Unveiling the Efficacy, Safety, and Tolerability of Anti-Interleukin-1 Treatment in Monogenic and Multifactorial Autoinflammatory Diseases

 ,
, 


 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. IL-1 Blockade in Autoinflammatory Diseases
3. Cryopyrin-Associated Periodic Syndrome
4. Tumor Necrosis Factor Receptor-Associated Periodic Syndrome
5. Familial Mediterranean Fever
6. Hyperimmunoglobulin D Syndrome/Mevalonate Kinase Deficiency
7. Additional Evidence on IL-1 Inhibition in Autoinflammatory Diseases
8. Systemic Juvenile Idiopathic Arthritis
9. Kawasaki Disease
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Teague, M. Pediatric rheumatologic diseases: A review for primary care NPs. Nurse Pract. 2017, 42, 43–47. [Google Scholar] [CrossRef]
- Rigante, D. A systematic approach to autoinflammatory syndromes: A spelling booklet for the beginner. Expert Rev. Clin. Immunol. 2017, 13, 571–597. [Google Scholar] [CrossRef] [PubMed]
- Cantarini, L.; Vitale, A.; Lucherini, O.M.; De Clemente, C.; Caso, F.; Costa, L.; Emmi, G.; Silvestri, E.; Magnotti, F.; Maggio, M.C.; et al. The labyrinth of autoinflammatory disorders: A snapshot on the activity of a third-level center in Italy. Clin. Rheumatol. 2015, 34, 17–28. [Google Scholar] [CrossRef]
- Rigante, D.; Lopalco, G.; Vitale, A.; Lucherini, O.M.; Caso, F.; De Clemente, C.; Molinaro, F.; Messina, M.; Costa, L.; Atteno, M.; et al. Untangling the web of systemic autoinflammatory diseases. Mediat. Inflamm. 2014, 2014, 948154. [Google Scholar] [CrossRef] [PubMed]
- Cattalini, M.; Soliani, M.; Lopalco, G.; Rigante, D.; Cantarini, L. Systemic and organ involvement in monogenic autoinflammatory disorders: A global review filtered through internists’ lens. Intern. Emerg. Med. 2016, 11, 781–791. [Google Scholar] [CrossRef]
- Obici, L.; Merlini, G. Amyloidosis in autoinflammatory syndromes. Autoimmun. Rev. 2012, 12, 14–17. [Google Scholar] [CrossRef]
- Cimaz, R. Systemic-onset juvenile idiopathic arthritis. Autoimmun. Rev. 2016, 15, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.Y.; Schneider, R. Systemic juvenile idiopathic arthritis. Pediatr. Clin. N. Am. 2018, 65, 691–709. [Google Scholar] [CrossRef]
- Barut, K.; Adrovic, A.; Şahin, S.; Kasapçopu, Ö. Juvenile idiopathic arthritis. Balk. Med. J. 2017, 34, 90–101. [Google Scholar] [CrossRef]
- Patel, R.M.; Shulman, S.T. Kawasaki disease: A comprehensive review of treatment options. J. Clin. Pharm. 2015, 40, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Hersh, A.; von Scheven, E.; Yelin, E. Adult outcomes of childhood-onset rheumatic diseases. Nat. Rev. Rheumatol. 2011, 7, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) pathway. Sci. Signal. 2010, 3, cm1. [Google Scholar] [CrossRef] [PubMed]
- Cantarini, L.; Lopalco, G.; Cattalini, M.; Vitale, A.; Galeazzi, M.; Rigante, D. Interleukin-1: Ariadne’s Thread in autoinflammatory and autoimmune disorders. Isr. Med. Assoc. J. 2015, 17, 93–97. [Google Scholar] [PubMed]
- O’Neill, L.A.J. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol. Rev. 2008, 226, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Beesu, M.; Caruso, G.; Salyer, A.C.D.; Shukla, N.M.; Khetani, K.K.; Smith, L.J.; Fox, L.M.; Tanji, H.; Ohto, U.; Shimizu, T.; et al. Identification of a human Toll-Like Receptor (TLR) 8-specific agonist and a functional pan-TLR inhibitor in 2-aminoimidazoles. J. Med. Chem. 2016, 59, 3311–3330. [Google Scholar] [CrossRef] [PubMed]
- Federici, S.; Martini, A.; Gattorno, M. The Central Role of Anti-IL-1 Blockade in the Treatment of Monogenic and Multi-Factorial Autoinflammatory Diseases. Front. Immunol. 2013, 4, 351. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Simon, A.; Aksentijevich, I.; Kastner, D.L. Horror autoinflammaticus: The molecular pathophysiology of autoinflammatory disease (*). Annu. Rev. Immunol. 2009, 27, 621–668. [Google Scholar] [CrossRef]
- Toplak, N.; Blazina, Š.; Avčin, T. The role of IL-1 inhibition in systemic juvenile idiopathic arthritis: Current status and future perspectives. Drug Des. Dev. Ther. 2018, 12, 1633–1643. [Google Scholar] [CrossRef]
- Bettiol, A.; Silvestri, E.; Di Scala, G.; Amedei, A.; Becatti, M.; Fiorillo, C.; Lopalco, G.; Salvarani, C.; Cantarini, L.; Soriano, A.; et al. The right place of interleukin-1 inhibitors in the treatment of Behçet’s syndrome: A systematic review. Rheumatol. Int. 2019. [Google Scholar] [CrossRef]
- European Medicines Agency. Summary of Product Characteristics—Kineret. Available online: https://www.ema.europa.eu/documents/product-information/kineret-epar-product-information_en.pdf (accessed on 8 March 2019).
- Urien, S.; Bardin, C.; Bader-Meunier, B.; Mouy, R.; Compeyrot-Lacassagne, S.; Foissac, F.; Florkin, B.; Wouters, C.; Neven, B.; Treluyer, J.-M.; et al. Anakinra pharmacokinetics in children and adolescents with systemic-onset juvenile idiopathic arthritis and autoinflammatory syndromes. BMC Pharmacol. Toxicol. 2013, 14, 40. [Google Scholar] [CrossRef]
- European Medicines Agency. Summary of Product Characteristics—Ilaris. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/ilaris (accessed on 17 April 2019).
- Sun, H.; Van, L.M.; Floch, D.; Jiang, X.; Klein, U.R.; Abrams, K.; Sunkara, G. Pharmacokinetics and pharmacodynamics of canakinumab in patients with systemic juvenile idiopathic arthritis. J. Clin. Pharmacol. 2016, 56, 1516–1527. [Google Scholar] [CrossRef]
- Cantarini, L.; Lucherini, O.M.; Frediani, B.; Brizi, M.G.; Bartolomei, B.; Cimaz, R.; Galeazzi, M.; Rigante, D. Bridging the gap between the clinician and the patient with cryopyrin-associated periodic syndromes. Int. J. Immunopathol. Pharmacol. 2011, 24, 827–836. [Google Scholar] [CrossRef]
- Lachmann, H.J.; Kone-Paut, I.; Kuemmerle-Deschner, J.B.; Leslie, K.S.; Hachulla, E.; Quartier, P.; Gitton, X.; Widmer, A.; Patel, N.; Hawkins, P.N.; et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N. Engl. J. Med. 2009, 360, 2416–2425. [Google Scholar] [CrossRef] [PubMed]
- Kuemmerle-Deschner, J.B.; Ramos, E.; Blank, N.; Roesler, J.; Felix, S.D.; Jung, T.; Stricker, K.; Chakraborty, A.; Tannenbaum, S.; Wright, A.M.; et al. Canakinumab (ACZ885, a fully human IgG1 anti-IL-1β mAb) induces sustained remission in pediatric patients with cryopyrin-associated periodic syndrome (CAPS). Arthritis Res. Ther. 2011, 13, R34. [Google Scholar] [CrossRef]
- Kuemmerle-Deschner, J.B.; Hachulla, E.; Cartwright, R.; Hawkins, P.N.; Tran, T.A.; Bader-Meunier, B.; Hoyer, J.; Gattorno, M.; Gul, A.; Smith, J.; et al. Two-year results from an open-label, multicentre, phase III study evaluating the safety and efficacy of canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. Ann. Rheum. Dis. 2011, 70, 2095–2102. [Google Scholar] [CrossRef]
- Koné-Paut, I.; Lachmann, H.J.; Kuemmerle-Deschner, J.B.; Hachulla, E.; Leslie, K.S.; Mouy, R.; Ferreira, A.; Lheritier, K.; Patel, N.; Preiss, R.; et al. Sustained remission of symptoms and improved health-related quality of life in patients with cryopyrin-associated periodic syndrome treated with canakinumab: Results of a double-blind placebo-controlled randomized withdrawal study. Arthritis Res. Ther. 2011, 13, R202. [Google Scholar] [CrossRef] [PubMed]
- Imagawa, T.; Nishikomori, R.; Takada, H.; Takeshita, S.; Patel, N.; Kim, D.; Lheritier, K.; Heike, T.; Hara, T.; Yokota, S. Safety and efficacy of canakinumab in Japanese patients with phenotypes of cryopyrin-associated periodic syndrome as established in the first open-label, phase-3 pivotal study (24-week results). Clin. Exp. Rheumatol. 2013, 31, 302–309. [Google Scholar] [PubMed]
- Yokota, S.; Imagawa, T.; Nishikomori, R.; Takada, H.; Abrams, K.; Lheritier, K.; Heike, T.; Hara, T. Long-term safety and efficacy of canakinumab in cryopyrin-associated periodic syndrome: Results from an open-label, phase III pivotal study in Japanese patients. Clin. Exp. Rheumatol. 2016, 35 (Suppl. 108), 19–26. [Google Scholar]
- Kullenberg, T.; Löfqvist, M.; Leinonen, M.; Goldbach-Mansky, R.; Olivecrona, H. Long-term safety profile of anakinra in patients with severe cryopyrin-associated periodic syndromes. Rheumatology 2016, 55, 1499–1506. [Google Scholar] [CrossRef] [Green Version]
- Goldbach-Mansky, R.; Dailey, N.J.; Canna, S.W.; Gelabert, A.; Jones, J.; Rubin, B.I.; Kim, H.J.; Brewer, C.; Zalewski, C.; Wiggs, E.; et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N. Engl. J. Med. 2006, 355, 581–592. [Google Scholar] [CrossRef]
- Sibley, C.H.; Chioato, A.; Felix, S.; Colin, L.; Chakraborty, A.; Plass, N.; Rodriguez-Smith, J.; Brewer, C.; King, K.; Zalewski, C.; et al. A 24-month open-label study of canakinumab in neonatal-onset multisystem inflammatory disease. Ann. Rheum. Dis. 2015, 74, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Wikén, M.; Hallén, B.; Kullenberg, T.; Koskinen, L.O. Development and effect of antibodies to anakinra during treatment of severe CAPS: Sub-analysis of a long-term safety and efficacy study. Clin. Rheumatol. 2018, 37, 3381–3386. [Google Scholar] [CrossRef] [PubMed]
- De Benedetti, F.; Gattorno, M.; Anton, J.; Ben-Chetrit, E.; Frenkel, J.; Hoffman, H.M.; Koné-Paut, I.; Lachmann, H.J.; Ozen, S.; Simon, A.; et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N. Engl. J. Med. 2018, 378, 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Gattorno, M.; Pelagatti, M.A.; Meini, A.; Obici, L.; Barcellona, R.; Federici, S.; Buoncompagni, A.; Plebani, A.; Merlini, G.; Martini, A. Persistent efficacy of anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum. 2008, 58, 1516–1520. [Google Scholar] [CrossRef]
- Gattorno, M.; Obici, L.; Cattalini, M.; Tormey, V.; Abrams, K.; Davis, N.; Speziale, A.; Bhansali, S.G.; Martini, A.; Lachmann, H.J. Canakinumab treatment for patients with active recurrent or chronic TNF receptor-associated periodic syndrome (TRAPS): An open-label, phase II study. Ann. Rheum. Dis. 2017, 76, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Torene, R.; Nirmala, N.; Obici, L.; Cattalini, M.; Tormey, V.; Caorsi, R.; Starck-Schwertz, S.; Letzkus, M.; Hartmann, N.; Abrams, K.; et al. Canakinumab reverses overexpression of inflammatory response genes in tumour necrosis factor receptor-associated periodic syndrome. Ann. Rheum. Dis. 2017, 76, 303–309. [Google Scholar] [CrossRef]
- Brik, R.; Butbul-Aviel, Y.; Lubin, S.; Ben Dayan, E.; Rachmilewitz-Minei, T.; Tseng, L.; Hashkes, P.J. Canakinumab for the treatment of children with colchicine-resistant familial Mediterranean fever: A 6-month open-label, single-arm pilot study. Arthritis Rheumatol. 2014, 66, 3241–3243. [Google Scholar] [CrossRef] [PubMed]
- Gül, A.; Ozdogan, H.; Erer, B.; Ugurlu, S.; Kasapcopur, O.; Davis, N.; Sevgi, S. Efficacy and safety of canakinumab in adolescents and adults with colchicine-resistant familial Mediterranean fever. Arthritis Res. Ther. 2015, 17, 243. [Google Scholar] [CrossRef]
- Arostegui, J.I.; Anton, J.; Calvo, I.; Robles, A.; Iglesias, E.; López-Montesinos, B.; Banchereau, R.; Hong, S.; Joubert, Y.; Junge, G.; et al. Open-label phase II Study to assess the efficacy and safety of canakinumab treatment in active hyperimmunoglobulinemia D with periodic fever syndrome. Arthritis Rheumatol. 2017, 69, 1679–1688. [Google Scholar] [CrossRef]
- Caroli, F.; Pontillo, A.; D’Osualdo, A.; Travan, L.; Ceccherini, I.; Crovella, S.; Alessio, M.; Stabile, A.; Gattorno, M.; Tommasini, A.; et al. Clinical and genetic characterization of Italian patients affected by CINCA syndrome. Rheumatology 2007, 46, 473–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neven, B.; Marvillet, I.; Terrada, C.; Ferster, A.; Boddaert, N.; Couloignier, V.; Pinto, G.; Pagnier, A.; Bodemer, C.; Bodaghi, B.; et al. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2010, 62, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibley, C.H.; Plass, N.; Snow, J.; Wiggs, E.A.; Brewer, C.C.; King, K.A.; Zalewski, C.; Kim, H.J.; Bishop, R.; Hill, S.; et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: A cohort study to determine three- and five-year outcomes. Arthritis Rheum. 2012, 64, 2375–2386. [Google Scholar] [CrossRef] [PubMed]
- Tran, T. Muckle–Wells syndrome: Clinical perspectives. Open Access Rheumatol. Res. Rev. 2017, 9, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Kuemmerle-Deschner, J.B.; Tyrrell, P.N.; Koetter, I.; Wittkowski, H.; Bialkowski, A.; Tzaribachev, N.; Lohse, P.; Koitchev, A.; Deuter, C.; Foell, D.; et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum. 2011, 63, 840–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuemmerle-Deschner, J.B.; Wittkowski, H.; Tyrrell, P.N.; Koetter, I.; Lohse, P.; Ummenhofer, K.; Reess, F.; Hansmann, S.; Koitschev, A.; Deuter, C.; et al. Treatment of Muckle-Wells syndrome: Analysis of two IL-1-blocking regimens. Arthritis Res. Ther. 2013, 15, R64. [Google Scholar] [CrossRef]
- Dalgic, B.; Egritas, O.; Sari, S.; Cuisset, L. A variant Muckle-Wells syndrome with a novel mutation in CIAS1 gene responding to anakinra. Pediatr. Nephrol. 2007, 22, 1391–1394. [Google Scholar] [CrossRef]
- Maksimovic, L.; Stirnemann, J.; Caux, F.; Ravet, N.; Rouaghe, S.; Cuisset, L.; Letellier, E.; Grateau, G.; Morin, A.-S.; Fain, O. New CIAS1 mutation and anakinra efficacy in overlapping of Muckle-Wells and familial cold autoinflammatory syndromes. Rheumatology 2008, 47, 309–310. [Google Scholar] [CrossRef]
- Marchica, C.; Zawawi, F.; Basodan, D.; Scuccimarri, R.; Daniel, S.J. Resolution of unilateral sensorineural hearing loss in a pediatric patient with a severe phenotype of Muckle-Wells syndrome treated with Anakinra: A case report and review of the literature. J. Otolaryngol. Head Neck Surg. 2018, 47, 9. [Google Scholar] [CrossRef]
- Stew, B.T.; Fishpool, S.J.C.; Owens, D.; Quine, S. Muckle-Wells syndrome: A treatable cause of congenital sensorineural hearing loss. B-Ent 2013, 9, 161–163. [Google Scholar]
- Yamazaki, T.; Masumoto, J.; Agematsu, K.; Sawai, N.; Kobayashi, S.; Shigemura, T.; Yasui, K.; Koike, K. Anakinra improves sensory deafness in a Japanese patient with Muckle-Wells syndrome, possibly by inhibiting the cryopyrin inflammasome. Arthritis Rheum. 2008, 58, 864–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigante, D.; Lopalco, G.; Vitale, A.; Lucherini, O.M.; De Clemente, C.; Caso, F.; Emmi, G.; Costa, L.; Silvestri, E.; Andreozzi, L.; et al. Key facts and hot spots on tumor necrosis factor receptor-associated periodic syndrome. Clin. Rheumatol. 2014, 33, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Magnotti, F.; Vitale, A.; Rigante, D.; Lucherini, O.M.; Cimaz, R.; Muscari, I.; Granados Afonso de Faria, A.; Frediani, B.; Galeazzi, M.; Cantarini, L. The most recent advances in pathophysiology and management of tumour necrosis factor receptor-associated periodic syndrome (TRAPS): Personal experience and literature review. Clin. Exp. Rheumatol. 2013, 31, 141–149. [Google Scholar] [PubMed]
- Lopalco, G.; Rigante, D.; Vitale, A.; Frediani, B.; Iannone, F.; Cantarini, L. Tumor necrosis factor receptor-associated periodic syndrome managed with the couple canakinumab-alendronate. Clin. Rheumatol. 2015, 34, 807–809. [Google Scholar] [CrossRef]
- Cantarini, L.; Lopalco, G.; Vitale, A.; Caso, F.; Lapadula, G.; Iannone, F.; Galeazzi, M.; Rigante, D. Delights and let-downs in the management of tumor necrosis factor receptor-associated periodic syndrome: The canakinumab experience in a patient with a high-penetrance T50M TNFRSF1A variant. Int. J. Rheum. Dis. 2015, 18, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Rigante, D.; Lopalco, G.; Tarantino, G.; Compagnone, A.; Fastiggi, M.; Cantarini, L. Non-canonical manifestations of familial Mediterranean fever: A changing paradigm. Clin. Rheumatol. 2015, 34, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, M. Familial Mediterranean fever, review of the literature. Clin. Rheumatol. 2017, 36, 1707–1713. [Google Scholar] [CrossRef] [PubMed]
- Gülez, N.; Makay, B.; Sözeri, B. Long-Term Effectıveness and safety of canakınumab ın pedıatrıc famılıal Medıterranean fever patıents. Mod. Rheumatol. 2018, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Laskari, K.; Boura, P.; Dalekos, G.N.; Garyfallos, A.; Karokis, D.; Pikazis, D.; Settas, L.; Skarantavos, G.; Tsitsami, E.; Sfikakis, P.P. Long-term beneficial effect of canakinumab in colchicine-resistant familial Mediterranean fever. J. Rheumatol. 2017, 44, 102–109. [Google Scholar] [CrossRef]
- Başaran, Ö.; Uncu, N.; Çelikel, B.A.; Taktak, A.; Gür, G.; Cakar, N. Interleukin-1 targeting treatment in familial Mediterranean fever: An experience of pediatric patients. Mod. Rheumatol. 2015, 25, 621–624. [Google Scholar] [CrossRef]
- Cetin, P.; Sari, I.; Sozeri, B.; Cam, O.; Birlik, M.; Akkoc, N.; Onen, F.; Akar, S. Efficacy of Interleukin-1 Targeting Treatments in Patients with Familial Mediterranean Fever. Inflammation 2015, 38, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Özçakar, Z.B.; Özdel, S.; Yılmaz, S.; Kurt-Şükür, E.D.; Ekim, M.; Yalçınkaya, F. Anti-IL-1 treatment in familial Mediterranean fever and related amyloidosis. Clin. Rheumatol. 2016, 35, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Kuis, W.; Duran, M.; de Koning, T.J.; van Royen-Kerkhof, A.; Romeijn, G.J.; Frenkel, J.; Dorland, L.; de Barse, M.M.J.; Huijbers, W.A.R.; et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat. Genet. 1999, 22, 175–177. [Google Scholar] [CrossRef] [PubMed]
- Ter Haar, N.M.; Jeyaratnam, J.; Lachmann, H.J.; Simon, A.; Brogan, P.A.; Doglio, M.; Cattalini, M.; Anton, J.; Modesto, C.; Quartier, P.; et al. The phenotype and genotype of mevalonate kinase deficiency: A series of 114 cases from the Eurofever registry. Arthritis Rheumatol. 2016, 68, 2795–2805. [Google Scholar] [CrossRef] [PubMed]
- Bodar, E.J.; Kuijk, L.M.; Drenth, J.P.H.; van der Meer, J.W.M.; Simon, A.; Frenkel, J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann. Rheum. Dis. 2011, 70, 2155–2158. [Google Scholar] [CrossRef] [PubMed]
- van der Hilst, J.C.H.; Bodar, E.J.; Barron, K.S.; Frenkel, J.; Drenth, J.P.H.; van der Meer, J.W.M.; Simon, A. International HIDS Study Group Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine 2008, 87, 301–310. [Google Scholar] [CrossRef]
- Galeotti, C.; Meinzer, U.; Quartier, P.; Rossi-Semerano, L.; Bader-Meunier, B.; Pillet, P.; Koné-Paut, I. Efficacy of interleukin-1-targeting drugs in mevalonate kinase deficiency. Rheumatology 2012, 51, 1855–1859. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Yoshioka, K.; Nishikomori, R.; Sakai, H.; Abe, J.; Yamashita, Y.; Hiramoto, R.; Morimoto, A.; Ishii, E.; Arakawa, H.; et al. National survey of Japanese patients with mevalonate kinase deficiency reveals distinctive genetic and clinical characteristics. Mod. Rheumatol. 2018, 29, 181–187. [Google Scholar] [CrossRef]
- Ozen, S.; Kuemmerle-Deschner, J.B.; Cimaz, R.; Livneh, A.; Quartier, P.; Kone-Paut, I.; Zeft, A.; Spalding, S.; Gul, A.; Hentgen, V.; et al. International retrospective chart review of treatment patterns in severe familial Mediterranean fever, tumor necrosis factor receptor-associated periodic syndrome, and mevalonate kinase deficiency/hyperimmunoglobulinemia D syndrome. Arthritis Care Res. 2017, 69, 578–586. [Google Scholar] [CrossRef]
- Ter Haar, N.; Lachmann, H.; Özen, S.; Woo, P.; Uziel, Y.; Modesto, C.; Koné-Paut, I.; Cantarini, L.; Insalaco, A.; Neven, B.; et al. Treatment of autoinflammatory diseases: Results from the Eurofever Registry and a literature review. Ann. Rheum. Dis. 2013, 72, 678–685. [Google Scholar] [CrossRef]
- Vitale, A.; Insalaco, A.; Sfriso, P.; Lopalco, G.; Emmi, G.; Cattalini, M.; Manna, R.; Cimaz, R.; Priori, R.; Talarico, R.; et al. A snapshot on the on-label and off-label use of the interleukin-1 inhibitors in Italy among rheumatologists and pediatric rheumatologists: A nationwide multi-center retrospective observational study. Front. Pharmacol. 2016, 7, 380. [Google Scholar] [CrossRef]
- Rossi-Semerano, L.; Fautrel, B.; Wendling, D.; Hachulla, E.; Galeotti, C.; Semerano, L.; Touitou, I.; Koné-Paut, I. Tolerance and efficacy of off-label anti-interleukin-1 treatments in France: A nationwide survey. Orphanet J. Rare Dis. 2015, 10, 19. [Google Scholar] [CrossRef]
- Topaloglu, R.; Batu, E.D.; Orhan, D.; Ozen, S.; Besbas, N. Anti-interleukin 1 treatment in secondary amyloidosis associated with autoinflammatory diseases. Pediatr. Nephrol. 2016, 31, 633–640. [Google Scholar] [CrossRef]
- Gattorno, M.; Piccini, A.; Lasigliè, D.; Tassi, S.; Brisca, G.; Carta, S.; Delfino, L.; Ferlito, F.; Pelagatti, M.A.; Caroli, F.; et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2008, 58, 1505–1515. [Google Scholar] [CrossRef]
- Brachat, A.H.; Grom, A.A.; Wulffraat, N.; Brunner, H.I.; Quartier, P.; Brik, R.; McCann, L.; Ozdogan, H.; Rutkowska-Sak, L.; Schneider, R.; et al. Early changes in gene expression and inflammatory proteins in systemic juvenile idiopathic arthritis patients on canakinumab therapy. Arthritis Res. Ther. 2017, 19, 13. [Google Scholar] [CrossRef]
- Feist, E.; Quartier, P.; Fautrel, B.; Schneider, R.; Sfriso, P.; Efthimiou, P.; Cantarini, L.; Lheritier, K.; Leon, K.; Karyekar, C.S.; et al. Efficacy and safety of canakinumab in patients with Still’s disease: Exposure-response analysis of pooled systemic juvenile idiopathic arthritis data by age groups. Clin. Exp. Rheumatol. 2018, 36, 668–675. [Google Scholar]
- Grom, A.A.; Ilowite, N.T.; Pascual, V.; Brunner, H.I.; Martini, A.; Lovell, D.; Ruperto, N.; Abrams, K.; Leon, K.; Lheritier, K.; et al. Rate and clinical presentation of macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis treated with canakinumab. Arthritis Rheumatol. 2016, 68, 218–228. [Google Scholar] [CrossRef]
- Ilowite, N.; Porras, O.; Reiff, A.; Rudge, S.; Punaro, M.; Martin, A.; Allen, R.; Harville, T.; Sun, Y.-N.; Bevirt, T.; et al. Anakinra in the treatment of polyarticular-course juvenile rheumatoid arthritis: Safety and preliminary efficacy results of a randomized multicenter study. Clin. Rheumatol. 2009, 28, 129–137. [Google Scholar] [CrossRef]
- Kimura, Y.; Grevich, S.; Beukelman, T.; Morgan, E.; Nigrovic, P.A.; Mieszkalski, K.; Graham, T.B.; Ibarra, M.; Ilowite, N.; Klein-Gitelman, M.; et al. Pilot study comparing the Childhood Arthritis & Rheumatology Research Alliance (CARRA) systemic Juvenile Idiopathic Arthritis consensus treatment plans. Pediatr. Rheumatol. Online J. 2017, 15, 23. [Google Scholar]
- Quartier, P.; Allantaz, F.; Cimaz, R.; Pillet, P.; Messiaen, C.; Bardin, C.; Bossuyt, X.; Boutten, A.; Bienvenu, J.; Duquesne, A.; et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann. Rheum. Dis. 2011, 70, 747–754. [Google Scholar] [CrossRef]
- Ruperto, N.; Brunner, H.I.; Quartier, P.; Constantin, T.; Wulffraat, N.; Horneff, G.; Brik, R.; McCann, L.; Kasapcopur, O.; Rutkowska-Sak, L.; et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N. Engl. J. Med. 2012, 367, 2396–2406. [Google Scholar] [CrossRef] [PubMed]
- Ruperto, N.; Quartier, P.; Wulffraat, N.; Woo, P.; Ravelli, A.; Mouy, R.; Bader-Meunier, B.; Vastert, S.J.; Noseda, E.; D’Ambrosio, D.; et al. A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 2012, 64, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Ruperto, N.; Brunner, H.I.; Quartier, P.; Constantin, T.; Wulffraat, N.M.; Horneff, G.; Kasapcopur, O.; Schneider, R.; Anton, J.; Barash, J.; et al. Canakinumab in patients with systemic juvenile idiopathic arthritis and active systemic features: Results from the 5-year long-term extension of the phase III pivotal trials. Ann. Rheum. Dis. 2018, 77, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Hedrich, C.M.; Bruck, N.; Fiebig, B.; Gahr, M. Anakinra: A safe and effective first-line treatment in systemic onset juvenile idiopathic arthritis (SoJIA). Rheumatol. Int. 2012, 32, 3525–3530. [Google Scholar] [CrossRef]
- Horneff, G.; Schulz, A.C.; Klotsche, J.; Hospach, A.; Minden, K.; Foeldvari, I.; Trauzeddel, R.; Ganser, G.; Weller-Heinemann, F.; Haas, J.P. Experience with etanercept, tocilizumab and interleukin-1 inhibitors in systemic onset juvenile idiopathic arthritis patients from the BIKER registry. Arthritis Res. Ther. 2017, 19, 256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearsley-Fleet, L.; Beresford, M.W.; Davies, R.; De Cock, D.; Baildam, E.; Foster, H.E.; Southwood, T.R.; Thomson, W.; Hyrich, K.L. Short-term outcomes in patients with systemic juvenile idiopathic arthritis treated with either tocilizumab or anakinra. Rheumatology 2019, 58, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Lequerré, T.; Quartier, P.; Rosellini, D.; Alaoui, F.; De Bandt, M.; Mejjad, O.; Koné-Paut, I.; Michel, M.; Dernis, E.; Khellaf, M.; et al. Interleukin-1 receptor antagonist (anakinra) treatment in patients with systemic-onset juvenile idiopathic arthritis or adult onset Still disease: Preliminary experience in France. Ann. Rheum. Dis. 2008, 67, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Nigrovic, P.A.; Mannion, M.; Prince, F.H.M.; Zeft, A.; Rabinovich, C.E.; van Rossum, M.A.J.; Cortis, E.; Pardeo, M.; Miettunen, P.M.; Janow, G.; et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: Report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011, 63, 545–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardeo, M.; Pires Marafon, D.; Insalaco, A.; Bracaglia, C.; Nicolai, R.; Messia, V.; De Benedetti, F. Anakinra in systemic juvenile idiopathic arthritis: A single-center experience. J. Rheumatol. 2015, 42, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Pontikaki, I.; Gattinara, M.; Ardoino, I.; Donati, C.; Boracchi, P.; Meroni, P.L.; Gerloni, V. Drug survival and reasons for discontinuation of the first course of biological therapy in 301 juvenile idiopathic arthritis patients. Reumatismo 2014, 65, 278–285. [Google Scholar] [CrossRef]
- Vastert, S.J.; de Jager, W.; Noordman, B.J.; Holzinger, D.; Kuis, W.; Prakken, B.J.; Wulffraat, N.M. Effectiveness of first-line treatment with recombinant interleukin-1 receptor antagonist in steroid-naive patients with new-onset systemic juvenile idiopathic arthritis: Results of a prospective cohort study. Arthritis Rheumatol. 2014, 66, 1034–1043. [Google Scholar] [CrossRef]
- Marrani, E.; Burns, J.C.; Cimaz, R. How should we classify Kawasaki disease? Front. Immunol. 2018, 9, 2974. [Google Scholar] [CrossRef]
- Principi, N.; Rigante, D.; Esposito, S. The role of infection in Kawasaki syndrome. J. Infect. 2013, 67, 1–10. [Google Scholar] [CrossRef]
- De Rosa, G.; Pardeo, M.; Rigante, D. Current recommendations for the pharmacologic therapy in Kawasaki syndrome and management of its cardiovascular complications. Eur. Rev. Med. Pharmacol. Sci. 2007, 11, 301–308. [Google Scholar]
- Rigante, D.; Valentini, P.; Rizzo, D.; Leo, A.; De Rosa, G.; Onesimo, R.; De Nisco, A.; Angelone, D.F.; Compagnone, A.; Delogu, A.B. Responsiveness to intravenous immunoglobulins and occurrence of coronary artery abnormalities in a single-center cohort of Italian patients with Kawasaki syndrome. Rheumatol. Int. 2010, 30, 841–846. [Google Scholar] [CrossRef] [Green Version]
- Rigante, D.; Andreozzi, L.; Fastiggi, M.; Bracci, B.; Natale, M.F.; Esposito, S. Critical overview of the risk scoring systems to predict non-responsiveness to intravenous immunoglobulin in Kawasaki syndrome. Int. J. Mol. Sci. 2016, 17, 278. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Agrawal, D.K. Kawasaki disease: Etiopathogenesis and novel treatment strategies. Expert Rev. Clin. Immunol. 2017, 13, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Blonz, G.; Lacroix, S.; Benbrik, N.; Warin-Fresse, K.; Masseau, A.; Trewick, D.; Hamidou, M.; Stephan, J.-L.; Néel, A. Severe late-onset Kawasaki disease successfully treated with anakinra. J. Clin. Rheumatol. 2018, 1. [Google Scholar] [CrossRef]
- Cohen, S.; Tacke, C.E.; Straver, B.; Meijer, N.; Kuipers, I.M.; Kuijpers, T.W. A child with severe relapsing Kawasaki disease rescued by IL-1 receptor blockade and extracorporeal membrane oxygenation. Ann. Rheum. Dis. 2012, 71, 2059–2061. [Google Scholar] [CrossRef] [PubMed]
- Guillaume, M.-P.; Reumaux, H.; Dubos, F. Usefulness and safety of anakinra in refractory Kawasaki disease complicated by coronary artery aneurysm. Cardiol. Young 2018, 28, 739–742. [Google Scholar] [CrossRef]
- Sánchez-Manubens, J.; Gelman, A.; Franch, N.; Teodoro, S.; Palacios, J.R.; Rudi, N.; Rivera, J.; Antón, J. A child with resistant Kawasaki disease successfully treated with anakinra: A case report. BMC Pediatr. 2017, 17, 102. [Google Scholar] [CrossRef] [PubMed]
- Shafferman, A.; Birmingham, J.D.; Cron, R.Q. High dose Anakinra for treatment of severe neonatal Kawasaki disease: A case report. Pediatr. Rheumatol. Online J. 2014, 12, 26. [Google Scholar] [CrossRef]
- Koné-Paut, I.; Cimaz, R.; Herberg, J.; Bates, O.; Carbasse, A.; Saulnier, J.P.; Maggio, M.C.; Anton, J.; Piram, M. The use of interleukin 1 receptor antagonist (anakinra) in Kawasaki disease: A retrospective cases series. Autoimmun. Rev. 2018, 17, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, C.; Knight, A.; Nordström, D.; Pettersson, T.; Fransson, J.; Florin-Robertsson, E.; Pilström, B. Injection-site reactions upon Kineret (anakinra) administration: Experiences and explanations. Rheumatol. Int. 2012, 32, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Sota, J.; Vitale, A.; Insalaco, A.; Sfriso, P.; Lopalco, G.; Emmi, G.; Cattalini, M.; Manna, R.; Cimaz, R.; Priori, R.; et al. Safety profile of the interleukin-1 inhibitors anakinra and canakinumab in real-life clinical practice: A nationwide multicenter retrospective observational study. Clin. Rheumatol. 2018, 37, 2233–2240. [Google Scholar] [CrossRef]


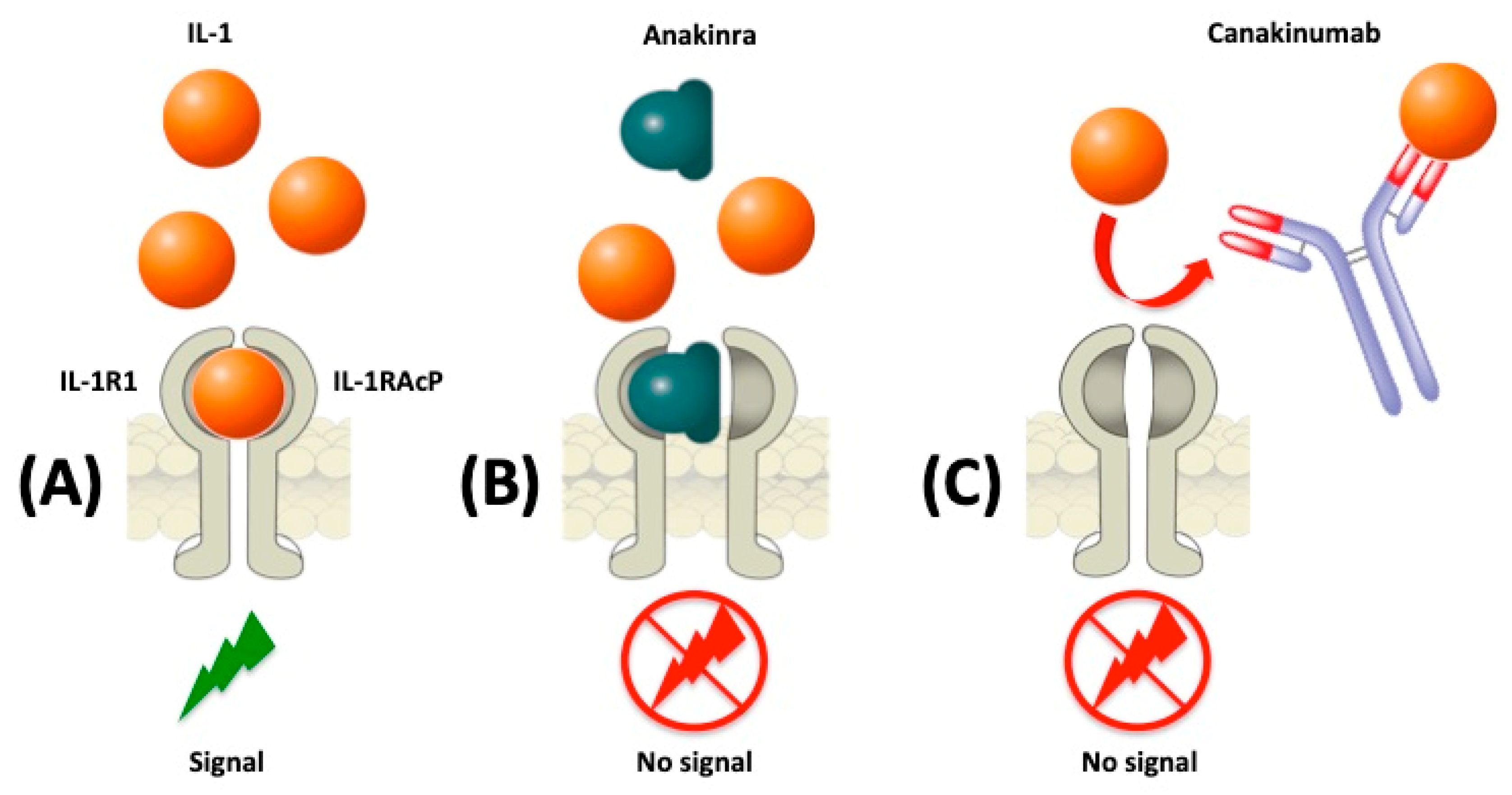{kind=link}
| Authors | Title | Study Design | Population | Drug |
|---|---|---|---|---|
| Cryopyrin-Associated Periodic Syndrome (Including CINCA/NOMID and MWS) | ||||
| Goldbach-Mansky et al., 2006 [33] | Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. | Clinical trial | Pediatric + adults (total n = 18) | ANA (1 to 2 mg/kg/day s.c.) |
| Goldbach-Mansky et al., 2006 [33] | Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. | Clinical trial | Pediatric (n = 15) + adults (n = 3) | ANA (1 to 2 mg/kg/day s.c.) |
| Imagawa et al., 2013 [30] | Safety and efficacy of Canakinumab in Japanese patients with phenotypes of cryopyrin-associated periodic syndrome as established in the first open-label, phase-3 pivotal study (24-week results). | Phase-3 pivotal study | Pediatric + adults (total n = 19) | CANA (150 mg s.c. (or 2 mg/kg for patients with a body weight ≤ 40 kg) every 8 weeks for 24 weeks) |
| Koné-Paut et al., 2011 [29] | Sustained remission of symptoms and improved health-related quality of life in patients with cryopyrin-associated periodic syndrome treated with Canakinumab: Results of a double-blind placebo-controlled randomized withdrawal study. | Double-blind placebo-controlled randomized withdrawal study | Pediatric + adults (total n = 35) | CANA (150 mg s.c. at 8-week intervals) |
| Kuemmerle-Deschner et al., 2011 [27] | Canakinumab (ACZ885, a fully human IgG1 anti-IL-1β mAb) induces sustained remission in pediatric patients with cryopyrin-associated periodic syndrome (CAPS). | Phase II, open-label study | Pediatric (n = 7) | CANA (2 mg/kg s.c. for patients ≤ 40 kg or 150 mg s.c. for patients > 40 kg; re-dosing upon each relapse) |
| Kuemmerle-Deschner et al., 2011 [28] | Two-year results from an open-label multicentre phase III study evaluating the safety and efficacy of Canakinumab in patients with cryopyrin-associated periodic syndrome across different severity phenotypes. | Open-label, multicentre, phase III study | Pediatric (n = 47) + adults (n = 119) | CANA (s.c. 150 mg or 2 mg/kg for patients ≤ 40 kg every 8 weeks for up to 2 years) |
| Kullenberg et al., 2016 [32] | Long-term safety profile of Anakinra in patients with severe cryopyrin-associated periodic syndrome. | Prospective, open-label, single center trial | Pediatric (n = 36) + adults (n = 7) | ANA (starting dose 0.5 to 2.5 mg/kg/day) |
| Lachmann et al., 2009 [26] | Use of Canakinumab in the cryopyrin-associated periodic syndrome. | Double-blind, placebo-controlled, randomized withdrawal study | Pediatric (n = 4) + adults (n = 31) | CANA (part 1: 150 mg s.c.; part 2: Either 150 mg of CANA or placebo every 8 weeks for up to 24 week) |
| Sibley et al., 2017 [34] | A 24-month open-label study of Canakinumab in neonatal-onset multisystem inflammatory disease. | Open-label study | Pediatric (n = 2) + adults (n = 4) | CANA (150 mg (or 2 mg/kg in patients ≤ 40 kg) or 300 mg (or 4 mg/kg) with escalation up to 600 mg (or 8 mg/kg) every 4 weeks) |
| Wikén et al., 2018 [35] | Development and effect of antibodies to Anakinra during treatment of severe CAPS: Sub-analysis of a long-term safety and efficacy study. | Post hoc analysis on data from a prospective, open-label, single-center, clinical cohort study | Pediatric (n = 36) + adults (n = 7) | ANA (1.0 to 2.4 mg/kg/day s.c., increased to 2.0 to 5.0 mg/kg/day according to clinical need (median 3.1 mg/kg/day)) |
| Yokota et al., 2017 [31] | Long-term safety and efficacy of Canakinumab in cryopyrin-associated periodic syndrome: results from an open-label, phase III pivotal study in Japanese patients. | Open-label, phase III pivotal study | Pediatric + adults (n = 35) | CANA (2 to 8 mg/kg s.c. every 8 weeks) |
| Tumor Necrosis Factor Receptor-Associated Periodic Syndrome | ||||
| De Benedetti et al., 2018 [36] | Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. | Clinical trial | Pediatric (TRAPS n = 27) + adult (TRAPS n = 19) | CANA (initial phase 150 mg/4 weeks) |
| Gattorno et al., 2008 [37] | Persistent efficacy of Anakinra in patients with tumor necrosis factor receptor-associated periodic syndrome. | Clinical trial | Pediatric (n = 4) + adult (n = 1) | ANA (1.5 mg/kg/day) |
| Gattorno et al., 2017 [38] | Canakinumab treatment for patients with active recurrent or chronic TNF receptor-associated periodic syndrome (TRAPS): An open-label, phase II study. | Open-label, phase II study | Pediatric (n = 6) + adults (n = 14) | CANA (150 mg every 4 weeks for 4 months (2 mg/kg for patients ≤ 40 kg)) |
| Torene et al., 2017 [39] | Canakinumab reverses overexpression of inflammatory response genes in tumor necrosis factor receptor-associated periodic syndrome. | Open-label, multicentre, proof-of-concept study (gene analysis) | Pediatric (n.a.) + adults (n.a.) | CANA (150 mg every 4 weeks for 4 months) |
| Familial Mediterranean Fever | ||||
| Brik et al., 2014 [40] | Canakinumab for the treatment of children with colchicine-resistant familial Mediterranean fever: A 6-month open-label, single-arm pilot study. | Open-label, single-arm pilot study | Pediatric (n = 7) | CANA (2 mg/kg (maximum 150 mg); the dose was doubled to 4 mg/kg (maximum 300 mg) if an attack occurred between day 1 and day 29) |
| De Benedetti et al., 2018 [36] | Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. | Clinical trial | Pediatric (crFMF n = 29) + adult (crFMF n = 34) | CANA (initial phase 150 mg every 4 weeks) |
| Gül et al., 2015 [41] | Efficacy and safety of Canakinumab in adolescents and adults with colchicine-resistant familial Mediterranean fever. | Open-label pilot study | Pediatric (n.a.) + adults (n.a.) | CANA (3 injections 150 mg s.c. injections every 4 weeks) |
| Hyperimmunoglobulin D Syndrome/Mevalonate Kinase Deficiency | ||||
| Arostegui et al., 2017 [42] | Open-label phase II study to assess the efficacy and safety of Canakinumab treatment in active hyperimmunoglobulinemia D with periodic fever syndrome. | Clinical trial | Pediatric (n = 6) + adults (n = 3) | CANA (300 mg or 4 mg/kg every 6 weeks) |
| De Benedetti et al., 2018 [36] | Canakinumab for the treatment of autoinflammatory recurrent fever syndromes. | Clinical trial | Pediatric (MKD n = 54) + adult (MKD n = 18) | CANA (initial phase 150 mg/4 weeks) |
| Systemic Juvenile Idiopathic Arthritis | ||||
|---|---|---|---|---|
| Authors | Title | Study Design | Population | Drug |
| Brachat et al., 2017 [77] | Early changes in gene expression and inflammatory proteins in systemic juvenile idiopathic arthritis patients on Canakinumab therapy. | Gene expression analysis (data from the two phase-3 trials evaluating CANA for SJIA) | Pediatric (n.a.) | CANA (Trial 1: Patients were randomly assigned to a single s.c. dose of CANA (4 mg/kg) or placebo. Trial 2: Open-label phase (s.c. CANA 4 mg/kg every 4 weeks for up to 32 weeks) + withdrawal phase) |
| Feist et al., 2018 [78] | Efficacy and safety of Canakinumab in patients with Still’s disease: Exposure-response analysis of pooled systemic juvenile idiopathic arthritis data by age groups. | Pooled results of clinical trials | Pediatric (n = 216) + adolescents (n = 56) + adult (n = 29) | CANA (Study 1: Single s.c. CANA at 4 mg/kg (maximum of 300 mg) or placebo; Study 2: CANA 4 mg/kg (maximum dose of 300 mg) every 4 weeks for up to 8 months + second double-blind randomized placebo-controlled phase; Study 3: S.c. CANA 4 mg/kg every 4 weeks for 12 weeks (patients had received CANA in either Study 1 or Study 2, with an additional cohort of CANA-naïve patients); Study 4: Dose-ranging study (0.5–9 mg/kg) |
| Gattorno et al., 2008 [76] | The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis. | Clinical study | Pediatric (n = 22) | ANA (starting dosage of 1 mg/kg/day, s.c. (maximum 100 mg)) |
| Grom et al., 2016 [79] | Rate and clinical presentation of macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis treated with Canakinumab. | Pooled analysis | Pediatric (n = 21) | CANA (n.a.) |
| Ilowite et al., 2009 [80] | Anakinra in the treatment of polyarticular-course juvenile rheumatoid arthritis: Safety and preliminary efficacy results of a randomized multicenter study. | 2-week open-label run-in phase | Pediatric (n = 86) | ANA (1 mg/kg daily, maximum 100 mg/day) |
| Kimura et al., 2017 [81] | Pilot study comparing the Childhood Arthritis & Rheumatology Research Alliance (CARRA) systemic juvenile idiopathic arthritis consensus treatment plans. | Pilot interventional study | Pediatric (n = 30; IL-1 inhibitors n = 12) | ANA (CANA) (median initial dose of ANA 2.93 (IQR 2–3.6)) |
| Quartier et al., 2011 [82] | A multicentre randomized double-blind placebo-controlled trial with the interleukin-1 receptor antagonist Anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). | Multicentre, randomized, double-blind, placebo-controlled trial | Pediatric (n = 24) | ANA (2 mg/kg s.c. daily, maximum 100 mg) |
| Ruperto et al. (trial 1), 2012 [83] | Two randomized trials of Canakinumab in systemic juvenile idiopathic arthritis. | 2 phase III trials | Pediatric (n = 84 + 177) | CANA (s.c., 4 mg/kg per month (maximum dose, 300 mg)) |
| Ruperto et al. (trial 2), 2012 [84] | A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of Canakinumab in systemic juvenile idiopathic arthritis with active systemic features. | Phase II, multicenter, open-label, dosage-escalation study | Pediatric (n = 23) | CANA (single s.c. dose of 0.5 to 9 mg/kg) |
| Ruperto et al., 2018 [85] | Canakinumab in patients with systemic juvenile idiopathic arthritis and active systemic features: Results from the 5-year long-term extension of the phase III pivotal trials. | 5-year long-term extension of the phase III pivotal trials. | Pediatric (n = 177; 144 in the long-term extension phase) | CANA (4 mg/kg s.c. every 4 weeks (maximum dose 300 mg); in the long-term extension, tapered to 2 mg/kg every 4 weeks in patients who were glucocorticoid free as per physicians’ judgement) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bettiol, A.; Lopalco, G.; Emmi, G.; Cantarini, L.; Urban, M.L.; Vitale, A.; Denora, N.; Lopalco, A.; Cutrignelli, A.; Lopedota, A.; et al. Unveiling the Efficacy, Safety, and Tolerability of Anti-Interleukin-1 Treatment in Monogenic and Multifactorial Autoinflammatory Diseases. Int. J. Mol. Sci. 2019, 20, 1898. https://doi.org/10.3390/ijms20081898
Bettiol A, Lopalco G, Emmi G, Cantarini L, Urban ML, Vitale A, Denora N, Lopalco A, Cutrignelli A, Lopedota A, et al. Unveiling the Efficacy, Safety, and Tolerability of Anti-Interleukin-1 Treatment in Monogenic and Multifactorial Autoinflammatory Diseases. International Journal of Molecular Sciences. 2019; 20(8):1898. https://doi.org/10.3390/ijms20081898
Chicago/Turabian StyleBettiol, Alessandra, Giuseppe Lopalco, Giacomo Emmi, Luca Cantarini, Maria Letizia Urban, Antonio Vitale, Nunzio Denora, Antonio Lopalco, Annalisa Cutrignelli, Angela Lopedota, and et al. 2019. "Unveiling the Efficacy, Safety, and Tolerability of Anti-Interleukin-1 Treatment in Monogenic and Multifactorial Autoinflammatory Diseases" International Journal of Molecular Sciences 20, no. 8: 1898. https://doi.org/10.3390/ijms20081898
APA StyleBettiol, A., Lopalco, G., Emmi, G., Cantarini, L., Urban, M. L., Vitale, A., Denora, N., Lopalco, A., Cutrignelli, A., Lopedota, A., Venerito, V., Fornaro, M., Vannacci, A., Rigante, D., Cimaz, R., & Iannone, F. (2019). Unveiling the Efficacy, Safety, and Tolerability of Anti-Interleukin-1 Treatment in Monogenic and Multifactorial Autoinflammatory Diseases. International Journal of Molecular Sciences, 20(8), 1898. https://doi.org/10.3390/ijms20081898