1. Introduction
Diabetes is an ubiquitously spread disease with a worldwide occurrence exceeding 387 million in 2014, and expected to reach 592 million by the year 2035 [
1]. Among the total number of cases, more than 90% are represented by the type 2 diabetes mellitus (T2DM), characterized with impaired insulin action and/or insulin secretion [
2]. As the onset of the early metabolic alterations (insulin tolerance and hyperglycaemia) is asymptomatic, vascular diabetic complications are often revealed at the moment of T2DM diagnosis [
3]. Hence, early discovery of T2DM, or even preceding changes in glucose metabolism, would essentially increase therapy efficiency and reduce the costs required for the treatment of diabetic complications [
4]. Obviously, even minor changes in blood glucose concentrations might affect the rates of blood protein glycation [
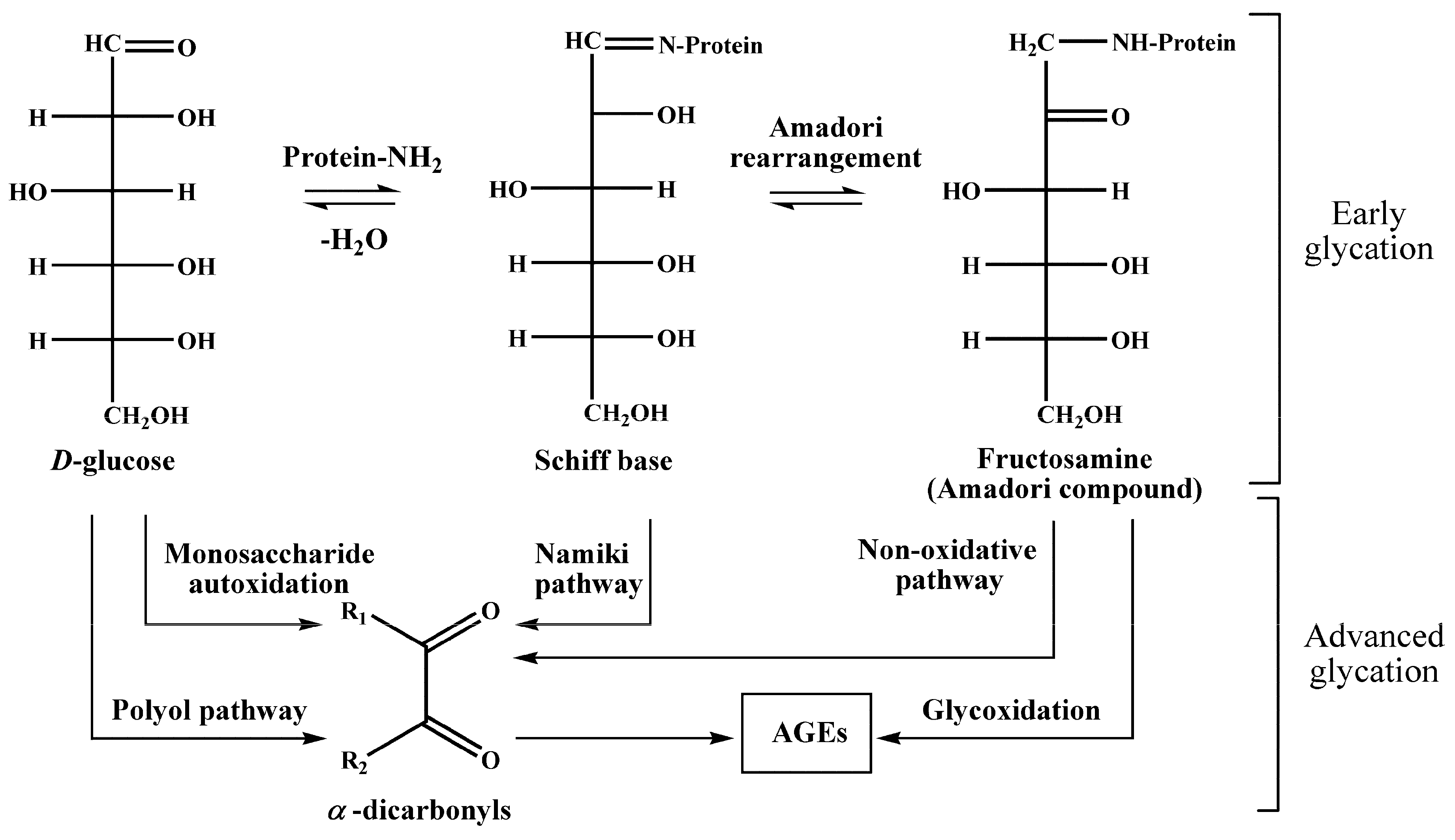
5], i.e., interaction of glucose with free amino groups of proteins, yielding fructosylamines, also known as Amadori compounds [
6] (
Figure 1). Their further degradation results in the formation of a highly heterogenic group of advanced glycation end products (AGEs) [
7], known for their pro-inflammatory impact in the development of vascular diabetic complications [
8,
9]. However, although AGEs are well-established biomarkers of diabetic complications [
10], Amadori compounds might suit better for glycemic control and early diagnostics of T2DM [
11].
Fasting plasma glucose (FPG) is the preferred diagnostic parameter, but an increase of blood glucose is also detected in patients suffering from other diseases besides diabetes mellitus [
12]. Currently, the blood content of hemoglobin isoform, HbA
1c, glycated by the N-terminus of its α-chain (˃6.5% of the total hemoglobin fraction), is one of the principle diagnostic criteria of this disease [
13] and an efficient tool of long-term (60–90 days) glycemic control [
14]. However, HbA
1c does not deliver any information about short-term alterations in plasma glucose concentrations accompanying the onset of metabolic syndrome [
15]. In contrast, short-living plasma proteins provide a good possibility to decrease the time dimensions of glycemic control. Thus, human serum albumin (HSA), the major blood plasma protein with a half-life of 21 days, can be used as a marker of T2DM [
16]. Its global glycation rates can be quantitatively assigned by an array of enzymatic [
17], colorimetric [
18], immunochemical [
19], electrophoretic [
20], and chromatographic [
21] methods. Importantly, the levels of HSA glycation vary from 1% and 10% in healthy individuals to 20% to 90% in patients with diabetes [
22]. However, as the glycation rates at individual lysyl residues in the HSA molecule differ essentially [
23], the sensitivity of the potential glycation sites to short-term changes of blood glucose levels might also be different.
In this context, the monitoring of glycation rates at specific glycation sites might be advantageous in comparison to the quantification of global glycation levels. Therefore during the last decade, mass spectrometry was intensively employed in the establishment of such techniques [
24,
25,
26]. In all cases, analytics relied on the bottom-up proteomic approach, based on nano-scaled liquid chromatography-mass spectrometry (nanoLC-MS) and tandem mass spectrometry (MS/MS). Thus, we showed that only some HSA lysyl residues are differentially glycated in plasma of T2DM patients, whereas glycation rates at other potential modification sites seemed to not be affected [
27]. Hence, tryptic peptides, representing differentially glycated sites, might be considered as prospective T2DM biomarkers. For some of them, it was additionally confirmed by absolute quantification using internal standardization with dabsylated bi-labeled Amadori-modified peptides [
28] or
13C,
15N synthetic analogs of specifically glycated peptides [
29]. As was shown recently, one of the plasma glycation sites (namely K
141 in haptoglobin) might provide an additional diagnostic tool in combination with well-established T2DM markers, such as fasting plasma glucose (FPG) and HbA
1c. The main advantage of combining two markers—HbA
1c and glycated haptoglobin, K
141—is the simultaneous consideration of two proteins with different half-life times, i.e., 3 to 4 and 2 to 4 days, respectively. It makes this biomarker is sensitive to long- and short-term fluctuations of blood glucose concentrations. The set of glycated K
141 of haptoglobin and HbA
1c provided a sensitivity of 94%, a specificity of 98%, and an accuracy of 96% to identify T2DM [
30].
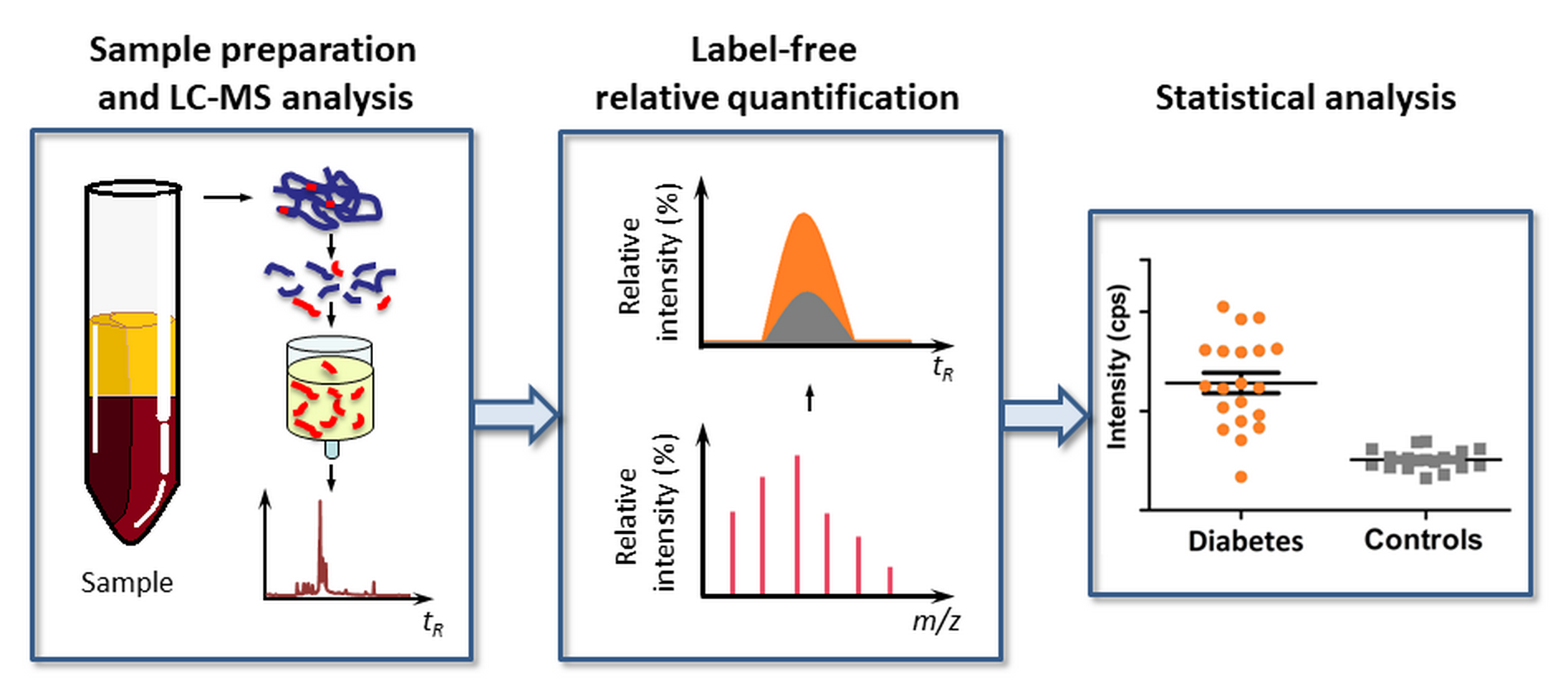
In this context, it is logical to assume that a biomarker strategy, based on multiple specific glycation sites in plasma proteins, could essentially increase the efficiency of glycemic control and disease prediction. Indeed, the involvement of several glycated proteins with different half-life times (τ1/2) allows several time segments in the glycemic control to be addressed without additional analyses. Moreover, this approach might decrease the impact of individual glycation sites in the overall dispersion of the data, when larger cohorts are considered. Therefore, here, we present a mass spectrometry-based biomarker approach relying on multiple glycation sites. We demonstrate its applicability to different time points in the span of glycemic control. Based on the results of linear discriminant analysis (LDA) performed for the cohorts of T2DM patients and individuals without diabetes, we characterize a set of Amadori-modified peptides with a diagnosis accuracy of up to 92%.
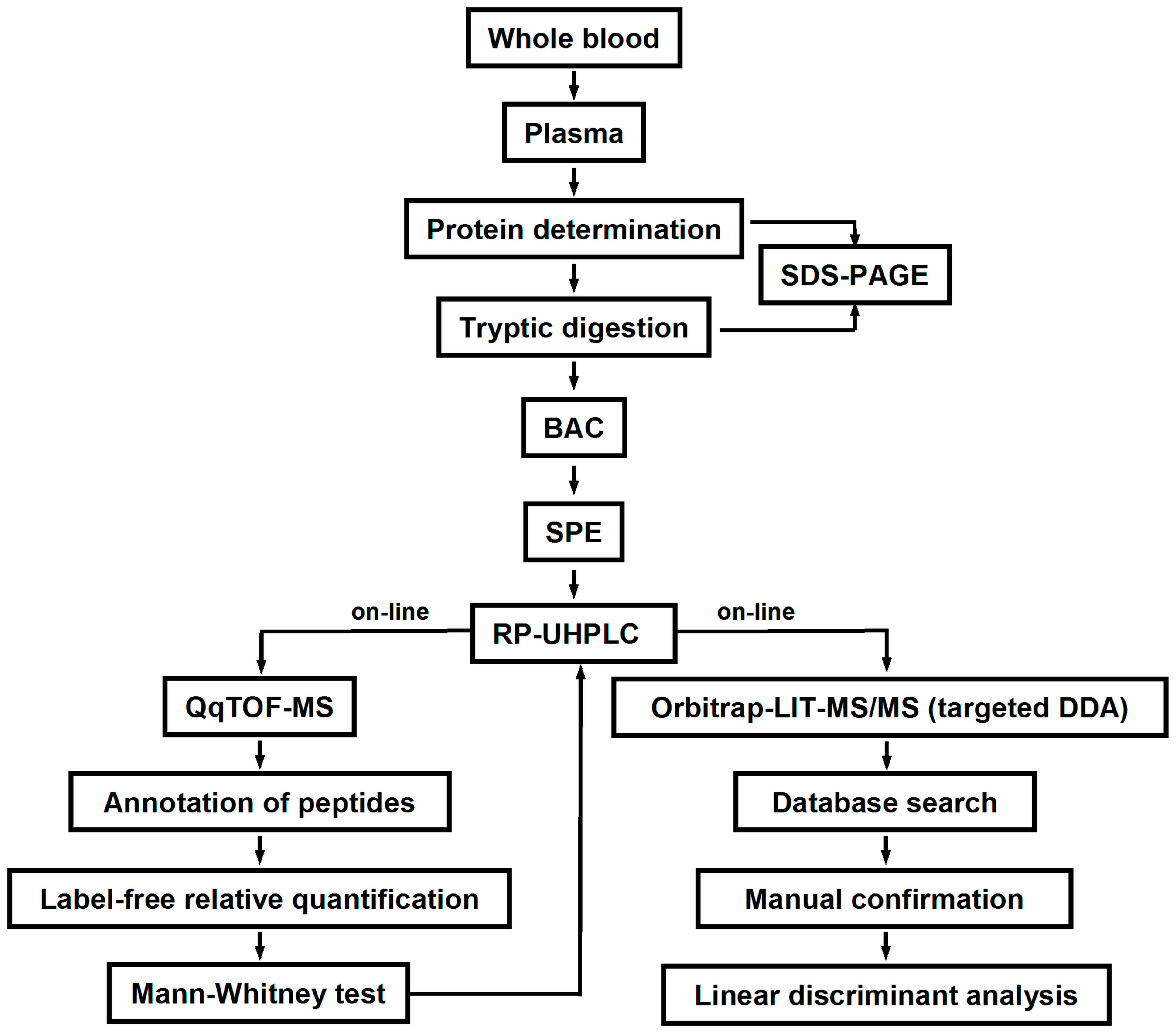
4. Materials and Methods
4.1. Reagents
Unless stated otherwise, materials were obtained from the following manufacturers. AMRESCO LLC (Fountain Parkway, Solon, OH, USA): ammonium persulfate (ACS grade), glycine (biotechnology grade), N,Nʹ-methylene-bis-acrylamide (ultra-pure grade), tris(hydroxymethyl)aminomethan (tris, ultra-pure grade); Aptec Diagnostics nv (Saint-Petersburg, Russia): Albumin kit; Becton Dickinson (Moscow, Russia): BD Vacutainer® Plus Plastic K2EDTA tubes; Carl Roth GmbH & Co (Karlsruhe, Germany): acetonitrile (≥99.95%, LC-MS grade), formic acid (≥98%, p.a., ACS), ethanol (≥99.8%), methanol (HPLC Ultra Gradient Grade), sodium dodecyl sulfate (SDS) (>99%), tris-(2-carboxyethyl)-phosphine hydrochloride (TCEP, ≥98%); Component-Reactiv (Moscow, Russia): phosphoric acid (analytical quality); PanReac AppliChem (Darmstadt, Germany): acrylamide (2K Standard Grade), glycerol (ACS grade); QIAGEN GmbH (Hilden, Germany): 1 mL polypropylene columns; Reachem (Moscow, Russia): Hydrochloric acid (analytical quality); Roche Diagnostics (Saint-Petersburg, Russia): Precinorm U plus Control, Precipath U plus Control; SERVA Electrophoresis GmbH (Heidelberg, Germany): Coomassi Brilliant Blue G-250, 2-mercaptoethanol (research grade), trypsin NB (sequencing grade, modified from porcine pancreas); Thermo Fisher Scientific (Waltham, MA, USA): PageRuler™ Unstained Protein Ladder #26614, PageRuler™ Prestained Protein Ladder #26616; Vekton (Saint-Petersburg, Russia): ammonia hydroxide (ACS grade); Waters GmbH (Eschborn, Germany): Oasis HLB cartridges (10 mg, 30 µm particle size). All other chemicals were purchased from Sigma-Aldrich Chemie GmbH (Taufkirchen, Germany). Water was purified in house (resistance >16 mΩ/cm) on a water conditioning and purification system “UVOI-MF-1-NA(18)-N” (Mediana-Filtr, Moscow, Russia).
4.2. Setup of Experimental Cohorts
The T2DM patient (
n = 20) and normoglycemic control (
n = 18) groups comprised non-smoking female volunteers aged 45 to 75 years (63.4 ± 7.9 and 60.7 ± 4.7, respectively), who were not receiving hormone replacement therapy and had no clinically manifested diabetes complications (
Table 4 and
Table 5). The control individuals’ HbA
1C levels did not exceed 6.5%, and did not have diagnosed diabetes and anti-hyperglycemic therapy in their medical history. All participants provided written informed consent. The study was approved 02-03-2015 by the Local Ethical Committee of the Federal Almazov North-West Medical Research Centre, Saint-Petersburg, Russian Federation, and was performed in agreement with the Declaration of Helsinki.
4.3. Blood Sampling and Plasma Isolation
The blood samples (approximately 10 mL each) were collected in polypropylene tubes coated with etylenediaminetetraacetic acid (Becton Dickinson, Franklin Lakes, NJ, USA). Plasma was separated by centrifugation (1200×
g, 15 min, 4 °C) and transferred to 1.5 mL polypropylene tubes. The total plasma protein contents were determined by Bradford assay in a 96-well microtiter plate format as described by Greifenhagen and co-workers [
51]. The precision of protein determination was verified by SDS-PAGE according to an established protocol [
11]. Average densities across individual lanes (expressed in arbitrary units) were determined by a ChemiDoc XRS imaging system controlled by Quantity One
® 1-D analysis software (Bio-Rad Laboratories Ltd., Moscow, Russia). Thereby, for inter-gel normalization, the first and the last plasma protein samples loaded on each gel were replicated in the previous and following gels, respectively (
Figure S1). For the calculation of RSDs, the densities of individual lines were normalized to the gel average value. Individual plasma samples were split into aliquots of 20 µL, and stored at −80 °C. Alternatively, 30 µL of each T2DM sample were combined to obtain a representative pool of diabetic material. The plasma contents of human serum albumin were determined calorimetrically at 628 nm after a color reaction with bromocresol purple (Clinical Chemistry Analyzer CA90, Furuno Electric Co. LTD, Nishinomiya, Japan) using an Albumin kit (Aptec Diagnostics nv) and the control serum kits, Precinorm U and Precipath U (Roche Diagnostics). The plasma HbA
1c level was measured using the commercial kit, BioRad (Hercules, CA, USA), on a hemoglobin analyzer BioRad D-10 (Bio-Rad Laboratories Inc, Hercules, CA, USA).
4.4. Tryptic Digestion
The plasma proteins were digested according to Frolov and co-workers [
27] with slight modifications. Briefly, aliquots of plasma containing 150 μg of protein were diluted with 100 mmol/L ammonium bicarbonate buffer (pH 8.0), complemented with 10 µL of SDS (0.5% in water,
w/
v) and 10 µL of 50 mmol/L TCEP in 100 mmol/L ammonium bicarbonate buffer, and incubated for 30 min at 37 °C under continuous shaking (450 rpm). Afterwards, the samples were cooled to room temperature (RT), and 11 µL of 100 mmol/L iodoacetamide in 100 mmol/L ammonium bicarbonate buffer were added, and alkylation of free sulfhydryls was performed during 15 min in darkness at RT. After completion of the incubation, the proteins were sequentially digested at 37 °C with trypsin (25 μg/mL in 100 mmol/L ammonium bicarbonate buffer) taken in the 1:20 and 1:40 (
w/
w) enzyme-protein ratio for 5 and 12 h, respectively, under continuous shaking (450 rpm).
The completeness of the digest was verified by SDS-PAGE as described by Schmidt and co-workers [
52] with modifications. Briefly, aliquots of digested samples containing 5 μg of protein were diluted with sample buffer (65.8 mmol/L Tris-HCl, pH 6.8, 20% (
v/
v) glycerol, 2% SDS, 10% (
v/
v) β-mercaptoethanol, 0.05% (
v/
v) bromophenol blue) at least 2-fold and heated at 95 °C for 5 min. Afterwards, the samples were separated on a polyacrylamide gel (T = 12.00%, C = 2.65%), and stained with colloidal Coomassie Brilliant Blue G 250 dye. The digests were frozen and stored at −80 °C before further analysis.
4.5. Boronic Acid Affinity Chromatography
Enrichment of glycated peptides was performed according to Soboleva and co-workers [
28] with slight modifications. In detail, the pH of tryptic digests was adjusted to 8.0 with 25% (
v/
v) ammonia hydroxide using indicator paper (Lachema, Brno, Czech Republic), before 400 μL of ice cold (4 °C) loading buffer (250 mmol/L ammonium acetate, 50 mmol/L magnesium acetate, pH 8.1) were added. The samples were loaded on 1 mL polypropylene gravity flow columns packed with m-aminophenylboronic acid (mAPBA) agarose, and unbound peptides were washed out with 12 mL of ice-cold (4 °C) loading buffer. Afterwards, glycated peptides were sequentially eluted with 0.1 and 0.2 mol/L warm (37 °C) acetic acid (8 and 2 mL, respectively). The eluates were combined, and loaded on Oasis HLB SPE cartridges, installed on the VacElut 12 Manifold (Agilent Technologies, Moscow, Russia), pre-conditioned with 1 mL of methanol, and pre-equilibrated with 2 mL of 0.1% (
v/
v) aqueous (aq.) formic acid. After a wash with 2 mL of 0.1% (
v/
v) formic acid, peptides were eluted in a step gradient of 40%, 60%, and 80% acetonitrile (0.33 mL each), as described by Spiller et al. [
29]. The SPE-eluates were combined, dried under vacuum by a CentriVap Vacuum Concentrator (Labconco, Kansas City, MO, USA), and stored at −20 °C for further analysis.
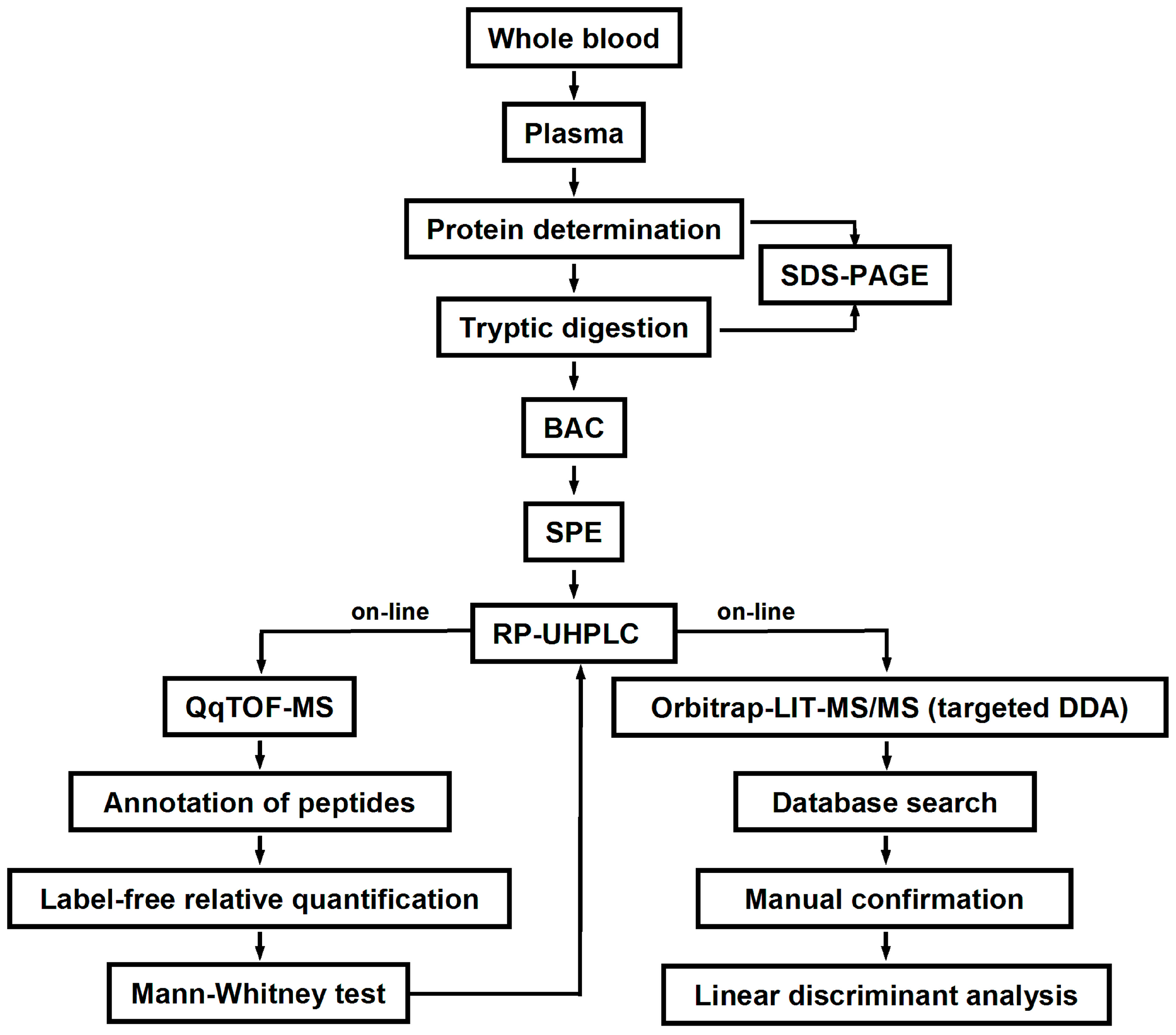
4.6. LC-MS Analysis
The dried eluates were reconstituted in 40 µL of 3% (
v/
v) acetonitrile in aq. 0.1% (%
v/
v) formic acid, and 8 µL of the obtained solutions were loaded on a ZORBAX SB column (C18, ID 0.3 mm, length 150 mm, particle size 3.5 μm, Agilent Technologies, Moscow, Russia) using an Agilent 1200 Compact liquid chromatograph equipped with an Agilent 1200 Infinity autosampler and Agilent 1260 Infinity capillary pump (Agilent Technologies, Moscow, Russia). The eluents, A and B, were 4% and 90% acetonitrile, respectively, both containing 0.1% (
v/
v) formic acid. After a 5-min isocratic step (0% eluent B), glycated peptides were eluted at the flow rate of 5 µL/min at 25 °C in sequential linear gradients to 45% and to 100% eluent B in 30 and 2 min, respectively. The column effluents were introduced on-line in an Agilent 6538 Ultra High Definition Accurate-Mass Q-TOF quadrupole-time of flight (QqTOF) mass spectrometer via a dual electrospray ionization (ESI) source (Agilent Technologies, Moscow, Russia). The instrument was operated in the positive ion mode under the settings summarized in
Table S7 and controlled by MassHunter Workstation software (Agilent Technologies, Moscow, Russia). Analyte annotation and label free quantification relied on TOF-MS scans acquired in the mass range of 400–2000
m/z. Thereby, a pooled enriched tryptic digest, obtained from T2DM patients, was used as an external QC, injected after each eighth sample. Prospective Amadori-modified peptides were annotated by t
Rs and exact
m/z values (mass accuracy better than 3 ppm). Relative abundances of individual prospectively glycated peptides were calculated by integration of characteristic extracted ion chromatograms (XICs,
m/z ± 0.02) built for the annotated
m/z values at specific t
Rs (quantitative analysis tool of the MassHunter Workstation software).
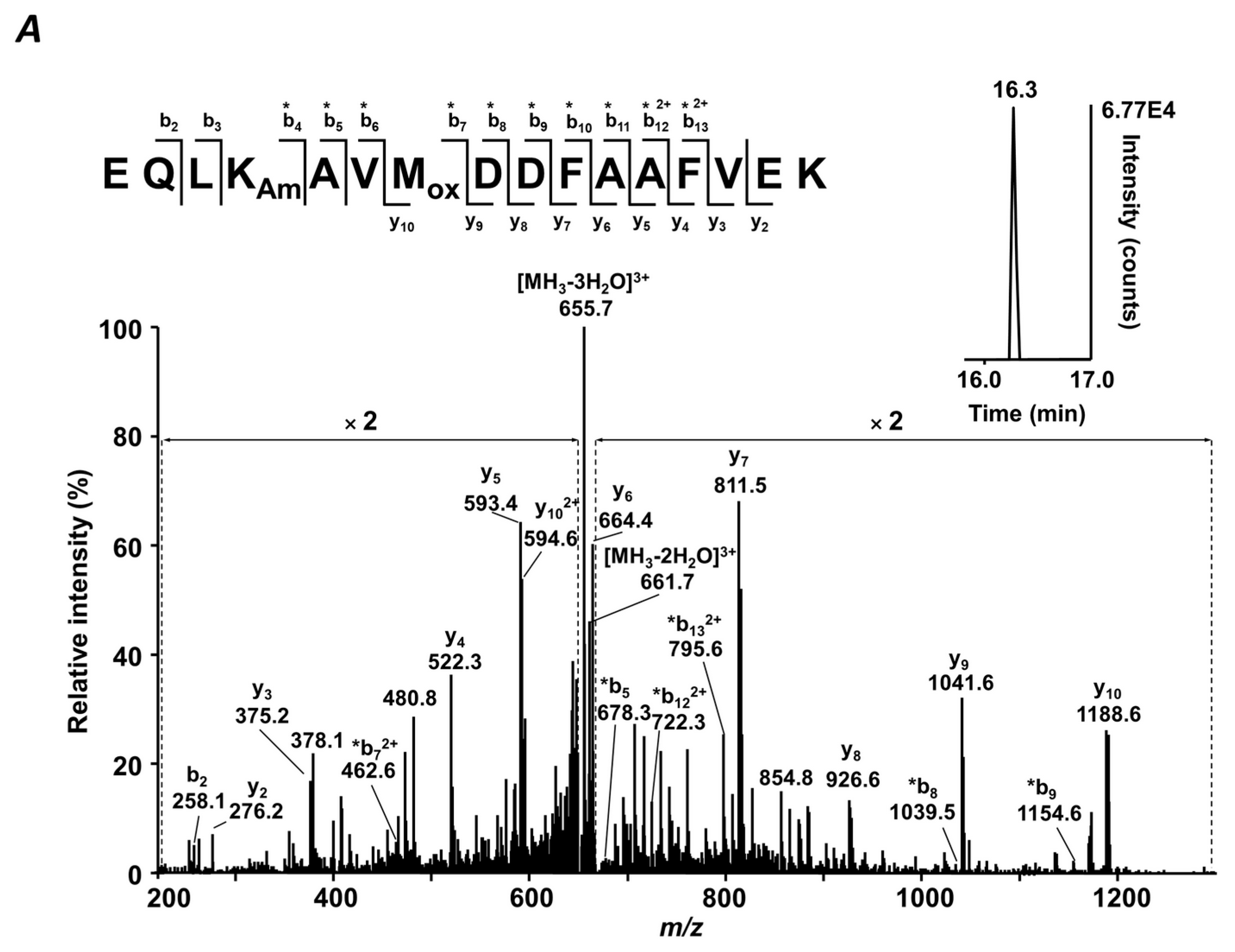
4.7. MS/MS Analysis
The sequences and glycation status of differentially abundant glycated peptides were confirmed by MS/MS using a combination of DDA and targeted MS/MS experiments using an LTQ-Orbitrap Velos Pro mass spectrometer. For this, a digest of pooled T2DM plasma (1.5 µg) was loaded on the Acclaim® PepMap100 pre-column (C18-phase, ID 75 μm, length 2 cm, particle size 3 µm), and separated on the EASY-Spray ES803 C18 column (500 × 0.075 mm, 2 µm particle size, 40 °C) using an EASY-nLC 1000 nano liquid chromatography system controlled by the Xcalibur 2.1.0 software. Eluents A and B were water and acetonitrile, respectively, both containing 0.1% (
v/
v) formic acid. The analytes were eluted at the flow rate of 0.3 µL/min as follows: 5% B (0–15 min), 5% to 40% B (15–195 min), 40% to 80% B (195–197 min). The column effluent was transferred on-line to MS (operated in positive ion mode and controlled by Xcalibur 2.1.0 software) via an EASY-spray ion source. DDA analyses relied on survey Orbitrap-MS scans followed by dependent linear ion trap (LIT) ones (collision induced dissociation (CID) fragmentation, He, 35% normalized collision energy) acquired for the 20 most intense signals with charge states of two or more. For targeted MS/MS, 10 LIT MS2 sub-experiments per liquid chromatography-tandem mass-spectrometry (LC-MS/MS) run were acquired for the two most intense quasi-molecular ions of each peptide, not fragmented in DDA experiments. The MS settings are summarized in
Table S8. Tandem mass spectra were searched against a FASTA file containing sequences of the proteins annotated by QqTOF-MS using the Mascot search engine within Proteome Discoverer 1.4 software (Thermo-Fisher Scientific, Bremen, Germany) with the following settings: Peptide tolerance—7 ppm, MS/MS match tolerance—0.8 Da, 3 missed cleavage sites per peptide. Glycated peptides were annotated by their t
Rs,
m/
z values, and isotopic patterns. The results were filtered with consideration of peptide confidence (medium), rank (one), false positive rate (0.05), and post-translational modifications—carbamidomethylation (C), oxidation (M), and glycation (K).
4.8. Statistical Analysis
The statistical significance of differences in the relative abundances of specific glycated peptides detected in the plasma protein digests obtained from T2DM patients and healthy individuals was determined by the Mann-Whitney
U test [
53]. Holm-Bonferroni correction was applied to adjust significance in terms of multiple hypothesis tests [
46]. Since the intensities observed for the signals representing individual charge states of the same peptides demonstrated high correlation, these values were averaged for further processing. To distinguish the T2DM patients from the age-matched controls without diabetes, a linear discriminant analysis (LDA) approach was applied as described by Venables and Ripley [
54]. All LDA-related procedures were performed in R (MASS package); all other calculations were performed in MS Excel with RealStatisticsaddon (
http://www.real-statistics.com).
4.9. Generation of the LDA Model
Because the initial correlation analysis of the feature space showed a high degree of multicollinearity between factors (see
Supplementary Figure S6 for a factor cross-correlation plot), we employed two strategies for feature space optimization prior to LDA calculation: (i) PCA-based transformation of the feature space, and (ii) generation of an orthogonal set of original features by the removal of factors with high variance inflation factor (VIF) values [
55]. The latter can be calculated for any variable in the set (1) and is a numeric measure of to which extent the variable in question can be predicted by the linear combination of other variables, with the most commonly used critical VIF values being 5 and 10, corresponding to the multiple correlation of 90% and 95% between the tested variables and the rest variables in the set.
where:
VIFi—variance inflation factor for variable
i;
—coefficient of determination, i.e., the proportion of the variance in the variable,
I, which is predictable based on another independent variable
The application of PCA was straightforward and only the PCs responsible for 95% of the original sample variance were selected. The corresponding values were estimated for each sample, and the resulting reduced feature set was subjected to further processing with LDA. The VIF-based filtering relied on an iterative procedure, employing a random subset of two features (a test sub-set), and defined in the beginning of each calculation run. Afterwards, further features were randomly selected from the original set and sequentially included in the test subset in each new analysis run. Then, individual features with the VIF values above a selected cutoff (set as described above [
43]) were iteratively removed from the test subset in the order of decreasing values. This procedure was further applied in this way to test all initial features.
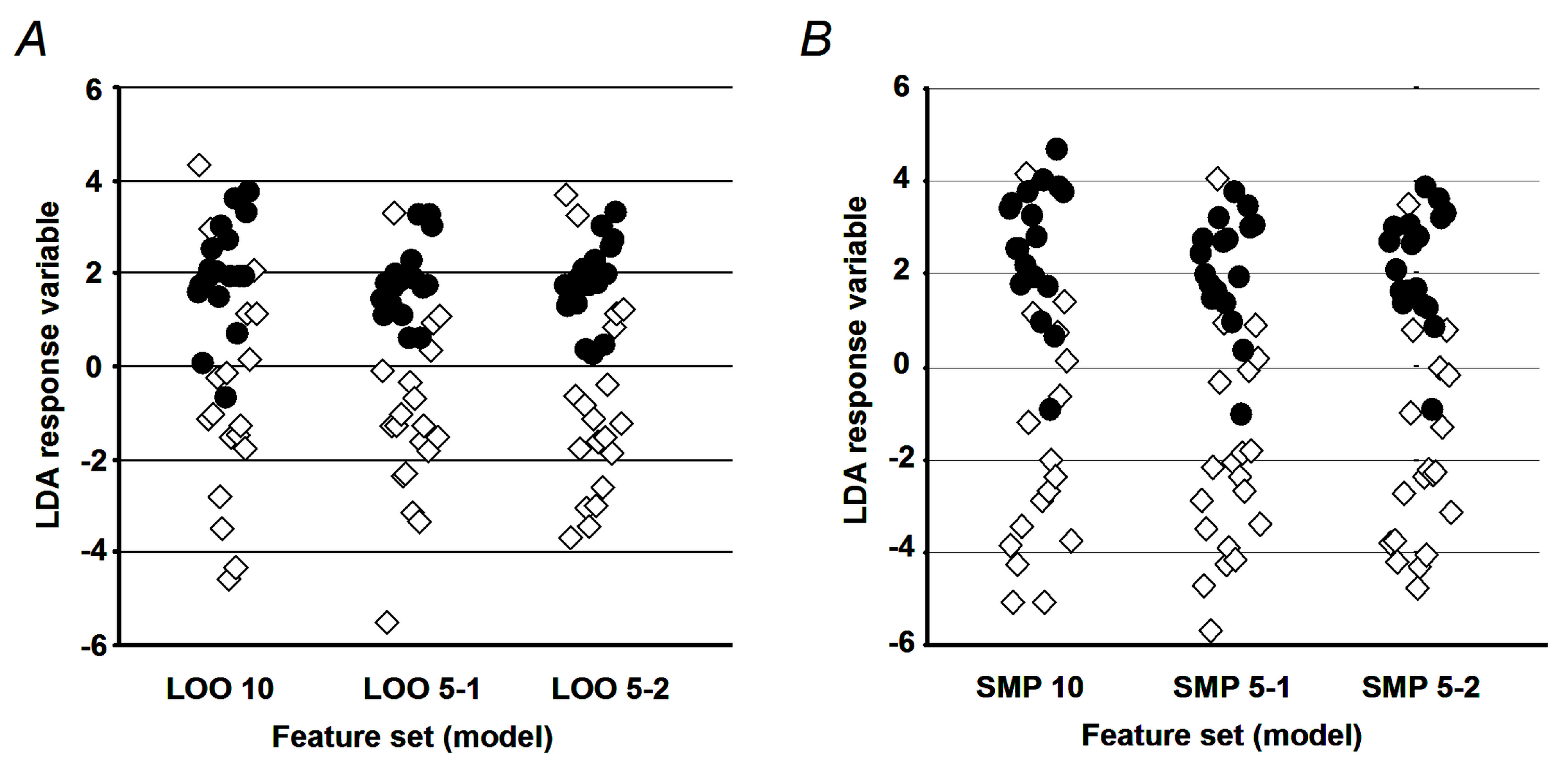
4.10. Validation of the LDA Model
To assess the model performance, two complementary approaches were employed. In terms of the first one, accuracy, sensitivity, and specificity were determined for the established set of diagnostic peptides. To avoid the overfitting problem [
56], parameterization of the LDA model was performed in silico with a set of 400 calculated sample patterns (200 for each cohort), obtained by the sub-sampling feature distributions of original samples as described by Politis and co-authors [
57]. Thereby, all selected training samples shared less than 25% of the factor values, obtained with any of the original (verification) vectors. The second approach relied on the so-called leave-one-out cross-validation procedure [
58]. It employed a set of iterations, where at each run one sample from the original sample set was taken for verification, whereas the other samples were used for parameterization of the LDA model. The performance characteristics (i.e., accuracy, sensitivity, and specificity) were calculated from the set of individual iteration outcomes as described elsewhere [
59,
60]. For the PCA-generated feature subset, only the leave-one-out cross-validation procedure was performed.
5. Conclusions
T2DM is one of the most widely spread metabolic disorders. Typically, the first stages of the disease are slow and are not accompanied by any clinically manifested symptoms. Because of this reason, T2DM is most often discovered at the step of complications, which makes therapy less efficient and more expensive. Unfortunately, HbA1c—a recognized T2DM marker—delivers information about changes in glycaemic status over three months, and, hence, is insensitive to short-term glucose excursions preceding the disease. Thus, control of glycemic status over various, especially short, periods of time might increase the rates of early T2DM discovery. In this context, our approach might bring the desired “time dimension” in glycemic control. Indeed, the integrated biomarker, proposed here, not only covers three weeks before blood sampling, but also indicates a continuous character of the glycation process throughout this period. Secondly, although our integrated biomarker relies on multiple glycation sites, all required information is acquired in one experiment, which is advantageous in comparison to the approaches employing several tests. Finally, the proposed marker has potential for further optimization. Thus, it can be “tuned” for shorter times by excluding relatively long-living proteins. On the the other hand, implementation of immunoaffinity depletion might essentially increase the pattern of marker peptides, and, hence, the selection of marker proteins and the reliability of the marker.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}