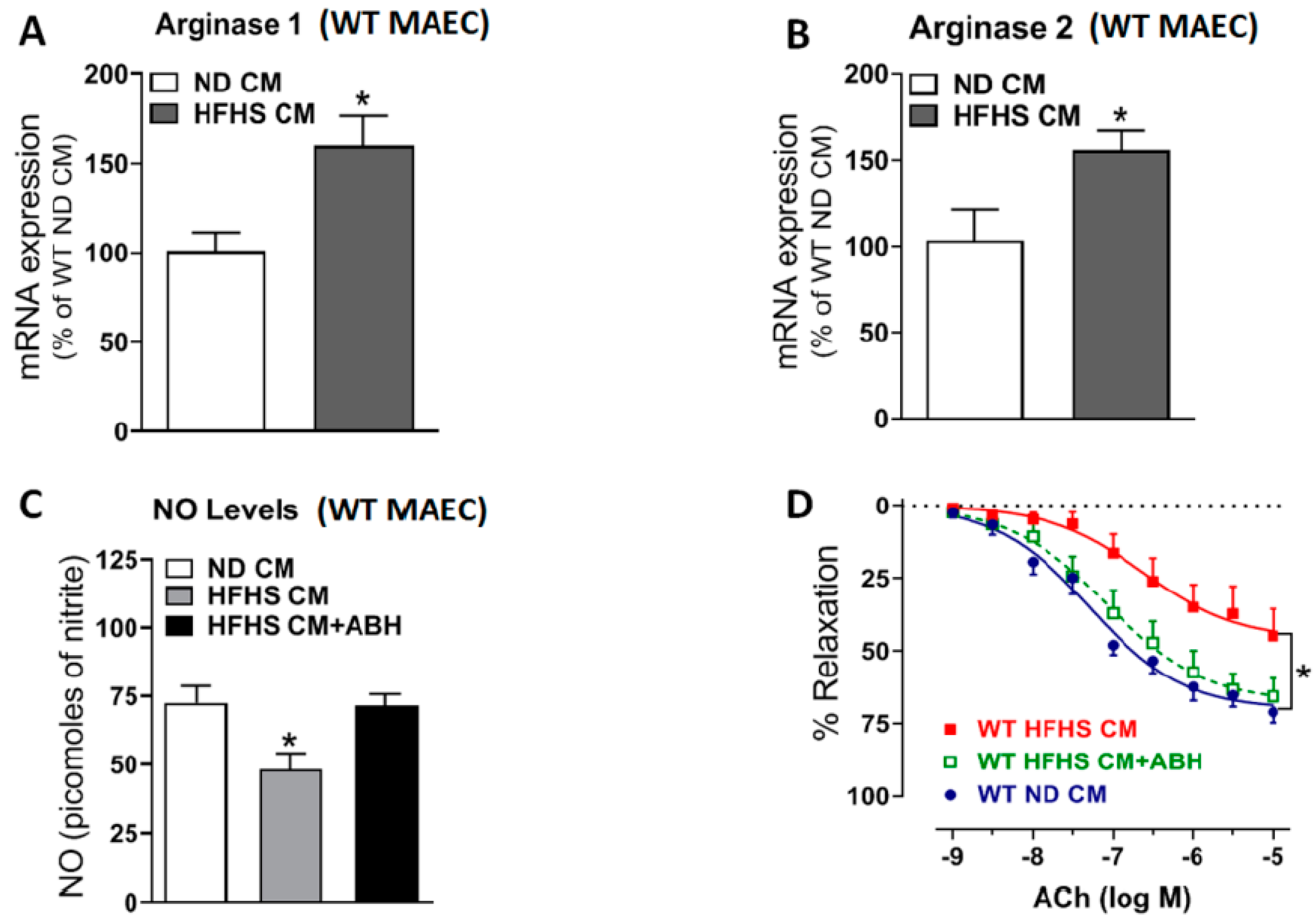
2.9. Arginase Inhibition Prevented Vascular Endothelial Dysfunction Induced by Conditioned Medium (CM) from VAT of WT HFHS Mice
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Body Weight, Body Composition, Fasting Blood Glucose, and Serum Insulin
4.3. Indirect Calorimetry
4.4. Tissue Harvest
4.5. Flow Cytometry Analysis
4.6. Quantitative Reverse Transcription-PCR (Q-PCR)
4.7. Adipose Tissue Western Blot
4.8. Preadipocyte Isolation and Adipogenic Differentiation In Vitro
4.9. Determination of Plasma Lipid Peroxide Levels and NO
4.10. Vascular Function
4.11. Conditioned Media (CM) Preparation from VAT and Endothelial Cell Treatment
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Atawia, R.T.; Bunch, K.L.; Toque, H.A.; Caldwell, R.B.; Caldwell, R.W. Mechanisms of obesity-induced metabolic and vascular dysfunctions. Front. Biosci. Landmark 2019, 24, 890–934. [Google Scholar]
- Shuster, A.; Patlas, M.; Pinthus, J.; Mourtzakis, M. The clinical importance of visceral adiposity: A critical review of methods for visceral adipose tissue analysis. Br. J. Radiol. 2012, 85, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sturm, W.; Sandhofer, A.; Engl, J.; Laimer, M.; Molnar, C.; Kaser, S.; Weiss, H.; Tilg, H.; Ebenbichler, C.F.; Patsch, J.R. Influence of visceral obesity and liver fat on vascular structure and function in obese subjects. Obesity 2009, 17, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Obin, M.S. Obesity and the role of adipose tissue in inflammation and metabolism. Am. J. Clin. Nutr. 2006, 83, 461S–465S. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, A.; Korac, A.; Buzadzic, B.; Stancic, A.; Otasevic, V.; Ferdinandy, P.; Daiber, A.; Korac, B. Targeting the no/superoxide ratio in adipose tissue: Relevance to obesity and diabetes management. Br. J. Pharmacol. 2017, 174, 1570–1590. [Google Scholar] [CrossRef]
- Castro, A.V.; Kolka, C.M.; Kim, S.P.; Bergman, R.N. Obesity, insulin resistance and comorbidities? Mechanisms of association. Arq. Bras. Endocrinol. Metabol. 2014, 58, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Lafontan, M. Adipose tissue and adipocyte dysregulation. Diabetes Metab. 2014, 40, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Gornicka, A.; Berk, M.P.; Thapaliya, S.; Dixon, L.J.; Kashyap, S.; Schauer, P.R.; Feldstein, A.E. Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J. Biol. Chem. 2010, 285, 3428–3438. [Google Scholar] [CrossRef]
- Jung, U.J.; Choi, M.-S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed]
- Flachs, P.; Rossmeisl, M.; Kuda, O.; Kopecky, J. Stimulation of mitochondrial oxidative capacity in white fat independent of ucp1: A key to lean phenotype. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2013, 1831, 986–1003. [Google Scholar] [CrossRef]
- Daval, M.; Foufelle, F.; Ferré, P. Functions of amp-activated protein kinase in adipose tissue. J. Physiol. 2006, 574, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.W.; Rodriguez, P.C.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.B. Arginase: A multifaceted enzyme important in health and disease. Physiol. Rev. 2018, 98, 641–665. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.J.; Platt, D.H.; Tawfik, H.E.; Labazi, M.; El-Remessy, A.B.; Bartoli, M.; Caldwell, R.B.; Caldwell, R.W. Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ. Res. 2008, 102, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Kövamees, O.; Shemyakin, A.; Checa, A.; Wheelock, C.E.; Lundberg, J.O.; Östenson, C.-G.; Pernow, J. Arginase inhibition improves microvascular endothelial function in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 2016, 101, 3952–3958. [Google Scholar] [CrossRef]
- Hu, H.; Moon, J.; Chung, J.H.; Kim, O.Y.; Yu, R.; Shin, M.-J. Arginase inhibition ameliorates adipose tissue inflammation in mice with diet-induced obesity. Biochem. Biophys. Res. Commun. 2015, 464, 840–847. [Google Scholar] [CrossRef]
- Ming, X.F.; Rajapakse, A.G.; Yepuri, G.; Xiong, Y.; Carvas, J.M.; Ruffieux, J.; Scerri, I.; Wu, Z.; Popp, K.; Li, J.; et al. Arginase ii promotes macrophage inflammatory responses through mitochondrial reactive oxygen species, contributing to insulin resistance and atherogenesis. J. Am. Heart Assoc. 2012, 1, e000992. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, A.; Yao, L.; Xu, Z.; Toque, H.A.; Chen, J.; Atawia, R.T.; Fouda, A.Y.; Bagi, Z.; Lucas, R.; Caldwell, R.B.; et al. Obesity-induced vascular dysfunction and arterial stiffening requires endothelial cell arginase 1. Cardiovasc. Res. 2017, 113, 1664–1676. [Google Scholar] [CrossRef]
- Yao, L.; Bhatta, A.; Xu, Z.; Chen, J.; Toque, H.A.; Chen, Y.; Xu, Y.; Bagi, Z.; Lucas, R.; Huo, Y.; et al. Obesity-induced vascular inflammation involves elevated arginase activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R560–R571. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yepuri, G.; Necetin, S.; Montani, J.-P.; Ming, X.-F.; Yang, Z. Arginase-ii promotes tumor necrosis factor-α release from pancreatic acinar cells causing β-cell apoptosis in aging. Diabetes 2017, 66, 1636–1649. [Google Scholar] [CrossRef]
- Ryoo, S.; Gupta, G.; Benjo, A.; Lim, H.K.; Camara, A.; Sikka, G.; Lim, H.K.; Sohi, J.; Santhanam, L.; Soucy, K.; et al. Endothelial arginase ii: A novel target for the treatment of atherosclerosis. Circ. Res. 2008, 102, 923–932. [Google Scholar] [CrossRef]
- Navarro, L.A.; Wree, A.; Povero, D.; Berk, M.P.; Eguchi, A.; Ghosh, S.; Papouchado, B.G.; Erzurum, S.C.; Feldstein, A.E. Arginase 2 deficiency results in spontaneous steatohepatitis: A novel link between innate immune activation and hepatic de novo lipogenesis. J. Hepatol. 2015, 62, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Lim, E.-J.; Yoon, Y.-S.; Ahn, Y.-H.; Park, E.-M.; Kim, H.-S.; Kang, J.L. Liver x receptor and stat1 cooperate downstream of gas6/mer to induce anti-inflammatory arginase 2 expression in macrophages. Sci. Rep. 2016, 6, 29673. [Google Scholar] [CrossRef]
- Jiang, C.; Qu, A.; Matsubara, T.; Chanturiya, T.; Jou, W.; Gavrilova, O.; Shah, Y.M.; Gonzalez, F.J. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet–fed mice. Diabetes 2011, 60, 2484–2495. [Google Scholar] [CrossRef]
- Xue, J.; Nelin, L.D.; Chen, B. Hypoxia induces arginase ii expression and increases viable human pulmonary artery smooth muscle cell numbers via ampkα1 signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L568–L578. [Google Scholar] [CrossRef]
- Kleiner, S.; Mepani, R.J.; Laznik, D.; Ye, L.; Jurczak, M.J.; Jornayvaz, F.R.; Estall, J.L.; Bhowmick, D.C.; Shulman, G.I.; Spiegelman, B.M. Development of insulin resistance in mice lacking pgc-1α in adipose tissues. Proc. Natl. Acad. Sci. USA 2012, 109, 9635–9640. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Auwerx, J. Pgc-1alpha, sirt1 and ampk, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Root-Mccaig, J.; Castellani, L.; Kemp, B.E.; Steinberg, G.R.; Wright, D.C. Evidence for the role of ampk in regulating pgc-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity 2014, 22, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Horke, S.; Habermeier, A.; Closs, E.I.; Reifenberg, G.; Gericke, A.; Mikhed, Y.; Munzel, T.; Daiber, A.; Forstermann, U.; et al. Uncoupling of endothelial nitric oxide synthase in perivascular adipose tissue of diet-induced obese mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 78–85. [Google Scholar] [CrossRef]
- Moon, J.; Do, H.J.; Cho, Y.; Shin, M.-J. Arginase inhibition ameliorates hepatic metabolic abnormalities in obese mice. PLoS ONE 2014, 9, e103048. [Google Scholar] [CrossRef]
- Liu, C.; Rajapakse, A.G.; Riedo, E.; Fellay, B.; Bernhard, M.-C.; Montani, J.-P.; Yang, Z.; Ming, X.-F. Targeting arginase-ii protects mice from high-fat-diet-induced hepatic steatosis through suppression of macrophage inflammation. Sci. Rep. 2016, 6, 20405. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, H.; Nittinger, J.; Engel, S.; Fürst, P. Thermogenesis of white adipocytes: A novel method allowing long-term microcalomrimetric investigations. J. Biochem. Biophys. Methods 1991, 23, 181–187. [Google Scholar] [CrossRef]
- Kelley, D.E. Skeletal muscle fat oxidation: Timing and flexibility are everything. J. Clin. Investig. 2005, 115, 1699–1702. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Divoux, A.; Tordjman, J.; Lacasa, D.; Veyrie, N.; Hugol, D.; Aissat, A.; Basdevant, A.; Guerre-Millo, M.; Poitou, C.; Zucker, J.D.; et al. Fibrosis in human adipose tissue: Composition, distribution, and link with lipid metabolism and fat mass loss. Diabetes 2010, 59, 2817–2825. [Google Scholar] [CrossRef]
- Halberg, N.; Khan, T.; Trujillo, M.E.; Wernstedt-Asterholm, I.; Attie, A.D.; Sherwani, S.; Wang, Z.V.; Landskroner-Eiger, S.; Dineen, S.; Magalang, U.J.; et al. Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol. 2009, 29, 4467–4483. [Google Scholar] [CrossRef]
- Pellegrinelli, V.; Heuvingh, J.; Du Roure, O.; Rouault, C.; Devulder, A.; Klein, C.; Lacasa, M.; Clément, E.; Lacasa, D.; Clément, K. Human adipocyte function is impacted by mechanical cues. J. Pathol. 2014, 233, 183–195. [Google Scholar] [CrossRef]
- Wellen, K.E.; Hotamisligil, G.S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Investig. 2003, 112, 1785–1788. [Google Scholar] [CrossRef]
- Fjeldborg, K.; Pedersen, S.B.; Møller, H.J.; Christiansen, T.; Bennetzen, M.; Richelsen, B. Human adipose tissue macrophages are enhanced but changed to an anti-inflammatory profile in obesity. J. Immunol. Res. 2014, 2014, 309548. [Google Scholar] [CrossRef]
- Fujisaka, S.; Usui, I.; Bukhari, A.; Ikutani, M.; Oya, T.; Kanatani, Y.; Tsuneyama, K.; Nagai, Y.; Takatsu, K.; Urakaze, M.; et al. Regulatory mechanisms for adipose tissue m1 and m2 macrophages in diet-induced obese mice. Diabetes 2009, 58, 2574–2582. [Google Scholar] [CrossRef]
- Choi, M.-S.; Kim, Y.-J.; Kwon, E.-Y.; Ryoo, J.Y.; Kim, S.R.; Jung, U.J. High-fat diet decreases energy expenditure and expression of genes controlling lipid metabolism, mitochondrial function and skeletal system development in the adipose tissue, along with increased expression of extracellular matrix remodelling-and inflammation-related genes. Br. J. Nutr. 2015, 113, 867–877. [Google Scholar] [PubMed]
- Rupasinghe, H.V.; Sekhon-Loodu, S.; Mantso, T.; Panayiotidis, M.I. Phytochemicals in regulating fatty acid β-oxidation: Potential underlying mechanisms and their involvement in obesity and weight loss. Pharmacol. Ther. 2016, 165, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Rogge, M.M. The role of impaired mitochondrial lipid oxidation in obesity. Biol. Res. Nurs. 2009, 10, 356–373. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Leuenberger, N.; Tan, M.J.; Yan, Y.W.; Chen, Y.; Kambadur, R.; Wahli, W.; Tan, N.S. Smad3 deficiency in mice protects against insulin resistance and obesity induced by a high-fat diet. Diabetes 2011, 60, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Malandrino, M.I.; Fucho, R.; Weber, M.; Calderon-Dominguez, M.; Mir, J.F.; Valcarcel, L.; Escoté, X.; Gómez-Serrano, M.; Peral, B.; Salvadó, L.; et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E756–E769. [Google Scholar] [CrossRef] [PubMed]
- Barberá, M.J.; Schlüter, A.; Pedraza, N.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome proliferator-activated receptor alpha activates transcription of the brown fat uncoupling protein-1 gene. A link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. J. Biol. Chem. 2001, 276, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Ruschke, K.; Fishbein, L.; Dietrich, A.; Kloting, N.; Tonjes, A.; Oberbach, A.; Fasshauer, M.; Jenkner, J.; Schon, M.R.; Stumvoll, M.; et al. Gene expression of ppargamma and pgc-1alpha in human omental and subcutaneous adipose tissues is related to insulin resistance markers and mediates beneficial effects of physical training. Eur. J. Endocrinol. 2010, 162, 515–523. [Google Scholar] [CrossRef]
- Gustafson, B.; Jack, M.M.; Cushman, S.W.; Smith, U. Adiponectin gene activation by thiazolidinediones requires pparγ2, but not c/ebpα—Evidence for differential regulation of the ap2 and adiponectin genes. Biochem. Biophys. Res. Commun. 2003, 308, 933–939. [Google Scholar] [CrossRef]
- Wu, X.; Motoshima, H.; Mahadev, K.; Stalker, T.J.; Scalia, R.; Goldstein, B.J. Involvement of amp-activated protein kinase in glucose uptake stimulated by the globular domain of adiponectin in primary rat adipocytes. Diabetes 2003, 52, 1355–1363. [Google Scholar] [CrossRef]
- Caballero, A.E. Endothelial dysfunction in obesity and insulin resistance: A road to diabetes and heart disease. Obes. Res. 2003, 11, 1278–1289. [Google Scholar] [CrossRef]
- Roberts, A.C.; Porter, K.E. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diabetes Vasc. Disease Res. 2013, 10, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Serra, D.; Mera, P.; Malandrino, M.I.; Mir, J.F.; Herrero, L. Mitochondrial fatty acid oxidation in obesity. Antioxid. Redox Signal. 2013, 19, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Jain, S.K. Obesity, oxidative stress, adipose tissue dysfunction, and the associated health risks: Causes and therapeutic strategies. Metab. Syndr. Relat. Disord. 2015, 13, 423–444. [Google Scholar] [CrossRef]
- Yesilbursa, D.; Serdar, Z.; Serdar, A.; Sarac, M.; Coskun, S.; Jale, C. Lipid peroxides in obese patients and effects of weight loss with orlistat on lipid peroxides levels. Int. J. Obes. 2005, 29, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Rajapakse, A.G.; Montani, J.-P.; Yang, Z.; Ming, X.-F. P38 mitogen-activated protein kinase is involved in arginase-ii-mediated enos-uncoupling in obesity. Cardiovasc. Diabetol. 2014, 13, 113. [Google Scholar] [CrossRef]
- Lee, Y.; Ka, S.-O.; Cha, H.-N.; Chae, Y.-N.; Kim, M.-K.; Park, S.-Y.; Bae, E.J.; Park, B.-H. Myeloid sirtuin 6 deficiency causes insulin resistance in high-fat diet-fed mice by eliciting macrophage polarization toward an m1 phenotype. Diabetes 2017, 66, 2659–2668. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Zhang, Y.; Liu, Q.; Shen, J.; Pu, S.; Cheng, S.; Chen, L.; Li, H.; Wu, T.; Li, R.; et al. Fat-specific sirt6 ablation sensitizes mice to high-fat diet-induced obesity and insulin resistance by inhibiting lipolysis. Diabetes 2017, 66, 1159–1171. [Google Scholar] [CrossRef]
- Crowe, S.; Turpin, S.M.; Ke, F.; Kemp, B.E.; Watt, M.J. Metabolic remodeling in adipocytes promotes ciliary neurotrophic factor-mediated fat loss in obesity. Endocrinology 2008, 149, 2546–2556. [Google Scholar] [CrossRef] [PubMed]
- Romero, M.J.; Iddings, J.A.; Platt, D.H.; Ali, M.I.; Cederbaum, S.D.; Stepp, D.W.; Caldwell, R.B.; Caldwell, R.W. Diabetes-induced vascular dysfunction involves arginase i. Am. J. Physiol. Heart Circ. Physiol. 2011, 302, H159–H166. [Google Scholar] [CrossRef] [PubMed]
- Winzell, M.S.; Ahrén, B. The high-fat diet–fed mouse: A model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 2004, 53, S215–S219. [Google Scholar] [CrossRef]
- Wang, C.Y.; Liao, J.K. A mouse model of diet-induced obesity and insulin resistance. Methods Mol. Biol. (Clifton NJ) 2012, 821, 421–433. [Google Scholar]
- Bjursell, M.; Gerdin, A.-K.; Ploj, K.; Svensson, D.; Svensson, L.; Oscarsson, J.; Snaith, M.; Törnell, J.; Bohlooly-y, M. Melanin-concentrating hormone receptor 1 deficiency increases insulin sensitivity in obese leptin-deficient mice without affecting body weight. Diabetes 2006, 55, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Vroegrijk, I.O.; van Diepen, J.A.; van den Berg, S.A.; Romijn, J.A.; Havekes, L.M.; van Dijk, K.W.; Darland, G.; Konda, V.; Tripp, M.L.; Bland, J.S.; et al. Meta060 protects against diet-induced obesity and insulin resistance in a high-fat-diet fed mouse. Nutrition 2013, 29, 276–283. [Google Scholar] [CrossRef]
- Chatterjee, T.K.; Idelman, G.; Blanco, V.; Blomkalns, A.L.; Piegore, M.G., Jr.; Weintraub, D.S.; Kumar, S.; Rajsheker, S.; Manka, D.; Rudich, S.M.; et al. Histone deacetylase 9 is a negative regulator of adipogenic differentiation. J. Biol. Chem. 2011, 286, 27836–27847. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.P.; Yun, C.H.; Lee, G.W.; Park, A.; Kim, Y.M.; Jang, M.H. Tlr9 regulates adipose tissue inflammation and obesity-related metabolic disorders. Obesity 2015, 23, 2199–2206. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.J.; Rios, L.; Trivedi, P.; D’Souza, K.; Cowie, A.; Nzirorera, C.; Webster, D.; Brunt, K.; Legare, J.F.; Hassan, A.; et al. Validation of optimal reference genes for quantitative real time pcr in muscle and adipose tissue for obesity and diabetes research. Sci. Rep. 2017, 7, 3612. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, T.K.; Stoll, L.L.; Denning, G.M.; Harrelson, A.; Blomkalns, A.L.; Idelman, G.; Rothenberg, F.G.; Neltner, B.; Romig-Martin, S.A.; Dickson, E.W.; et al. Proinflammatory phenotype of perivascular adipocytes: Influence of high-fat feeding. Circ. Res. 2009, 104, 541–549. [Google Scholar] [CrossRef]
- Han, C.Y.; Kargi, A.Y.; Omer, M.; Chan, C.K.; Wabitsch, M.; O’brien, K.D.; Wight, T.N.; Chait, A. Differential effect of saturated and unsaturated free fatty acids on the generation of monocyte adhesion and chemotactic factors by adipocytes: Dissociation of adipocyte hypertrophy from inflammation. Diabetes 2010, 59, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, M.; Mihara, M. Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal. Biochem. 1978, 86, 271–278. [Google Scholar] [CrossRef]
- Shin, S.H.; Kim, J.; Heo, S.C.; Kwon, Y.W.; Kim, Y.M.; Kim, I.-S.; Lee, T.G.; Kim, J.H. Proteomic identification of betaig-h3 as a lysophosphatidic acid-induced secreted protein of human mesenchymal stem cells: Paracrine activation of a549 lung adenocarcinoma cells by betaig-h3. Mol. Cell. Proteomics 2012, 11, M111.012385. [Google Scholar] [CrossRef]
- Park, H.; Kim, M.; Kwon, G.T.; Lim, D.Y.; Yu, R.; Sung, M.K.; Lee, K.W.; Daily, J.W., III; Park, J.H.Y. A high-fat diet increases angiogenesis, solid tumor growth, and lung metastasis of ct26 colon cancer cells in obesity-resistant balb/c mice. Mol. Carcinog. 2012, 51, 869–880. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Gene Symbol | Gene Name | Forward | Reverse |
|---|---|---|---|
| TNF-α | Tumor necrosis factor alpha | GCACCACCATCAAGGACTCA | TCGAGGCTCCAGTGAATTCG |
| MCP-1 | Monocyte chemoattractant protein 1 | GGCTCAGCCAGATGCAGTTAA | CCTACTCATTGGGATCATCTTGCT |
| A 1 | Arginase 1 | TTGGGTGGATGCTCACACTG | TTGCCCATGCAGATTCCC |
| Adiponectin | Adiponectin | AAGGACAAGGCCGTTCTCT | TATGGGTAGTTGCAGTCAGTTGG |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha | AACCACACCCACAGGATCAGA | TCTTCGCTTTATTGCTCCATGA |
| PPAR-ɣ | Peroxisome proliferator-activated receptor gamma | CAAGAATACCAAAGTGCGATCAA | GAGCTGGGTCTTTTCAGAATAATAAG |
| CD36 | Cluster of differentiation 36 | TTGTACCTATACTGTGGCTAAATGAGA | CTTGTGTTTTGAACATTTCTGCTT |
| LPL | Lipoprotein lipase | CTGCTGGCGTAGCAGGAAGT | GCTGGAAAGTGCCTCCATTG |
| PPAR-α | Peroxisome Proliferator Activated Receptor alpha | GCGTACGGCAATGGCTTTAT | ACAGAACGGCTTCCTCAGGTT |
| PPAR-δ | Peroxisome Proliferator Activated Receptor Delta | CCTCGGGCTTCCACTACG | CACTTGTTGCGGTTCTTCTTC |
| Ucp-2 | uncoupling protein 2 | GCCCGGGCTGGTGGTGGTC | CCCCGAAGGCAGAAGTGAAGTGG |
| Acox-1 | Acyl-CoA oxidase 1 | GCCAAGGCGACCTGAGTGAGC | ACCGCAAGCCATCCGACATTC |
| MCAD | Medium-chain acyl-CoA dehydrogenase | AACACTTACTATGCCTCGATTGCA | CCATAGCCTCCGAAAATCTGAA |
| LCAD | Long-chain acyl-CoA dehydrogenase | ATGGCAAAATACTGGGCATC | TCTTGCGATCAGCTCTTTCA |
| Ndufa1 | NADH:Ubiquinone Oxidoreductase Subunit A1 | ACATCCACAAATTCACCAACGG | AGCGATTGACTCCAGAGATACG |
| COX-8b | Cytochrome c oxidase subunit 8B | GAACCATGAAGCCAACGACT | GCGAAGTTCACAGTGGTTCC |
| Atp5b | ATP Synthase F1 Subunit Beta | CATTGGTGATGGTATTGCGC | TCCCAAACACGACAACTCC |
| UCP-1 | Uncoupling protein 1 | TCTTCTCAGCCGGAGTTTCAGCTT | ACCTTGGATCTGAAGGCGGACTTT |
| HPRT | Hypoxanthine Phosphoribosyltransferase 1 | GAAAGACTTGCTCGAGATGTCATG | CACACAGAGGGCCACAATGT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atawia, R.T.; Toque, H.A.; Meghil, M.M.; Benson, T.W.; Yiew, N.K.H.; Cutler, C.W.; Weintraub, N.L.; Caldwell, R.B.; Caldwell, R.W. Role of Arginase 2 in Systemic Metabolic Activity and Adipose Tissue Fatty Acid Metabolism in Diet-Induced Obese Mice. Int. J. Mol. Sci. 2019, 20, 1462. https://doi.org/10.3390/ijms20061462
Atawia RT, Toque HA, Meghil MM, Benson TW, Yiew NKH, Cutler CW, Weintraub NL, Caldwell RB, Caldwell RW. Role of Arginase 2 in Systemic Metabolic Activity and Adipose Tissue Fatty Acid Metabolism in Diet-Induced Obese Mice. International Journal of Molecular Sciences. 2019; 20(6):1462. https://doi.org/10.3390/ijms20061462
Chicago/Turabian StyleAtawia, Reem T., Haroldo A. Toque, Mohamed M. Meghil, Tyler W. Benson, Nicole K. H. Yiew, Christopher W. Cutler, Neal L. Weintraub, Ruth B. Caldwell, and Robert W. Caldwell. 2019. "Role of Arginase 2 in Systemic Metabolic Activity and Adipose Tissue Fatty Acid Metabolism in Diet-Induced Obese Mice" International Journal of Molecular Sciences 20, no. 6: 1462. https://doi.org/10.3390/ijms20061462
APA StyleAtawia, R. T., Toque, H. A., Meghil, M. M., Benson, T. W., Yiew, N. K. H., Cutler, C. W., Weintraub, N. L., Caldwell, R. B., & Caldwell, R. W. (2019). Role of Arginase 2 in Systemic Metabolic Activity and Adipose Tissue Fatty Acid Metabolism in Diet-Induced Obese Mice. International Journal of Molecular Sciences, 20(6), 1462. https://doi.org/10.3390/ijms20061462