Disabling the Protease DDI2 Attenuates the Transcriptional Activity of NRF1 and Potentiates Proteasome Inhibitor Cytotoxicity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
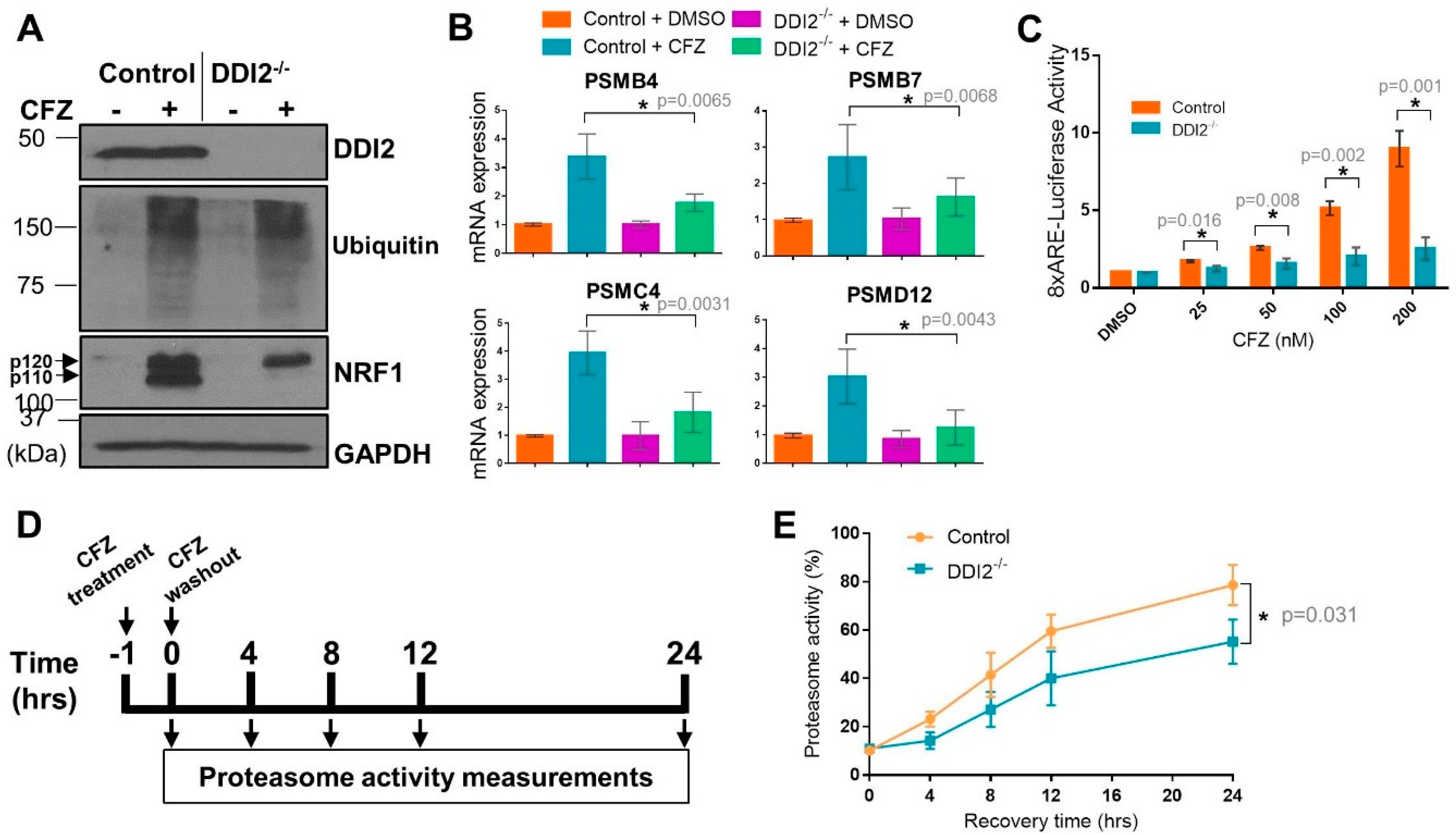
2.1. DDI2 Is Required for NRF1-Mediated Proteasome Bounce-Back Response
2.2. NRF1 Is Proteolytically Processed by DDI2 in the Cytosol
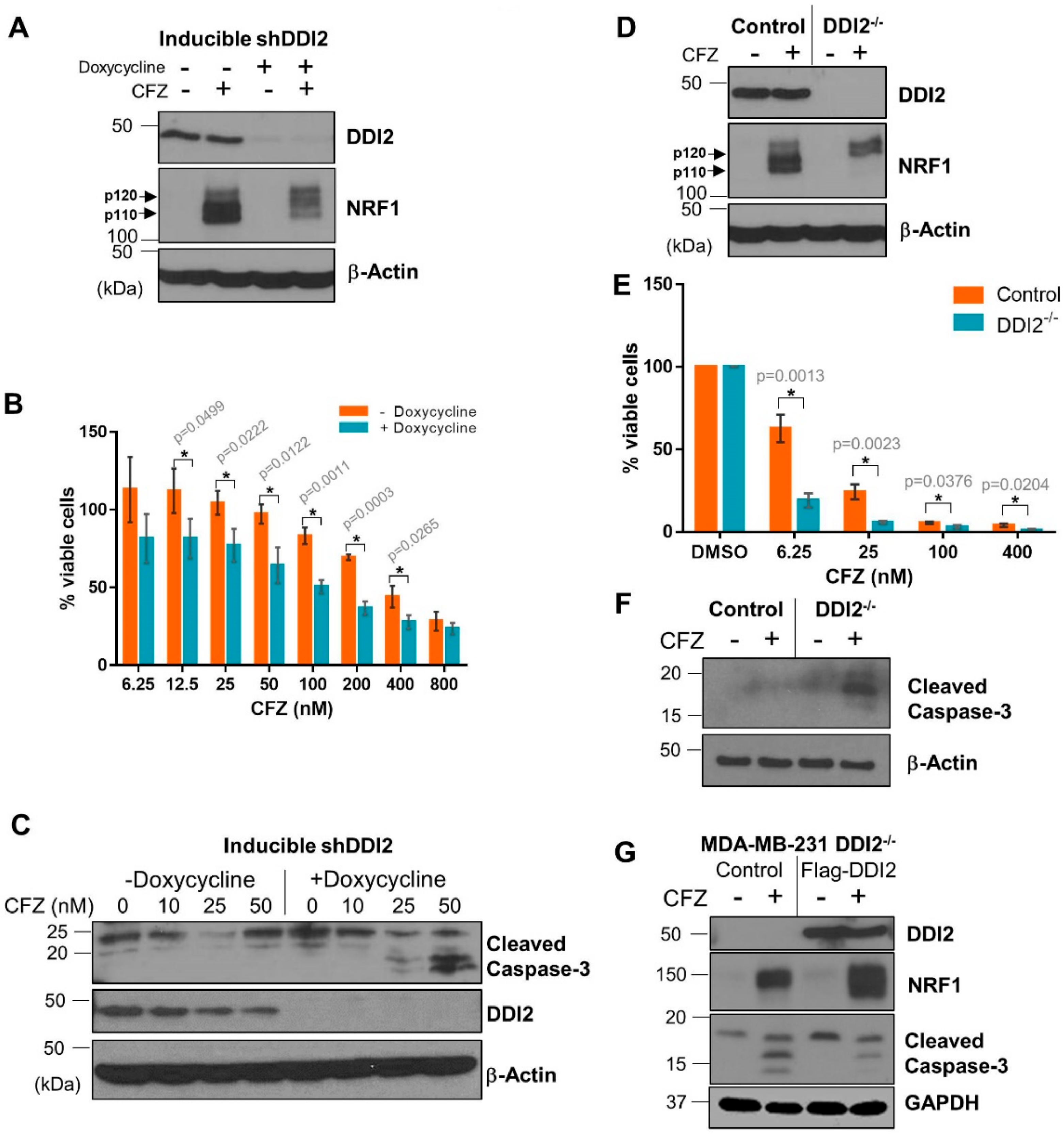
2.3. DDI2-Deficient MDA-MB-231 Cells Are More Sensitive to CFZ-Induced Apoptosis
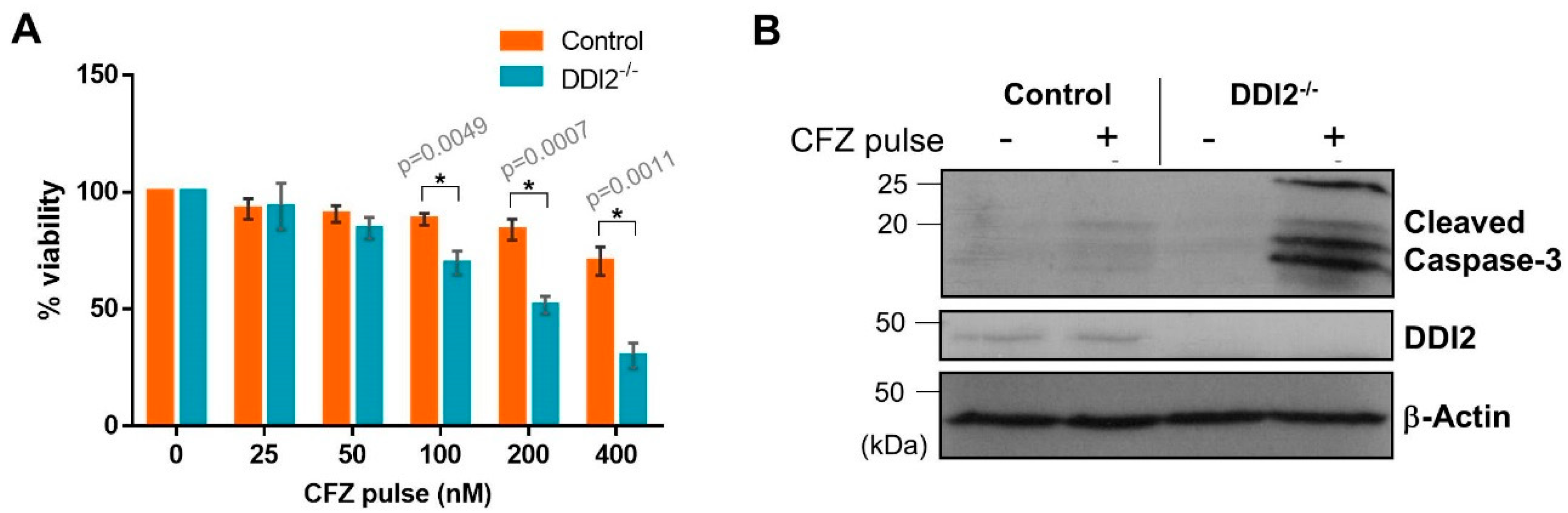
2.4. DDI2-Deficient MDA-MB-231 Cells Are More Sensitive to CFZ-Induced Apoptosis in an In Vitro Model of Rapid Clearance
2.5. Protease-Dead DDI2 Acts in a Dominant Negative Fashion to Inhibit NRF1 Processing and Sensitize MDA-MB-231 Cells to CFZ-Induced Apoptosis
3. Discussion
4. Materials and Methods
4.1. Constructs
4.2. Cell Lines and Culture Conditions
4.3. Western Blot Analysis
4.4. Quantitative Reverse Transcription PCR
4.5. Luciferase Assay
4.6. Proteasome Activity Recovery Assay
4.7. Subcellular Fractionation
4.8. Co-Immunoprecipitation Assay
4.9. Cell Viability Assays
Author Contributions
Funding
Conflicts of Interest
References
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef] [PubMed]
- Kleiger, G.; Mayor, T. Perilous journey: A tour of the ubiquitin-proteasome system. Trends Cell Biol. 2014, 24, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Tomaszewski, J.E. Proteasome inhibitors. Biochem. Pharmacol. 2015, 96, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Albornoz, N.; Bustamante, H.; Soza, A.; Burgos, P. Cellular Responses to Proteasome Inhibition: Molecular Mechanisms and Beyond. Int. J. Mol. Sci. 2019, 20, 3379. [Google Scholar] [CrossRef]
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharmacol. 2018, 81, 227–243. [Google Scholar] [CrossRef]
- Dou, Q.P.; Zonder, J.A. Overview of proteasome inhibitor-based anti-cancer therapies: Perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr. Cancer Drug Targets 2014, 14, 517–536. [Google Scholar] [CrossRef]
- Smith, D.C.; Kalebic, T.; Infante, J.R.; Siu, L.L.; Sullivan, D.; Vlahovic, G.; Kauh, J.S.; Gao, F.; Berger, A.J.; Tirrell, S.; et al. Phase 1 study of ixazomib, an investigational proteasome inhibitor, in advanced non-hematologic malignancies. Investig. New Drugs 2015, 33, 652–663. [Google Scholar] [CrossRef]
- Mitsiades, N.; Mitsiades, C.S.; Poulaki, V.; Chauhan, D.; Fanourakis, G.; Gu, X.; Bailey, C.; Joseph, M.; Libermann, T.A.; Treon, S.P.; et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14374–14379. [Google Scholar] [CrossRef]
- Lundgren, J.; Masson, P.; Mirzaei, Z.; Young, P. Identification and characterization of a Drosophila proteasome regulatory network. Mol. Cell. Biol. 2005, 25, 4662–4675. [Google Scholar] [CrossRef]
- Meiners, S.; Heyken, D.; Weller, A.; Ludwig, A.; Stangl, K.; Kloetzel, P.M.; Kruger, E. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of Mammalian proteasomes. J. Biol. Chem. 2003, 278, 21517–21525. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Seeger, M.; Koch, A.; Kruger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Varshavsky, A. RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: A negative feedback circuit. Proc. Natl. Acad. Sci. USA 2001, 98, 3056–3061. [Google Scholar] [CrossRef]
- Grimberg, K.B.; Beskow, A.; Lundin, D.; Davis, M.M.; Young, P. Basic leucine zipper protein Cnc-C is a substrate and transcriptional regulator of the Drosophila 26S proteasome. Mol. Cell. Biol. 2011, 31, 897–909. [Google Scholar] [CrossRef]
- Biswas, M.; Chan, J.Y. Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol. 2010, 244, 16–20. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; den Besten, W.; Deshaies, R.J. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife 2014, 3, e01856. [Google Scholar] [CrossRef]
- Tomlin, F.M.; Gerling-Driessen, U.I.M.; Liu, Y.C.; Flynn, R.A.; Vangala, J.R.; Lentz, C.S.; Clauder-Muenster, S.; Jakob, P.; Mueller, W.F.; Ordonez-Rueda, D.; et al. Inhibition of NGLY1 Inactivates the Transcription Factor Nrf1 and Potentiates Proteasome Inhibitor Cytotoxicity. ACS Cent. Sci. 2017, 3, 1143–1155. [Google Scholar] [CrossRef]
- Sha, Z.; Goldberg, A.L. Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr. Biol. 2014, 24, 1573–1583. [Google Scholar] [CrossRef]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. Elife 2016, 5, e18357. [Google Scholar] [CrossRef]
- Johnsen, O.; Skammelsrud, N.; Luna, L.; Nishizawa, M.; Prydz, H.; Kolsto, A.B. Small Maf proteins interact with the human transcription factor TCF11/Nrf1/LCR-F1. Nucleic Acids Res. 1996, 24, 4289–4297. [Google Scholar] [CrossRef] [PubMed]
- Vangala, J.R.; Radhakrishnan, S.K. Nrf1-mediated transcriptional regulation of the proteasome requires a functional TIP60 complex. J. Biol. Chem. 2019, 294, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Lehrbach, N.J.; Ruvkun, G. Proteasome dysfunction triggers activation of SKN-1A/Nrf1 by the aspartic protease DDI-1. Elife 2016, 5, e17721. [Google Scholar] [CrossRef] [PubMed]
- Auner, H.W.; Moody, A.M.; Ward, T.H.; Kraus, M.; Milan, E.; May, P.; Chaidos, A.; Driessen, C.; Cenci, S.; Dazzi, F.; et al. Combined inhibition of p97 and the proteasome causes lethal disruption of the secretory apparatus in multiple myeloma cells. PLoS ONE 2013, 8, e74415. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E.; Shalem, O.; Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11, 783–784. [Google Scholar] [CrossRef]
- Mathieson, T.; Franken, H.; Kosinski, J.; Kurzawa, N.; Zinn, N.; Sweetman, G.; Poeckel, D.; Ratnu, V.S.; Schramm, M.; Becher, I.; et al. Systematic analysis of protein turnover in primary cells. Nat. Commun. 2018, 9, 689. [Google Scholar] [CrossRef]
- Zhang, Y.; Hayes, J.D. Identification of topological determinants in the N-terminal domain of transcription factor Nrf1 that control its orientation in the endoplasmic reticulum membrane. Biochem. J. 2010, 430, 497–510. [Google Scholar] [CrossRef]
- Magnaghi, P.; D’Alessio, R.; Valsasina, B.; Avanzi, N.; Rizzi, S.; Asa, D.; Gasparri, F.; Cozzi, L.; Cucchi, U.; Orrenius, C.; et al. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol. 2013, 9, 548–556. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, J.; Kirk, C.; Fang, Y.; Alsina, M.; Badros, A.; Papadopoulos, K.; Wong, A.; Woo, T.; Bomba, D.; et al. Clinical pharmacokinetics, metabolism, and drug-drug interaction of carfilzomib. Drug Metab. Dispos. 2013, 41, 230–237. [Google Scholar] [CrossRef]
- Siva, M.; Svoboda, M.; Veverka, V.; Trempe, J.F.; Hofmann, K.; Kozisek, M.; Hexnerova, R.; Sedlak, F.; Belza, J.; Brynda, J.; et al. Human DNA-Damage-Inducible 2 Protein Is Structurally and Functionally Distinct from Its Yeast Ortholog. Sci. Rep. 2016, 6, 30443. [Google Scholar] [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS (Auckl. NZ) 2015, 7, 95. [Google Scholar] [CrossRef]
- Konvalinka, J.; Krausslich, H.G.; Muller, B. Retroviral proteases and their roles in virion maturation. Virology 2015, 479, 403–417. [Google Scholar] [CrossRef] [PubMed]
- Gabriely, G.; Kama, R.; Gelin-Licht, R.; Gerst, J.E. Different domains of the UBL-UBA ubiquitin receptor, Ddi1/Vsm1, are involved in its multiple cellular roles. Mol. Biol. Cell 2008, 19, 3625–3637. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Zhao, X.; Liu, S.; Cao, Q.; Yan, B.; Yang, J. MicroRNA-3607 inhibits the tumorigenesis of colorectal cancer by targeting DDI2 and regulating the DNA damage repair pathway. Apoptosis 2019, 24, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Conti, B.A.; Lach, F.P.; Smogorzewska, A. Removal of RTF2 from Stalled Replisomes Promotes Maintenance of Genome Integrity. Mol. Cell 2018, 69, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Yang, Y.; Wang, X.; Guan, S.; Fang, L.; Liu, F.; Walters, K.J.; Kaiser, P.; Huang, L. Characterization of Dynamic UbR-Proteasome Subcomplexes by In vivo Cross-linking (X) Assisted Bimolecular Tandem Affinity Purification (XBAP) and Label-free Quantitation. Mol. Cell. Proteomics 2016, 15, 2279–2292. [Google Scholar] [CrossRef]
- Hong, T.B.; Rahumatullah, A.; Yogarajah, T.; Ahmad, M.; Yin, K.B. Potential effects of chrysin on MDA-MB-231 cells. Int. J. Mol. Sci. 2010, 11, 1057–1069. [Google Scholar] [CrossRef]
- Khaw-On, P.; Pompimon, W.; Banjerdpongchai, R. Goniothalamin Induces Necroptosis and Anoikis in Human Invasive Breast Cancer MDA-MB-231 Cells. Int. J. Mol. Sci. 2019, 20, 3953. [Google Scholar] [CrossRef]
- Sayeed, M.A.; Bracci, M.; Ciarapica, V.; Malavolta, M.; Provinciali, M.; Pieragostini, E.; Gaetani, S.; Monaco, F.; Lucarini, G.; Rapisarda, V.; et al. Allyl Isothiocyanate Exhibits No Anticancer Activity in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 145. [Google Scholar] [CrossRef]
- Chen, C.; Wang, S.; Liu, P. Deferoxamine Enhanced Mitochondrial Iron Accumulation and Promoted Cell Migration in Triple-Negative MDA-MB-231 Breast Cancer Cells Via a ROS-Dependent Mechanism. Int. J. Mol. Sci. 2019, 20, 4952. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Northrop, A.; Vangala, J.R.; Feygin, A.; Radhakrishnan, S.K. Disabling the Protease DDI2 Attenuates the Transcriptional Activity of NRF1 and Potentiates Proteasome Inhibitor Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 327. https://doi.org/10.3390/ijms21010327
Northrop A, Vangala JR, Feygin A, Radhakrishnan SK. Disabling the Protease DDI2 Attenuates the Transcriptional Activity of NRF1 and Potentiates Proteasome Inhibitor Cytotoxicity. International Journal of Molecular Sciences. 2020; 21(1):327. https://doi.org/10.3390/ijms21010327
Chicago/Turabian StyleNorthrop, Amy, Janakiram R. Vangala, Alex Feygin, and Senthil K. Radhakrishnan. 2020. "Disabling the Protease DDI2 Attenuates the Transcriptional Activity of NRF1 and Potentiates Proteasome Inhibitor Cytotoxicity" International Journal of Molecular Sciences 21, no. 1: 327. https://doi.org/10.3390/ijms21010327
APA StyleNorthrop, A., Vangala, J. R., Feygin, A., & Radhakrishnan, S. K. (2020). Disabling the Protease DDI2 Attenuates the Transcriptional Activity of NRF1 and Potentiates Proteasome Inhibitor Cytotoxicity. International Journal of Molecular Sciences, 21(1), 327. https://doi.org/10.3390/ijms21010327