Pleiotropic Roles of Calmodulin in the Regulation of KRas and Rac1 GTPases: Functional Diversity in Health and Disease

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Interaction of Calmodulin with GTPases
2.1. RalA and RalB
2.2. Rab3A and Rab3D
2.3. Rac1 and Cdc42
2.4. Ric and Rin
2.5. Rad, Kir/Gem, and Rem
3. Ras-GTPases and their Post-Translational Modifications Fine-Tune Cellular Signalling
3.1. The Prominant Role of KRas in Human Cancers
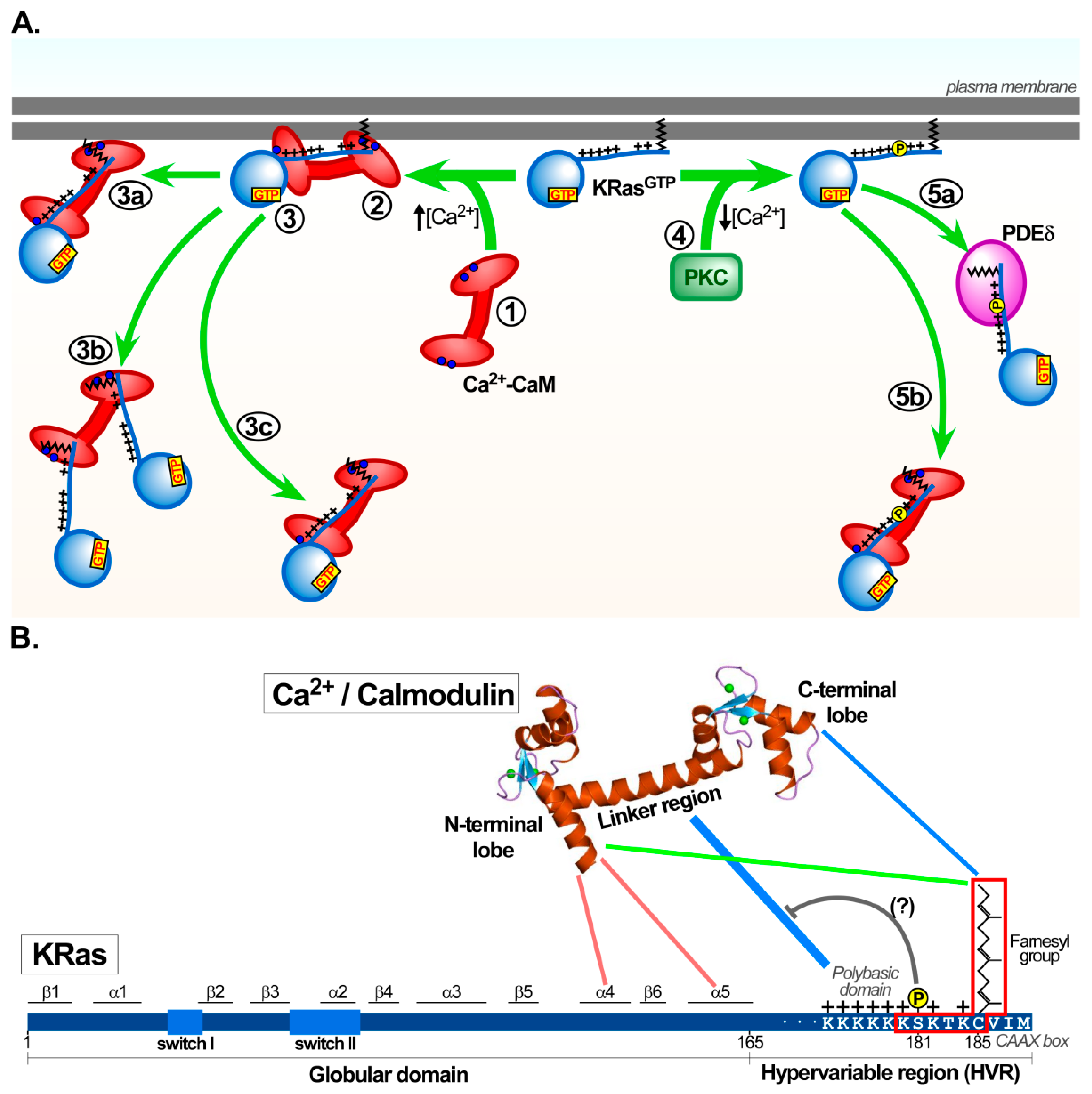
3.2. The Membrane Association of Ras Proteins
3.3. Domains Responsible for the Interaction of Calmodulin with KRas
3.3.1. Outcomes of KRas/calmodulin Interactions in Cell-Based Studies
3.3.2. Additional Insights on the Nucleotide Dependence of KRas/calmodulin Interaction
3.4. Calmodulin-Dependent KRas Activity—Diverse Outcomes for Cellular Signalling in Normal and Oncogenic Settings
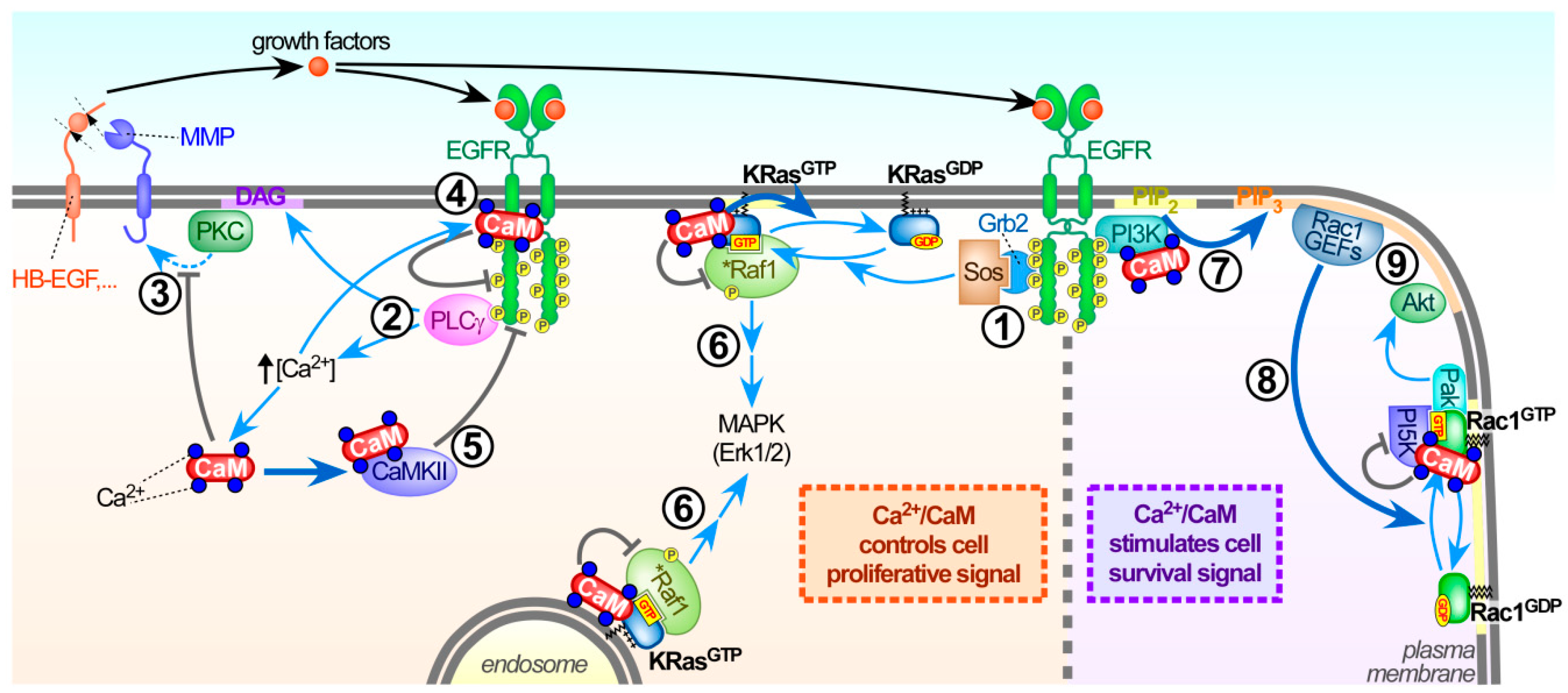
3.4.1. Calmodulin Regulates Several Guanine Nucleotide Exchange Factors (GEFs) and GTPase Activating Proteins (GAPs) in Neuronal Cells
3.4.2. Calmodulin Regulates Mitogen-Activated Protein Kinase (MAPK) and Phosphoinositide-3 Kinase (PI3K) Signalling in Non-neuronal Cells
3.4.3. Calmodulin and Ser181 KRas Phosphorylation
3.4.4. Alternative Models for Calmodulin-Dependent KRas Signalling
4. The Multiple Modes of Calmodulin to Influence Rac1 GTPase Activity
4.1. Overview
4.1.1. GEF-Mediated Rac1 Activation
4.1.2. Rac1 Membrane Association
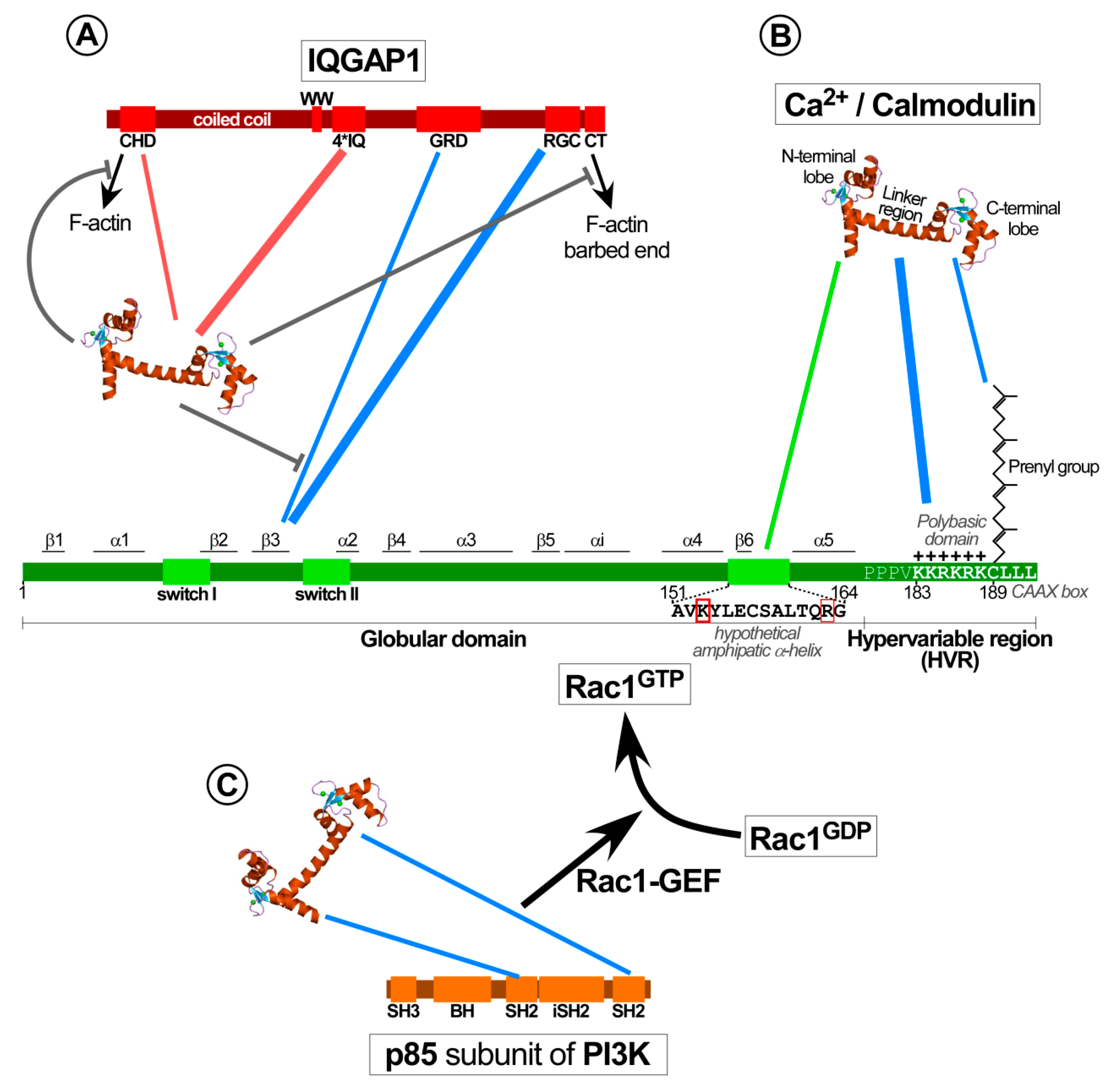
4.2. The Consequences of Direct Rac1/Calmodulin Interactions
4.2.1. Calmodulin Interacts with Active Rac1 in a Ca2+-Dependent Manner
4.2.2. Calmodulin Modulates the Interaction of IQGAP with Rac1 to Facilitate Cytoskeleton Rearrangements
4.3. Calmodulin Influences the Activation of Several Rac1-specific GEFs
5. Adding Another Layer of Complexity: Rac1 is a KRas Effector
6. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Potter, J.D.; Strang-Brown, P.; Walker, P.L.; Iida, S. Ca2+ binding to calmodulin. Methods Enzymol. 1983, 102, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Rhoads, A.R.; Friedberg, F. Sequence motifs for calmodulin recognition. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1997, 11, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.K.; Parameswaran, S. Calmodulin-binding proteins: A journey of 40 years. Cell Calcium 2018, 75, 89–100. [Google Scholar] [CrossRef] [PubMed]
- James, P.; Vorherr, T.; Carafoli, E. Calmodulin-binding domains: Just two faced or multi-faceted? Trends Biochem. Sci. 1995, 20, 38–42. [Google Scholar] [CrossRef]
- Cheney, R.E.; Mooseker, M.S. Unconventional myosins. Curr. Opin. Cell Biol. 1992, 4, 27–35. [Google Scholar] [CrossRef]
- Yap, K.L.; Kim, J.; Truong, K.; Sherman, M.; Yuan, T.; Ikura, M. Calmodulin target database. J. Struct. Funct. Genom. 2000, 1, 8–14. [Google Scholar] [CrossRef]
- McLaughlin, S.; Murray, D. Plasma membrane phosphoinositide organization by protein electrostatics. Nature 2005, 438, 605–611. [Google Scholar] [CrossRef]
- Oughtred, R.; Stark, C.; Breitkreutz, B.J.; Rust, J.; Boucher, L.; Chang, C.; Kolas, N.; O‘Donnell, L.; Leung, G.; McAdam, R.; et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res. 2019, 47, D529–D541. [Google Scholar] [CrossRef] [Green Version]
- Berggard, T.; Arrigoni, G.; Olsson, O.; Fex, M.; Linse, S.; James, P. 140 mouse brain proteins identified by Ca2+-calmodulin affinity chromatography and tandem mass spectrometry. J. Proteome Res. 2006, 5, 669–687. [Google Scholar] [CrossRef]
- O‘Day, D.H. CaMBOT: Profiling and characterizing calmodulin-binding proteins. Cell Signal. 2003, 15, 347–354. [Google Scholar] [CrossRef]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, S. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta 2007, 1773, 1177–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downward, J. Control of ras activation. Cancer Surv. 1996, 27, 87–100. [Google Scholar]
- Marshall, C.J. Ras effectors. Curr. Opin. Cell Biol. 1996, 8, 197–204. [Google Scholar] [CrossRef]
- Lopez-Alcala, C.; Alvarez-Moya, B.; Villalonga, P.; Calvo, M.; Bachs, O.; Agell, N. Identification of essential interacting elements in K-Ras/calmodulin binding and its role in K-Ras localization. J. Biol. Chem. 2008, 283, 10621–10631. [Google Scholar] [CrossRef] [Green Version]
- Abraham, S.J.; Nolet, R.P.; Calvert, R.J.; Anderson, L.M.; Gaponenko, V. The hypervariable region of K-Ras4B is responsible for its specific interactions with calmodulin. Biochemistry 2009, 48, 7575–7583. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.J.; Xu, L.R.; Liao, J.M.; Chen, J.; Liang, Y. Both the C-terminal polylysine region and the farnesylation of K-RasB are important for its specific interaction with calmodulin. PLoS ONE 2011, 6, e21929. [Google Scholar] [CrossRef] [Green Version]
- Villalonga, P.; Lopez-Alcala, C.; Bosch, M.; Chiloeches, A.; Rocamora, N.; Gil, J.; Marais, R.; Marshall, C.J.; Bachs, O.; Agell, N. Calmodulin binds to K-Ras, but not to H- or N-Ras, and modulates its downstream signaling. Mol. Cell. Biol. 2001, 21, 7345–7354. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, R.S.; Clough, R.R.; Bhullar, R.P. Ca2+/calmodulin binds and dissociates K-RasB from membrane. Biochem. Biophys. Res. Commun. 2003, 304, 655–660. [Google Scholar] [CrossRef]
- Xu, B.; Chelikani, P.; Bhullar, R.P. Characterization and functional analysis of the calmodulin-binding domain of Rac1 GTPase. PLoS ONE 2012, 7, e42975. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Quadras, M.; Gelabert-Baldrich, M.; Soriano-Castell, D.; Llado, A.; Rentero, C.; Calvo, M.; Pol, A.; Enrich, C.; Tebar, F. Rac1 and calmodulin interactions modulate dynamics of ARF6-dependent endocytosis. Traffic 2011, 12, 1879–1896. [Google Scholar] [CrossRef] [PubMed]
- Elsaraj, S.M.; Bhullar, R.P. Regulation of platelet Rac1 and Cdc42 activation through interaction with calmodulin. Biochim. Biophys. Acta 2008, 1783, 770–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, R.G.; Chen, J.; Kirn, D.; McCormick, F.; Symons, M. An essential role for Rac in Ras transformation. Nature 1995, 374, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.S.; Lourenco, F.C.; Olson, M.F. K-Ras mediated murine epidermal tumorigenesis is dependent upon and associated with elevated Rac1 activity. PLoS ONE 2011, 6, e17143. [Google Scholar] [CrossRef] [Green Version]
- Joneson, T.; White, M.A.; Wigler, M.H.; Bar-Sagi, D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science 1996, 271, 810–812. [Google Scholar] [CrossRef]
- Villalobo, A.; Berchtold, M.W. The role of calmodulin in tumor cell migration, invasiveness, and metastasis. Int. J. Mol. Sci. 2020, 21, 765. [Google Scholar] [CrossRef] [Green Version]
- Berchtold, M.W.; Villalobo, A. The many faces of calmodulin in cell proliferation, programmed cell death, autophagy, and cancer. Biochim. Biophys. Acta 2014, 1843, 398–435. [Google Scholar] [CrossRef]
- Alvarez-Moya, B.; Lopez-Alcala, C.; Drosten, M.; Bachs, O.; Agell, N. K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene 2010, 29, 5911–5922. [Google Scholar] [CrossRef] [Green Version]
- Coppola, T.; Perret-Menoud, V.; Luthi, S.; Farnsworth, C.C.; Glomset, J.A.; Regazzi, R. Disruption of Rab3-calmodulin interaction, but not other effector interactions, prevents Rab3 inhibition of exocytosis. EMBO J. 1999, 18, 5885–5891. [Google Scholar] [CrossRef] [Green Version]
- Fischer, R.; Wei, Y.; Anagli, J.; Berchtold, M.W. Calmodulin binds to and inhibits GTP binding of the ras-like GTPase Kir/Gem. J. Biol. Chem. 1996, 271, 25067–25070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Della, N.G.; Chew, C.E.; Zack, D.J. Rin, a neuron-specific and calmodulin-binding small G-protein, and Rit define a novel subfamily of ras proteins. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 6784–6794. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, R.S.; Bhullar, R.P. Rab3B in human platelet is membrane bound and interacts with Ca(2+)/calmodulin. Biochem. Biophys. Res. Commun. 2001, 289, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.L.; Khan, M.T.; Roufogalis, B.D. Identification and characterization of a calmodulin-binding domain in Ral-A, a Ras-related GTP-binding protein purified from human erythrocyte membrane. J. Biol. Chem. 1997, 272, 16002–16009. [Google Scholar] [CrossRef] [Green Version]
- Wes, P.D.; Yu, M.; Montell, C. RIC, a calmodulin-binding Ras-like GTPase. EMBO J. 1996, 15, 5839–5848. [Google Scholar] [CrossRef]
- Park, J.B.; Farnsworth, C.C.; Glomset, J.A. Ca2+/calmodulin causes Rab3A to dissociate from synaptic membranes. J. Biol. Chem. 1997, 272, 20857–20865. [Google Scholar] [CrossRef] [Green Version]
- Urano, T.; Emkey, R.; Feig, L.A. Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular transformation. EMBO J. 1996, 15, 810–816. [Google Scholar] [CrossRef]
- Clough, R.R.; Sidhu, R.S.; Bhullar, R.P. Calmodulin binds RalA and RalB and is required for the thrombin-induced activation of Ral in human platelets. J. Biol. Chem. 2002, 277, 28972–28980. [Google Scholar] [CrossRef] [Green Version]
- Sidhu, R.S.; Elsaraj, S.M.; Grujic, O.; Bhullar, R.P. Calmodulin binding to the small GTPase Ral requires isoprenylated Ral. Biochem. Biophys. Res. Commun. 2005, 336, 105–109. [Google Scholar] [CrossRef]
- Wang, K.L.; Roufogalis, B.D. Ca2+/calmodulin stimulates GTP binding to the ras-related protein ral-A. J. Biol. Chem. 1999, 274, 14525–14528. [Google Scholar] [CrossRef] [Green Version]
- van Dam, E.M.; Robinson, P.J. Ral: Mediator of membrane trafficking. Int. J. Biochem. Cell Biol. 2006, 38, 1841–1847. [Google Scholar] [CrossRef]
- Kajio, H.; Olszewski, S.; Rosner, P.J.; Donelan, M.J.; Geoghegan, K.F.; Rhodes, C.J. A low-affinity Ca2+-dependent association of calmodulin with the Rab3A effector domain inversely correlates with insulin exocytosis. Diabetes 2001, 50, 2029–2039. [Google Scholar] [CrossRef] [Green Version]
- Schluter, O.M.; Khvotchev, M.; Jahn, R.; Sudhof, T.C. Localization versus function of Rab3 proteins. Evidence for a common regulatory role in controlling fusion. J. Biol. Chem. 2002, 277, 40919–40929. [Google Scholar] [CrossRef] [Green Version]
- Geppert, M.; Sudhof, T.C. RAB3 and synaptotagmin: The yin and yang of synaptic membrane fusion. Annu. Rev. Neurosci. 1998, 21, 75–95. [Google Scholar] [CrossRef]
- Johannes, L.; Lledo, P.M.; Roa, M.; Vincent, J.D.; Henry, J.P.; Darchen, F. The GTPase Rab3a negatively controls calcium-dependent exocytosis in neuroendocrine cells. EMBO J. 1994, 13, 2029–2037. [Google Scholar] [CrossRef]
- Zhu, S.; Chim, S.M.; Cheng, T.; Ang, E.; Ng, B.; Lim, B.; Chen, K.; Qiu, H.; Tickner, J.; Xu, H.; et al. Calmodulin interacts with Rab3D and modulates osteoclastic bone resorption. Sci. Rep. 2016, 6, 37963. [Google Scholar] [CrossRef] [Green Version]
- Swart-Mataraza, J.M.; Li, Z.; Sacks, D.B. IQGAP1 is a component of Cdc42 signaling to the cytoskeleton. J. Biol. Chem. 2002, 277, 24753–24763. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.D.; Joyal, J.L.; Li, Z.; Sacks, D.B. IQGAP1 integrates Ca2+/calmodulin and Cdc42 signaling. J. Biol. Chem. 1999, 274, 464–470. [Google Scholar] [CrossRef] [Green Version]
- Fukata, M.; Kuroda, S.; Fujii, K.; Nakamura, T.; Shoji, I.; Matsuura, Y.; Okawa, K.; Iwamatsu, A.; Kikuchi, A.; Kaibuchi, K. Regulation of cross-linking of actin filament by IQGAP1, a target for Cdc42. J. Biol. Chem. 1997, 272, 29579–29583. [Google Scholar] [CrossRef] [Green Version]
- Joyal, J.L.; Annan, R.S.; Ho, Y.D.; Huddleston, M.E.; Carr, S.A.; Hart, M.J.; Sacks, D.B. Calmodulin modulates the interaction between IQGAP1 and Cdc42. Identification of IQGAP1 by nanoelectrospray tandem mass spectrometry. J. Biol. Chem. 1997, 272, 15419–15425. [Google Scholar] [CrossRef] [Green Version]
- Briggs, M.W.; Sacks, D.B. IQGAP1 as signal integrator: Ca2+, calmodulin, Cdc42 and the cytoskeleton. FEBS Lett. 2003, 542, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.W.; Li, Z.; Brown, M.D.; Sacks, D.B. IQGAP1 binds Rap1 and modulates its activity. J. Biol. Chem. 2007, 282, 20752–20762. [Google Scholar] [CrossRef] [Green Version]
- Baldassa, S.; Zippel, R.; Sturani, E. Depolarization-induced signaling to Ras, Rap1 and MAPKs in cortical neurons. Brain Res. Mol. Brain Res. 2003, 119, 111–122. [Google Scholar] [CrossRef]
- Hoshino, M.; Nakamura, S. The Ras-like small GTP-binding protein Rin is activated by growth factor stimulation. Biochem. Biophys. Res. Commun. 2002, 295, 651–656. [Google Scholar] [CrossRef]
- Hoshino, M.; Nakamura, S. Small GTPase Rin induces neurite outgrowth through Rac/Cdc42 and calmodulin in PC12 cells. J. Cell Biol. 2003, 163, 1067–1076. [Google Scholar] [CrossRef]
- Harrison, S.M.; Rudolph, J.L.; Spencer, M.L.; Wes, P.D.; Montell, C.; Andres, D.A.; Harrison, D.A. Activated RIC, a small GTPase, genetically interacts with the Ras pathway and calmodulin during Drosophila development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2005, 232, 817–826. [Google Scholar] [CrossRef]
- Kelly, K. The RGK family: A regulatory tail of small GTP-binding proteins. Trends Cell Biol. 2005, 15, 640–643. [Google Scholar] [CrossRef]
- Correll, R.N.; Pang, C.; Niedowicz, D.M.; Finlin, B.S.; Andres, D.A. The RGK family of GTP-binding proteins: Regulators of voltage-dependent calcium channels and cytoskeleton remodeling. Cell. Signal. 2008, 20, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Flynn, R.; Zamponi, G.W. Regulation of calcium channels by RGK proteins. Channels (Austin) 2010, 4, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Moyers, J.S.; Bilan, P.J.; Zhu, J.; Kahn, C.R. Rad and Rad-related GTPases interact with calmodulin and calmodulin-dependent protein kinase II. J. Biol. Chem. 1997, 272, 11832–11839. [Google Scholar] [CrossRef] [Green Version]
- Beguin, P.; Mahalakshmi, R.N.; Nagashima, K.; Cher, D.H.; Takahashi, A.; Yamada, Y.; Seino, Y.; Hunziker, W. 14-3-3 and calmodulin control subcellular distribution of Kir/Gem and its regulation of cell shape and calcium channel activity. J. Cell Sci. 2005, 118, 1923–1934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beguin, P.; Mahalakshmi, R.N.; Nagashima, K.; Cher, D.H.; Kuwamura, N.; Yamada, Y.; Seino, Y.; Hunziker, W. Roles of 14-3-3 and calmodulin binding in subcellular localization and function of the small G-protein Rem2. Biochem. J. 2005, 390, 67–75. [Google Scholar] [CrossRef]
- Beguin, P.; Nagashima, K.; Gonoi, T.; Shibasaki, T.; Takahashi, K.; Kashima, Y.; Ozaki, N.; Geering, K.; Iwanaga, T.; Seino, S. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature 2001, 411, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Mahalakshmi, R.N.; Ng, M.Y.; Guo, K.; Qi, Z.; Hunziker, W.; Beguin, P. Nuclear localization of endogenous RGK proteins and modulation of cell shape remodeling by regulated nuclear transport. Traffic 2007, 8, 1164–1178. [Google Scholar] [CrossRef]
- Moyers, J.S.; Zhu, J.; Kahn, C.R. Effects of phosphorylation on function of the Rad GTPase. Biochem. J. 1998, 333, 609–614. [Google Scholar] [CrossRef] [Green Version]
- Mahalakshmi, R.N.; Nagashima, K.; Ng, M.Y.; Inagaki, N.; Hunziker, W.; Beguin, P. Nuclear transport of Kir/Gem requires specific signals and importin alpha5 and is regulated by calmodulin and predicted serine phosphorylations. Traffic 2007, 8, 1150–1163. [Google Scholar] [CrossRef]
- Fivaz, M.; Meyer, T. Reversible intracellular translocation of KRas but not HRas in hippocampal neurons regulated by Ca2+/calmodulin. J. Cell Biol. 2005, 170, 429–441. [Google Scholar] [CrossRef]
- Agamasu, C.; Ghirlando, R.; Taylor, T.; Messing, S.; Tran, T.H.; Bindu, L.; Tonelli, M.; Nissley, D.V.; McCormick, F.; Stephen, A.G. KRAS prenylation is required for bivalent binding with calmodulin in a nucleotide-independent manner. Biophys. J. 2019, 116, 1049–1063. [Google Scholar] [CrossRef] [Green Version]
- Park, J.B.; Lee, J.Y.; Kim, J.W. Dissociation of RalA from synaptic membranes by Ca2+/calmodulin. Biochem. Biophys. Res. Commun. 1999, 263, 765–769. [Google Scholar] [CrossRef]
- Park, J.B.; Kim, J.S.; Lee, J.Y.; Kim, J.; Seo, J.Y.; Kim, A.R. GTP binds to Rab3A in a complex with Ca2+/calmodulin. Biochem. J. 2002, 362, 651–657. [Google Scholar] [CrossRef]
- Yunes, R.; Tomes, C.; Michaut, M.; De Blas, G.; Rodriguez, F.; Regazzi, R.; Mayorga, L.S. Rab3A and calmodulin regulate acrosomal exocytosis by mechanisms that do not require a direct interaction. FEBS Lett. 2002, 525, 126–130. [Google Scholar] [CrossRef] [Green Version]
- Boldt, K.; van Reeuwijk, J.; Lu, Q.; Koutroumpas, K.; Nguyen, T.M.; Texier, Y.; van Beersum, S.E.; Horn, N.; Willer, J.R.; Mans, D.A.; et al. An organelle-specific protein landscape identifies novel diseases and molecular mechanisms. Nat. Commun. 2016, 7, 11491. [Google Scholar] [CrossRef] [PubMed]
- Park, J.B. Regulation of GTP-binding state in RalA through Ca2+ and calmodulin. Exp. Mol. Med. 2001, 33, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Correll, R.N.; Pang, C.; Niedowicz, D.M.; Satin, J.; Andres, D.A. Calmodulin binding is dispensable for Rem-mediated Ca2+ channel inhibition. Mol. Cell. Biochem. 2008, 310, 103–110. [Google Scholar] [CrossRef]
- Marshall, C. How do small GTPase signal transduction pathways regulate cell cycle entry? Curr. Opin. Cell Biol. 1999, 11, 732–736. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Downward, J. Ras signalling and apoptosis. Curr. Opin. Genet. Dev. 1998, 8, 49–54. [Google Scholar] [CrossRef]
- Khosravi-Far, R.; Campbell, S.; Rossman, K.L.; Der, C.J. Increasing complexity of Ras signal transduction: Involvement of Rho family proteins. Adv. Cancer Res. 1998, 72, 57–107. [Google Scholar] [CrossRef]
- Rebollo, A.; Martinez, A.C. Ras proteins: Recent advances and new functions. Blood 1999, 94, 2971–2980. [Google Scholar] [CrossRef]
- Omerovic, J.; Hammond, D.E.; Clague, M.J.; Prior, I.A. Ras isoform abundance and signalling in human cancer cell lines. Oncogene 2008, 27, 2754–2762. [Google Scholar] [CrossRef] [Green Version]
- Bos, J.L. Ras oncogenes in human cancer: A review. Cancer Res. 1989, 49, 4682–4689. [Google Scholar]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Lakshman, B.; Messing, S.; Schmid, E.M.; Clogston, J.D.; Gillette, W.K.; Esposito, D.; Kessing, B.; Fletcher, D.A.; Nissley, D.V.; McCormick, F.; et al. Quantitative biophysical analysis defines key components modulating recruitment of the GTPase KRAS to the plasma membrane. J. Biol. Chem. 2019, 294, 2193–2207. [Google Scholar] [CrossRef] [Green Version]
- Newlaczyl, A.U.; Hood, F.E.; Coulson, J.M.; Prior, I.A. Decoding RAS isoform and codon-specific signalling. Biochem. Soc. Trans. 2014, 42, 742–746. [Google Scholar] [CrossRef] [Green Version]
- Hancock, J.F. Ras proteins: Different signals from different locations. Nat. Rev. Mol. Cell Biol. 2003, 4, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Muncke, C.; Parton, R.G.; Hancock, J.F. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 2003, 160, 165–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, T.; Harding, A.; Inder, K.; Plowman, S.; Parton, R.G.; Hancock, J.F. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat. Cell Biol. 2007, 9, 905–914. [Google Scholar] [CrossRef]
- Matallanas, D.; Sanz-Moreno, V.; Arozarena, I.; Calvo, F.; Agudo-Ibanez, L.; Santos, E.; Berciano, M.T.; Crespo, P. Distinct utilization of effectors and biological outcomes resulting from site-specific Ras activation: Ras functions in lipid rafts and Golgi complex are dispensable for proliferation and transformation. Mol. Cell. Biol. 2006, 26, 100–116. [Google Scholar] [CrossRef] [Green Version]
- Omerovic, J.; Laude, A.J.; Prior, I.A. Ras proteins: Paradigms for compartmentalised and isoform-specific signalling. Cell. Mol. Life Sci. 2007, 64, 2575–2589. [Google Scholar] [CrossRef] [Green Version]
- Henis, Y.I.; Hancock, J.F.; Prior, I.A. Ras acylation, compartmentalization and signaling nanoclusters (Review). Mol. Membr. Biol. 2009, 26, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, S.; Henis, Y.I. Interactions of Ras proteins with the plasma membrane and their roles in signaling. Cell Signal. 2008, 20, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Agudo-Ibanez, L.; Crespo, P. The Ras-ERK pathway: Understanding site-specific signaling provides hope of new anti-tumor therapies. Bioessays News Rev. Mol. Cell. Dev. Biol. 2010, 32, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hancock, J.F. Ras trafficking, localization and compartmentalized signalling. Semin. Cell Dev. Biol. 2012, 23, 145–153. [Google Scholar] [CrossRef] [Green Version]
- Tebar, F.; Enrich, C.; Rentero, C.; Grewal, T. GTPases Rac1 and Ras signaling from endosomes. Prog. Mol. Subcell. Biol. 2018, 57, 65–105. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, N.; Bar-Sagi, D.; Philips, M. Ras/MAPK signaling from endomembranes. Mol. Oncol. 2009, 3, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Heo, W.D.; Inoue, T.; Park, W.S.; Kim, M.L.; Park, B.O.; Wandless, T.J.; Meyer, T. PI(3,4,5)P3 and PI(4,5)P2 lipids target proteins with polybasic clusters to the plasma membrane. Science 2006, 314, 1458–1461. [Google Scholar] [CrossRef] [Green Version]
- Cho, K.J.; Park, J.H.; Piggott, A.M.; Salim, A.A.; Gorfe, A.A.; Parton, R.G.; Capon, R.J.; Lacey, E.; Hancock, J.F. Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. J. Biol. Chem. 2012, 287, 43573–43584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, K.J.; van der Hoeven, D.; Zhou, Y.; Maekawa, M.; Ma, X.; Chen, W.; Fairn, G.D.; Hancock, J.F. Inhibition of acid sphingomyelinase depletes cellular phosphatidylserine and mislocalizes K-Ras from the plasma membrane. Mol. Cell. Biol. 2016, 36, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Gulyas, G.; Radvanszki, G.; Matuska, R.; Balla, A.; Hunyady, L.; Balla, T.; Varnai, P. Plasma membrane phosphatidylinositol 4-phosphate and 4,5-bisphosphate determine the distribution and function of K-Ras4B but not H-Ras proteins. J. Biol. Chem. 2017, 292, 18862–18877. [Google Scholar] [CrossRef] [Green Version]
- Gelabert-Baldrich, M.; Soriano-Castell, D.; Calvo, M.; Lu, A.; Vina-Vilaseca, A.; Rentero, C.; Pol, A.; Grinstein, S.; Enrich, C.; Tebar, F. Dynamics of KRas on endosomes: Involvement of acidic phospholipids in its association. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 3023–3037. [Google Scholar] [CrossRef]
- Lu, A.; Tebar, F.; Alvarez-Moya, B.; Lopez-Alcala, C.; Calvo, M.; Enrich, C.; Agell, N.; Nakamura, T.; Matsuda, M.; Bachs, O. A clathrin-dependent pathway leads to KRas signaling on late endosomes en route to lysosomes. J. Cell Biol. 2009, 184, 863–879. [Google Scholar] [CrossRef] [Green Version]
- Miaczynska, M.; Bar-Sagi, D. Signaling endosomes: Seeing is believing. Curr. Opin. Cell Biol. 2010, 22, 535–540. [Google Scholar] [CrossRef] [Green Version]
- von Zastrow, M.; Sorkin, A. Signaling on the endocytic pathway. Curr. Opin. Cell Biol. 2007, 19, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Omerovic, J.; Prior, I.A. Compartmentalized signalling: Ras proteins and signalling nanoclusters. FEBS J. 2009, 276, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- Bivona, T.G.; Philips, M.R. Ras pathway signaling on endomembranes. Curr. Opin. Cell Biol. 2003, 15, 136–142. [Google Scholar] [CrossRef]
- Stasyk, T.; Huber, L.A. Spatio-temporal parameters of endosomal signaling in cancer: Implications for new treatment options. J. Cell. Biochem. 2016, 117, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Villalonga, P.; Lopez-Alcala, C.; Chiloeches, A.; Gil, J.; Marais, R.; Bachs, O.; Agell, N. Calmodulin prevents activation of Ras by PKC in 3T3 fibroblasts. J. Biol. Chem. 2002, 277, 37929–37935. [Google Scholar] [CrossRef] [Green Version]
- Garrido, E.; Lazaro, J.; Jaumot, M.; Agell, N.; Rubio-Martinez, J. Modeling and subtleties of K-Ras and Calmodulin interaction. PLoS Comput. Biol. 2018, 14, e1006552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperlich, B.; Kapoor, S.; Waldmann, H.; Winter, R.; Weise, K. Regulation of K-Ras4B membrane binding by calmodulin. Biophys. J. 2016, 111, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, H.; Banerjee, A.; Marcus, K.; Makowski, L.; Mattos, C.; Gaponenko, V.; Nussinov, R. The Structural basis of the farnesylated and methylated KRas4B interaction with calmodulin. Structure 2019, 27, 1647–1659. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.M.M.; Enomoto, M.; Back, S.I.; Lee, K.Y.; Gebregiworgis, T.; Ishiyama, N.; Ikura, M.; Marshall, C.B. Calmodulin disrupts plasma membrane localization of farnesylated KRAS4b by sequestering its lipid moiety. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J.; Chakrabarti, M.; Jang, H. A new view of Ras isoforms in cancers. Cancer Res. 2016, 76, 18–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashery, U.; Yizhar, O.; Rotblat, B.; Kloog, Y. Nonconventional trafficking of Ras associated with Ras signal organization. Traffic 2006, 7, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Muratcioglu, S.; Tsai, C.J.; Jang, H.; Gursoy, A.; Keskin, O. The key role of calmodulin in KRAS-Driven adenocarcinomas. Mol. Cancer Res. 2015, 13, 1265–1273. [Google Scholar] [CrossRef] [Green Version]
- Lu, S.; Jang, H.; Gu, S.; Zhang, J.; Nussinov, R. Drugging Ras GTPase: A comprehensive mechanistic and signaling structural view. Chem. Soc. Rev. 2016, 45, 4929–4952. [Google Scholar] [CrossRef] [Green Version]
- Cook, S.J.; Lockyer, P.J. Recent advances in Ca(2+)-dependent Ras regulation and cell proliferation. Cell Calcium 2006, 39, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Chavan, T.S.; Abraham, S.; Gaponenko, V. Application of reductive (1)(3)C-methylation of lysines to enhance the sensitivity of conventional NMR methods. Molecules 2013, 18, 7103–7119. [Google Scholar] [CrossRef] [Green Version]
- Bhagatji, P.; Leventis, R.; Rich, R.; Lin, C.J.; Silvius, J.R. Multiple cellular proteins modulate the dynamics of K-ras association with the plasma membrane. Biophys. J. 2010, 99, 3327–3335. [Google Scholar] [CrossRef] [Green Version]
- Chandra, A.; Grecco, H.E.; Pisupati, V.; Perera, D.; Cassidy, L.; Skoulidis, F.; Ismail, S.A.; Hedberg, C.; Hanzal-Bayer, M.; Venkitaraman, A.R.; et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 2011, 14, 148–158. [Google Scholar] [CrossRef]
- Figueroa, C.; Taylor, J.; Vojtek, A.B. Prenylated Rab acceptor protein is a receptor for prenylated small GTPases. J. Biol. Chem. 2001, 276, 28219–28225. [Google Scholar] [CrossRef] [Green Version]
- Barcelo, C.; Paco, N.; Beckett, A.J.; Alvarez-Moya, B.; Garrido, E.; Gelabert, M.; Tebar, F.; Jaumot, M.; Prior, I.; Agell, N. Oncogenic K-ras segregates at spatially distinct plasma membrane signaling platforms according to its phosphorylation status. J. Cell Sci. 2013, 126, 4553–4559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavan, T.S.; Jang, H.; Khavrutskii, L.; Abraham, S.J.; Banerjee, A.; Freed, B.C.; Johannessen, L.; Tarasov, S.G.; Gaponenko, V.; Nussinov, R.; et al. High-affinity interaction of the K-Ras4B hypervariable region with the Ras active site. Biophys. J. 2015, 109, 2602–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Moya, B.; Barcelo, C.; Tebar, F.; Jaumot, M.; Agell, N. CaM interaction and Ser181 phosphorylation as new K-Ras signaling modulators. Small Gtpases 2011, 2, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moodie, S.A.; Willumsen, B.M.; Weber, M.J.; Wolfman, A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260, 1658–1661. [Google Scholar] [CrossRef] [PubMed]
- Koide, H.; Satoh, T.; Nakafuku, M.; Kaziro, Y. GTP-dependent association of Raf-1 with Ha-Ras: Identification of Raf as a target downstream of Ras in mammalian cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8683–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, T.S.; Shapiro, P.S.; Ahn, N.G. Signal transduction through MAP kinase cascades. Adv. Cancer Res. 1998, 74, 49–139. [Google Scholar] [CrossRef]
- Robinson, M.J.; Cobb, M.H. Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 1997, 9, 180–186. [Google Scholar] [CrossRef]
- Alessi, D.R.; Cohen, P. Mechanism of activation and function of protein kinase B. Curr. Opin. Genet. Dev. 1998, 8, 55–62. [Google Scholar] [CrossRef]
- Coffer, P.J.; Jin, J.; Woodgett, J.R. Protein kinase B (c-Akt): A multifunctional mediator of phosphatidylinositol 3-kinase activation. Biochem. J. 1998, 335, 1–13. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Zippel, R.; Balestrini, M.; Lomazzi, M.; Sturani, E. Calcium and calmodulin are essential for Ras-GRF1-mediated activation of the Ras pathway by lysophosphatidic acid. Exp. Cell Res. 2000, 258, 403–408. [Google Scholar] [CrossRef]
- Gotoh, T.; Tian, X.; Feig, L.A. Prenylation of target GTPases contributes to signaling specificity of Ras-guanine nucleotide exchange factors. J. Biol. Chem. 2001, 276, 38029–38035. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.S.; Manzerra, P.; Kennedy, M.B. Regulation of the neuron-specific Ras GTPase-activating protein, synGAP, by Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 2004, 279, 17980–17988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norum, J.H.; Methi, T.; Mattingly, R.R.; Levy, F.O. Endogenous expression and protein kinase A-dependent phosphorylation of the guanine nucleotide exchange factor Ras-GRF1 in human embryonic kidney 293 cells. FEBS J. 2005, 272, 2304–2316. [Google Scholar] [CrossRef]
- Tian, X.; Feig, L.A. Age-dependent participation of Ras-GRF proteins in coupling calcium-permeable AMPA glutamate receptors to Ras/Erk signaling in cortical neurons. J. Biol. Chem. 2006, 281, 7578–7582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnsworth, C.L.; Freshney, N.W.; Rosen, L.B.; Ghosh, A.; Greenberg, M.E.; Feig, L.A. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 1995, 376, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Feig, L.A. Regulation of neuronal function by Ras-GRF exchange factors. Genes Cancer 2011, 2, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkup, W.G.T.; Washburn, L.; Sweredoski, M.J.; Carlisle, H.J.; Graham, R.L.; Hess, S.; Kennedy, M.B. Phosphorylation of synaptic GTPase-activating protein (synGAP) by Ca2+/calmodulin-dependent protein kinase II (CaMKII) and cyclin-dependent kinase 5 (CDK5) alters the ratio of its GAP activity toward Ras and Rap GTPases. J. Biol. Chem. 2015, 290, 4908–4927. [Google Scholar] [CrossRef] [Green Version]
- Rockliffe, N.; Gawler, D. Differential mechanisms of glutamate receptor regulation of SynGAP in cortical neurones. FEBS Lett. 2006, 580, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Tebar, F.; Villalonga, P.; Sorkina, T.; Agell, N.; Sorkin, A.; Enrich, C. Calmodulin regulates intracellular trafficking of epidermal growth factor receptor and the MAPK signaling pathway. Mol. Biol. Cell 2002, 13, 2057–2068. [Google Scholar] [CrossRef] [Green Version]
- Tebar, F.; Llado, A.; Enrich, C. Role of calmodulin in the modulation of the MAPK signalling pathway and the transactivation of epidermal growth factor receptor mediated by PKC. FEBS Lett. 2002, 517, 206–210. [Google Scholar] [CrossRef] [Green Version]
- Moreto, J.; Vidal-Quadras, M.; Pol, A.; Santos, E.; Grewal, T.; Enrich, C.; Tebar, F. Differential involvement of H- and K-Ras in Raf-1 activation determines the role of calmodulin in MAPK signaling. Cell. Signal. 2009, 21, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Moreto, J.; Llado, A.; Vidal-Quadras, M.; Calvo, M.; Pol, A.; Enrich, C.; Tebar, F. Calmodulin modulates H-Ras mediated Raf-1 activation. Cell Signal. 2008, 20, 1092–1103. [Google Scholar] [CrossRef]
- Bosch, M.; Gil, J.; Bachs, O.; Agell, N. Calmodulin inhibitor W13 induces sustained activation of ERK2 and expression of p21(cip1). J. Biol. Chem. 1998, 273, 22145–22150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshiki, S.; Matsunaga-Udagawa, R.; Aoki, K.; Kamioka, Y.; Kiyokawa, E.; Matsuda, M. Ras and calcium signaling pathways converge at Raf1 via the Shoc2 scaffold protein. Mol. Biol. Cell 2010, 21, 1088–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcheva, M.M.; Szucs, M.; Wang, D.; Sadee, W.; Coscia, C.J. Mu-Opioid receptor-mediated ERK activation involves calmodulin-dependent epidermal growth factor receptor transactivation. J. Biol. Chem. 2001, 276, 33847–33853. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Rodriguez, E.; Esparis-Ogando, A.; Montero, J.C.; Yuste, L.; Pandiella, A. Stimulation of cleavage of membrane proteins by calmodulin inhibitors. Biochem. J. 2000, 346, 359–367. [Google Scholar] [CrossRef]
- Dong, J.; Wiley, H.S. Trafficking and proteolytic release of epidermal growth factor receptor ligands are modulated by their membrane-anchoring domains. J. Biol. Chem. 2000, 275, 557–564. [Google Scholar] [CrossRef] [Green Version]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar] [CrossRef]
- Li, H.; Ruano, M.J.; Villalobo, A. Endogenous calmodulin interacts with the epidermal growth factor receptor in living cells. FEBS Lett. 2004, 559, 175–180. [Google Scholar] [CrossRef]
- San Jose, E.; Benguria, A.; Geller, P.; Villalobo, A. Calmodulin inhibits the epidermal growth factor receptor tyrosine kinase. J. Biol. Chem. 1992, 267, 15237–15245. [Google Scholar]
- Martin-Nieto, J.; Villalobo, A. The human epidermal growth factor receptor contains a juxtamembrane calmodulin-binding site. Biochemistry 1998, 37, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Feinmesser, R.L.; Wicks, S.J.; Taverner, C.J.; Chantry, A. Ca2+/calmodulin-dependent kinase II phosphorylates the epidermal growth factor receptor on multiple sites in the cytoplasmic tail and serine 744 within the kinase domain to regulate signal generation. J. Biol. Chem. 1999, 274, 16168–16173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bivona, T.G.; Quatela, S.E.; Bodemann, B.O.; Ahearn, I.M.; Soskis, M.J.; Mor, A.; Miura, J.; Wiener, H.H.; Wright, L.; Saba, S.G.; et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol. Cell 2006, 21, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Melien, O.; Christoffersen, T.; Sioud, M. Evidence for the involvement of Gi2 in activation of extracellular signal-regulated kinases in hepatocytes. BMC Cell Biol. 2001, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Egea, J.; Espinet, C.; Soler, R.M.; Peiro, S.; Rocamora, N.; Comella, J.X. Nerve growth factor activation of the extracellular signal-regulated kinase pathway is modulated by Ca(2+) and calmodulin. Mol. Cell. Biol. 2000, 20, 1931–1946. [Google Scholar] [CrossRef] [Green Version]
- Buchanan, F.G.; Elliot, C.M.; Gibbs, M.; Exton, J.H. Translocation of the Rac1 guanine nucleotide exchange factor Tiam1 induced by platelet-derived growth factor and lysophosphatidic acid. J. Biol. Chem. 2000, 275, 9742–9748. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I.N.; Elliott, C.M.; Buchanan, F.G.; Downes, C.P.; Exton, J.H. Ca2+/calmodulin-dependent protein kinase II regulates Tiam1 by reversible protein phosphorylation. J. Biol. Chem. 1999, 274, 12753–12758. [Google Scholar] [CrossRef] [Green Version]
- Perez-Garcia, M.J.; Cena, V.; de Pablo, Y.; Llovera, M.; Comella, J.X.; Soler, R.M. Glial cell line-derived neurotrophic factor increases intracellular calcium concentration. Role of calcium/calmodulin in the activation of the phosphatidylinositol 3-kinase pathway. J. Biol. Chem. 2004, 279, 6132–6142. [Google Scholar] [CrossRef] [Green Version]
- Joyal, J.L.; Burks, D.J.; Pons, S.; Matter, W.F.; Vlahos, C.J.; White, M.F.; Sacks, D.B. Calmodulin activates phosphatidylinositol 3-kinase. J. Biol. Chem. 1997, 272, 28183–28186. [Google Scholar] [CrossRef] [Green Version]
- Fischer, R.; Julsgart, J.; Berchtold, M.W. High affinity calmodulin target sequence in the signalling molecule PI 3-kinase. FEBS Lett. 1998, 425, 175–177. [Google Scholar] [CrossRef] [Green Version]
- Ballester, R.; Furth, M.E.; Rosen, O.M. Phorbol ester- and protein kinase C-mediated phosphorylation of the cellular Kirsten ras gene product. J. Biol. Chem. 1987, 262, 2688–2695. [Google Scholar] [PubMed]
- Eisenberg, S.; Giehl, K.; Henis, Y.I.; Ehrlich, M. Differential interference of chlorpromazine with the membrane interactions of oncogenic K-Ras and its effects on cell growth. J. Biol. Chem. 2008, 283, 27279–27288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philips, M.R. Ras hitchhikes on PDE6delta. Nat. Cell Biol. 2012, 14, 128–129. [Google Scholar] [CrossRef] [Green Version]
- Liao, J.; Planchon, S.M.; Wolfman, J.C.; Wolfman, A. Growth factor-dependent AKT activation and cell migration requires the function of c-K(B)-Ras versus other cellular ras isoforms. J. Biol. Chem. 2006, 281, 29730–29738. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Wang, G.; Tsai, C.J.; Jang, H.; Lu, S.; Banerjee, A.; Zhang, J.; Gaponenko, V. Calmodulin and PI3K Signaling in KRAS Cancers. Trends Cancer 2017, 3, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras promotes tumorigenicity through suppression of non-canonical Wnt signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef] [Green Version]
- Liou, J.S.; Chen, J.S.; Faller, D.V. Characterization of p21Ras-mediated apoptosis induced by protein kinase C inhibition and application to human tumor cell lines. J. Cell. Physiol. 2004, 198, 277–294. [Google Scholar] [CrossRef]
- Xia, S.; Chen, Z.; Forman, L.W.; Faller, D.V. PKCdelta survival signaling in cells containing an activated p21Ras protein requires PDK1. Cell Signal. 2009, 21, 502–508. [Google Scholar] [CrossRef] [Green Version]
- Xia, S.; Forman, L.W.; Faller, D.V. Protein kinase C delta is required for survival of cells expressing activated p21RAS. J. Biol. Chem. 2007, 282, 13199–13210. [Google Scholar] [CrossRef] [Green Version]
- Barcelo, C.; Etchin, J.; Mansour, M.R.; Sanda, T.; Ginesta, M.M.; Sanchez-Arevalo Lobo, V.J.; Real, F.X.; Capella, G.; Estanyol, J.M.; Jaumot, M.; et al. Ribonucleoprotein HNRNPA2B1 interacts with and regulates oncogenic KRAS in pancreatic ductal adenocarcinoma cells. Gastroenterology 2014, 147, 882–892.e888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcelo, C.; Paco, N.; Morell, M.; Alvarez-Moya, B.; Bota-Rabassedas, N.; Jaumot, M.; Vilardell, F.; Capella, G.; Agell, N. Phosphorylation at Ser-181 of oncogenic KRAS is required for tumor growth. Cancer Res. 2014, 74, 1190–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, A.L.; Hall, A. Rho GTPases and their effector proteins. Biochem. J. 2000, 348, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Didsbury, J.; Weber, R.F.; Bokoch, G.M.; Evans, T.; Snyderman, R. Rac, a novel ras-related family of proteins that are botulinum toxin substrates. J. Biol. Chem. 1989, 264, 16378–16382. [Google Scholar] [PubMed]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, E.E.; Mulloy, J.C.; Zheng, Y. Rac1 GTPase: A “Rac“ of all trades. Cell. Mol. Life Sci. 2009, 66, 370–374. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006, 16, 522–529. [Google Scholar] [CrossRef]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Bustelo, X.R.; Sauzeau, V.; Berenjeno, I.M. GTP-binding proteins of the Rho/Rac family: Regulation, effectors and functions in vivo. Bioessays News Rev. Mol. Cell. Dev. Biol. 2007, 29, 356–370. [Google Scholar] [CrossRef] [Green Version]
- Bolis, A.; Corbetta, S.; Cioce, A.; de Curtis, I. Differential distribution of Rac1 and Rac3 GTPases in the developing mouse brain: Implications for a role of Rac3 in Purkinje cell differentiation. Eur. J. Neurosci. 2003, 18, 2417–2424. [Google Scholar] [CrossRef]
- Li, Q.; Ho, C.S.; Marinescu, V.; Bhatti, H.; Bokoch, G.M.; Ernst, S.A.; Holz, R.W.; Stuenkel, E.L. Facilitation of Ca(2+)-dependent exocytosis by Rac1-GTPase in bovine chromaffin cells. J. Physiol. 2003, 550, 431–445. [Google Scholar] [CrossRef] [PubMed]
- Hong-Geller, E.; Cerione, R.A. Cdc42 and Rac stimulate exocytosis of secretory granules by activating the IP(3)/calcium pathway in RBL-2H3 mast cells. J. Cell Biol. 2000, 148, 481–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marei, H.; Malliri, A. Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small Gtpases 2017, 8, 139–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hordijk, P.L.; ten Klooster, J.P.; van der Kammen, R.A.; Michiels, F.; Oomen, L.C.; Collard, J.G. Inhibition of invasion of epithelial cells by Tiam1-Rac signaling. Science 1997, 278, 1464–1466. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. RHO-GTPases and cancer. Nat. Rev. Cancer 2002, 2, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.A.; Khokhlatchev, A.; Stippec, S.; White, M.A.; Cobb, M.H. Differential effects of PAK1-activating mutations reveal activity-dependent and -independent effects on cytoskeletal regulation. J. Biol. Chem. 1998, 273, 28191–28198. [Google Scholar] [CrossRef] [Green Version]
- Yang, N.; Higuchi, O.; Ohashi, K.; Nagata, K.; Wada, A.; Kangawa, K.; Nishida, E.; Mizuno, K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature 1998, 393, 809–812. [Google Scholar] [CrossRef]
- Vidal, C.; Geny, B.; Melle, J.; Jandrot-Perrus, M.; Fontenay-Roupie, M. Cdc42/Rac1-dependent activation of the p21-activated kinase (PAK) regulates human platelet lamellipodia spreading: Implication of the cortical-actin binding protein cortactin. Blood 2002, 100, 4462–4469. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.A.; Zhou, S.; Eves, R.; Shen, L.; Jia, L.; Mak, A.S. Phosphorylation of cortactin by p21-activated kinase. Arch. Biochem. Biophys. 2006, 456, 183–193. [Google Scholar] [CrossRef]
- Sauvonnet, N.; Dujeancourt, A.; Dautry-Varsat, A. Cortactin and dynamin are required for the clathrin-independent endocytosis of gammac cytokine receptor. J. Cell Biol. 2005, 168, 155–163. [Google Scholar] [CrossRef]
- Grassart, A.; Meas-Yedid, V.; Dufour, A.; Olivo-Marin, J.C.; Dautry-Varsat, A.; Sauvonnet, N. Pak1 phosphorylation enhances cortactin-N-WASP interaction in clathrin-caveolin-independent endocytosis. Traffic 2010, 11, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Vetter, I.R.; Wittinghofer, A. The guanine nucleotide-binding switch in three dimensions. Science 2001, 294, 1299–1304. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, G.R.; Nassar, N.; Cerione, R.A. Structure of the Rho family GTP-binding protein Cdc42 in complex with the multifunctional regulator RhoGDI. Cell 2000, 100, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Marei, H.; Malliri, A. GEFs: Dual regulation of Rac1 signaling. Small Gtpases 2017, 8, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Olofsson, B. Rho guanine dissociation inhibitors: Pivotal molecules in cellular signalling. Cell. Signal. 1999, 11, 545–554. [Google Scholar] [CrossRef]
- DerMardirossian, C.; Bokoch, G.M. GDIs: Central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005, 15, 356–363. [Google Scholar] [CrossRef]
- Grizot, S.; Faure, J.; Fieschi, F.; Vignais, P.V.; Dagher, M.C.; Pebay-Peyroula, E. Crystal structure of the Rac1-RhoGDI complex involved in nadph oxidase activation. Biochemistry 2001, 40, 10007–10013. [Google Scholar] [CrossRef]
- Price, L.S.; Langeslag, M.; ten Klooster, J.P.; Hordijk, P.L.; Jalink, K.; Collard, J.G. Calcium signaling regulates translocation and activation of Rac. J. Biol. Chem. 2003, 278, 39413–39421. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.; Hall, A. Guanine nucleotide exchange factors for Rho GTPases: Turning on the switch. Genes Dev. 2002, 16, 1587–1609. [Google Scholar] [CrossRef] [Green Version]
- Eva, A.; Aaronson, S.A. Isolation of a new human oncogene from a diffuse B-cell lymphoma. Nature 1985, 316, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y. Dbl family guanine nucleotide exchange factors. Trends Biochem. Sci. 2001, 26, 724–732. [Google Scholar] [CrossRef]
- Cote, J.F.; Vuori, K. Identification of an evolutionarily conserved superfamily of DOCK180-related proteins with guanine nucleotide exchange activity. J. Cell Sci. 2002, 115, 4901–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, H.C.; Coadwell, W.J.; Stephens, L.R.; Hawkins, P.T. Phosphoinositide 3-kinase-dependent activation of Rac. FEBS Lett. 2003, 546, 93–97. [Google Scholar] [CrossRef] [Green Version]
- Palamidessi, A.; Frittoli, E.; Garre, M.; Faretta, M.; Mione, M.; Testa, I.; Diaspro, A.; Lanzetti, L.; Scita, G.; Di Fiore, P.P. Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell 2008, 134, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scita, G.; Di Fiore, P.P. The endocytic matrix. Nature 2010, 463, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Menard, L.; Parker, P.J.; Kermorgant, S. Receptor tyrosine kinase c-Met controls the cytoskeleton from different endosomes via different pathways. Nat. Commun. 2014, 5, 3907. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Lerida, I.; Sanchez-Perales, S.; Calvo, M.; Rentero, C.; Zheng, Y.; Enrich, C.; Del Pozo, M.A. A palmitoylation switch mechanism regulates Rac1 function and membrane organization. EMBO J. 2012, 31, 534–551. [Google Scholar] [CrossRef] [Green Version]
- Briggs, M.W.; Sacks, D.B. IQGAP proteins are integral components of cytoskeletal regulation. EMBO Rep. 2003, 4, 571–574. [Google Scholar] [CrossRef] [Green Version]
- Xu, B.; Bhullar, R.P. Regulation of Rac1 and Cdc42 activation in thrombin- and collagen-stimulated CHRF-288-11 cells. Mol. Cell. Biochem. 2011, 353, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Soriano-Castell, D.; Chavero, A.; Rentero, C.; Bosch, M.; Vidal-Quadras, M.; Pol, A.; Enrich, C.; Tebar, F. ROCK1 is a novel Rac1 effector to regulate tubular endocytic membrane formation during clathrin-independent endocytosis. Sci. Rep. 2017, 7, 6866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolias, K.F.; Hartwig, J.H.; Ishihara, H.; Shibasaki, Y.; Cantley, L.C.; Carpenter, C.L. Type Ialpha phosphatidylinositol-4-phosphate 5-kinase mediates Rac-dependent actin assembly. Curr. Biol. 2000, 10, 153–156. [Google Scholar] [CrossRef] [Green Version]
- Halstead, J.R.; Savaskan, N.E.; van den Bout, I.; Van Horck, F.; Hajdo-Milasinovic, A.; Snell, M.; Keune, W.J.; Ten Klooster, J.P.; Hordijk, P.L.; Divecha, N. Rac controls PIP5K localisation and PtdIns(4,5)P(2) synthesis, which modulates vinculin localisation and neurite dynamics. J. Cell Sci. 2010, 123, 3535–3546. [Google Scholar] [CrossRef] [Green Version]
- van Hennik, P.B.; ten Klooster, J.P.; Halstead, J.R.; Voermans, C.; Anthony, E.C.; Divecha, N.; Hordijk, P.L. The C-terminal domain of Rac1 contains two motifs that control targeting and signaling specificity. J. Biol. Chem. 2003, 278, 39166–39175. [Google Scholar] [CrossRef] [Green Version]
- van den Bout, I.; Divecha, N. PIP5K-driven PtdIns(4,5)P2 synthesis: Regulation and cellular functions. J. Cell Sci. 2009, 122, 3837–3850. [Google Scholar] [CrossRef]
- Kwiatkowska, K. One lipid, multiple functions: How various pools of PI(4,5)P(2) are created in the plasma membrane. Cell. Mol. Life Sci. 2010, 67, 3927–3946. [Google Scholar] [CrossRef]
- Apodaca, G.; Enrich, C.; Mostov, K.E. The calmodulin antagonist, W-13, alters transcytosis, recycling, and the morphology of the endocytic pathway in Madin-Darby canine kidney cells. J. Biol. Chem. 1994, 269, 19005–19013. [Google Scholar]
- Mostov, K.E.; Altschuler, Y.; Chapin, S.J.; Enrich, C.; Low, S.H.; Luton, F.; Richman-Eisenstat, J.; Singer, K.L.; Tang, K.; Weimbs, T. Regulation of protein traffic in polarized epithelial cells: The polymeric immunoglobulin receptor model. Cold Spring Harb. Symp. Quant. Biol. 1995, 60, 775–781. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.P.; Crossley, L.; Zhan, Q.; Huang, R.; Coffer, P.; Toker, A.; Robinson, D.; Badwey, J.A. Antagonists of calcium fluxes and calmodulin block activation of the p21-activated protein kinases in neutrophils. J. Immunol. 2001, 166, 2643–2650. [Google Scholar] [CrossRef] [Green Version]
- Filippi, B.M.; Mariggio, S.; Pulvirenti, T.; Corda, D. SRC-dependent signalling regulates actin ruffle formation induced by glycerophosphoinositol 4-phosphate. Biochim. Biophys. Acta 2008, 1783, 2311–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, A.E.; Roovers, R.C.; Collard, J.G. Regulation of Tiam1-Rac signalling. FEBS Lett. 2003, 546, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I.N.; Elliott, C.M.; Exton, J.H. Phospholipase C-gamma, protein kinase C and Ca2+/calmodulin-dependent protein kinase II are involved in platelet-derived growth factor-induced phosphorylation of Tiam1. FEBS Lett. 1998, 429, 229–233. [Google Scholar] [CrossRef] [Green Version]
- Nouri, K.; Timson, D.J.; Ahmadian, M.R. New model for the interaction of IQGAP1 with CDC42 and RAC1. Small Gtpases 2020, 11, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Bry, L.; Li, Z.; Sacks, D.B. IQGAP1 regulates Salmonella invasion through interactions with actin, Rac1, and Cdc42. J. Biol. Chem. 2007, 282, 30265–30272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuroda, S.; Fukata, M.; Nakagawa, M.; Kaibuchi, K. Cdc42, Rac1, and their effector IQGAP1 as molecular switches for cadherin-mediated cell-cell adhesion. Biochem. Biophys. Res. Commun. 1999, 262, 1–6. [Google Scholar] [CrossRef]
- Nouri, K.; Fansa, E.K.; Amin, E.; Dvorsky, R.; Gremer, L.; Willbold, D.; Schmitt, L.; Timson, D.J.; Ahmadian, M.R. IQGAP1 Interaction with RHO Family Proteins Revisited: Kinetic and equilibrium evidence for multiple distinct binding sites. J. Biol. Chem. 2016, 291, 26364–26376. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, S.; Fukata, M.; Kobayashi, K.; Nakafuku, M.; Nomura, N.; Iwamatsu, A.; Kaibuchi, K. Identification of IQGAP as a putative target for the small GTPases, Cdc42 and Rac1. J. Biol. Chem. 1996, 271, 23363–23367. [Google Scholar] [CrossRef] [Green Version]
- Brill, S.; Li, S.; Lyman, C.W.; Church, D.M.; Wasmuth, J.J.; Weissbach, L.; Bernards, A.; Snijders, A.J. The Ras GTPase-activating-protein-related human protein IQGAP2 harbors a potential actin binding domain and interacts with calmodulin and Rho family GTPases. Mol. Cell. Biol. 1996, 16, 4869–4878. [Google Scholar] [CrossRef] [Green Version]
- Mataraza, J.M.; Briggs, M.W.; Li, Z.; Entwistle, A.; Ridley, A.J.; Sacks, D.B. IQGAP1 promotes cell motility and invasion. J. Biol. Chem. 2003, 278, 41237–41245. [Google Scholar] [CrossRef] [Green Version]
- Mataraza, J.M.; Li, Z.; Jeong, H.W.; Brown, M.D.; Sacks, D.B. Multiple proteins mediate IQGAP1-stimulated cell migration. Cell Signal. 2007, 19, 1857–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.D.; Brown, M.D.; Sacks, D.B. IQGAPs in cancer: A family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009, 583, 1817–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, T.; Wang, S.; Noritake, J.; Sato, K.; Fukata, M.; Takefuji, M.; Nakagawa, M.; Izumi, N.; Akiyama, T.; Kaibuchi, K. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev. Cell 2004, 7, 871–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelikan-Conchaudron, A.; Le Clainche, C.; Didry, D.; Carlier, M.F. The IQGAP1 protein is a calmodulin-regulated barbed end capper of actin filaments: Possible implications in its function in cell migration. J. Biol. Chem. 2011, 286, 35119–35128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, W.J.; Bradley, C.A.; Hamilton, E.; Daly, C.; Mallon, T.; Timson, D.J. A calcium-dependent interaction between calmodulin and the calponin homology domain of human IQGAP1. Mol. Cell. Biochem. 2012, 371, 217–223. [Google Scholar] [CrossRef]
- Liu, J.; Kurella, V.B.; LeCour, L., Jr.; Vanagunas, T.; Worthylake, D.K. The IQGAP1 N-Terminus Forms Dimers, and the Dimer Interface Is Required for Binding F-Actin and Calcium-Bound Calmodulin. Biochemistry 2016, 55, 6433–6444. [Google Scholar] [CrossRef] [Green Version]
- Mateer, S.C.; McDaniel, A.E.; Nicolas, V.; Habermacher, G.M.; Lin, M.J.; Cromer, D.A.; King, M.E.; Bloom, G.S. The mechanism for regulation of the F-actin binding activity of IQGAP1 by calcium/calmodulin. J. Biol. Chem. 2002, 277, 12324–12333. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Sacks, D.B. Elucidation of the interaction of calmodulin with the IQ motifs of IQGAP1. J. Biol. Chem. 2003, 278, 4347–4352. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Li, Z.; Jang, H.; Hedman, A.C.; Sacks, D.B.; Nussinov, R. Ca(2+)-Dependent switch of calmodulin interaction mode with tandem IQ motifs in the scaffolding protein IQGAP1. Biochemistry 2019, 58, 4903–4911. [Google Scholar] [CrossRef]
- Li, Q.; Stuenkel, E.L. Calcium negatively modulates calmodulin interaction with IQGAP1. Biochem. Biophys. Res. Commun. 2004, 317, 787–795. [Google Scholar] [CrossRef]
- Brown, M.D.; Sacks, D.B. IQGAP1 in cellular signaling: Bridging the GAP. Trends Cell Biol. 2006, 16, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Bry, L.; Li, Z.; Sacks, D.B. Actin pedestal formation by enteropathogenic Escherichia coli is regulated by IQGAP1, calcium, and calmodulin. J. Biol. Chem. 2008, 283, 35212–35222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, I.N.; Batty, I.H.; Prescott, A.R.; Gray, A.; Kular, G.S.; Stewart, H.; Downes, C.P. Inositol phospholipids regulate the guanine-nucleotide-exchange factor Tiam1 by facilitating its binding to the plasma membrane and regulating GDP/GTP exchange on Rac1. Biochem. J. 2004, 382, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Sander, E.E.; van Delft, S.; ten Klooster, J.P.; Reid, T.; van der Kammen, R.A.; Michiels, F.; Collard, J.G. Matrix-dependent Tiam1/Rac signaling in epithelial cells promotes either cell-cell adhesion or cell migration and is regulated by phosphatidylinositol 3-kinase. J. Cell Biol. 1998, 143, 1385–1398. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Luby-Phelps, K.; Das, B.; Shu, X.; Xia, Y.; Mosteller, R.D.; Krishna, U.M.; Falck, J.R.; White, M.A.; Broek, D. Role of substrates and products of PI 3-kinase in regulating activation of Rac-related guanosine triphosphatases by Vav. Science 1998, 279, 558–560. [Google Scholar] [CrossRef]
- Swulius, M.T.; Waxham, M.N. Ca(2+)/calmodulin-dependent protein kinases. Cell. Mol. Life Sci. 2008, 65, 2637–2657. [Google Scholar] [CrossRef] [Green Version]
- Wayman, G.A.; Tokumitsu, H.; Davare, M.A.; Soderling, T.R. Analysis of CaM-kinase signaling in cells. Cell Calcium 2011, 50, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Herring, B.E.; Nicoll, R.A. Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc. Natl. Acad. Sci. USA 2016, 113, 2264–2269. [Google Scholar] [CrossRef] [Green Version]
- Saneyoshi, T.; Matsuno, H.; Suzuki, A.; Murakoshi, H.; Hedrick, N.G.; Agnello, E.; O‘Connell, R.; Stratton, M.M.; Yasuda, R.; Hayashi, Y. Reciprocal activation within a kinase-effector complex underlying persistence of structural LTP. Neuron 2019, 102, 1199–1210. [Google Scholar] [CrossRef]
- Saneyoshi, T.; Wayman, G.; Fortin, D.; Davare, M.; Hoshi, N.; Nozaki, N.; Natsume, T.; Soderling, T.R. Activity-dependent synaptogenesis: Regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron 2008, 57, 94–107. [Google Scholar] [CrossRef] [Green Version]
- Davare, M.A.; Saneyoshi, T.; Soderling, T.R. Calmodulin-kinases regulate basal and estrogen stimulated medulloblastoma migration via Rac1. J. Neuro-Oncol. 2011, 104, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Chung, L.W.; Siegal, G.P.; Zayzafoon, M. Alpha-CaMKII controls the growth of human osteosarcoma by regulating cell cycle progression. Lab. Investig. 2007, 87, 938–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolias, K.F.; Bikoff, J.B.; Burette, A.; Paradis, S.; Harrar, D.; Tavazoie, S.; Weinberg, R.J.; Greenberg, M.E. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron 2005, 45, 525–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okabe, T.; Nakamura, T.; Nishimura, Y.N.; Kohu, K.; Ohwada, S.; Morishita, Y.; Akiyama, T. RICS, a novel GTPase-activating protein for Cdc42 and Rac1, is involved in the beta-catenin-N-cadherin and N-methyl-D-aspartate receptor signaling. J. Biol. Chem. 2003, 278, 9920–9927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, S.Y.; Lin, T.E.; Wang, W.L.; Lee, P.L.; Tsai, M.C.; Chiu, J.J. Protein kinase C-delta and -beta coordinate flow-induced directionality and deformation of migratory human blood T-lymphocytes. J. Mol. Cell Biol. 2014, 6, 458–472. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.; Bracke, M.; Leitinger, B.; Porter, J.C.; Hogg, N. LFA-1-induced T cell migration on ICAM-1 involves regulation of MLCK-mediated attachment and ROCK-dependent detachment. J. Cell Sci. 2003, 116, 3123–3133. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Dudek, N.L.; Patel, T.B.; Muma, N.A. Transglutaminase-catalyzed transamidation: A novel mechanism for Rac1 activation by 5-hydroxytryptamine2A receptor stimulation. J. Pharmacol. Exp. Ther. 2008, 326, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Dudek, N.L.; Li, Q.; Muma, N.A. Phospholipase C, Ca2+, and calmodulin signaling are required for 5-HT2A receptor-mediated transamidation of Rac1 by transglutaminase. Psychopharmacology 2011, 213, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Fleming, I.N.; Gray, A.; Downes, C.P. Regulation of the Rac1-specific exchange factor Tiam1 involves both phosphoinositide 3-kinase-dependent and -independent components. Biochem. J. 2000, 351, 173–182. [Google Scholar] [CrossRef]
- Walsh, A.B.; Bar-Sagi, D. Differential activation of the Rac pathway by Ha-Ras and K-Ras. J. Biol. Chem. 2001, 276, 15609–15615. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.M.; Lambert, Q.T.; Reuther, G.W.; Malliri, A.; Siderovski, D.P.; Sondek, J.; Collard, J.G.; Der, C.J. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat. Cell Biol. 2002, 4, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Nimnual, A.S.; Yatsula, B.A.; Bar-Sagi, D. Coupling of Ras and Rac guanosine triphosphatases through the Ras exchanger Sos. Science 1998, 279, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; King, A.J.; Diaz, H.B.; Marshall, M.S. Regulation of the protein kinase Raf-1 by oncogenic Ras through phosphatidylinositol 3-kinase, Cdc42/Rac and Pak. Curr. Biol. 2000, 10, 281–284. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, J.; Miyamoto, Y.; Tanoue, A.; Shooter, E.M.; Chan, J.R. Ras activation of a Rac1 exchange factor, Tiam1, mediates neurotrophin-3-induced Schwann cell migration. Proc. Natl. Acad. Sci. USA 2005, 102, 14889–14894. [Google Scholar] [CrossRef] [Green Version]
- Itoh, R.E.; Kiyokawa, E.; Aoki, K.; Nishioka, T.; Akiyama, T.; Matsuda, M. Phosphorylation and activation of the Rac1 and Cdc42 GEF Asef in A431 cells stimulated by EGF. J. Cell Sci. 2008, 121, 2635–2642. [Google Scholar] [CrossRef] [Green Version]
- Murillo, M.M.; Rana, S.; Spencer-Dene, B.; Nye, E.; Stamp, G.; Downward, J. Disruption of the interaction of RAS with PI 3-kinase induces regression of EGFR-mutant-driven lung cancer. Cell Rep. 2018, 25, 3545–3553. [Google Scholar] [CrossRef] [Green Version]
- Khosravi-Far, R.; Solski, P.A.; Clark, G.J.; Kinch, M.S.; Der, C.J. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol. Cell. Biol. 1995, 15, 6443–6453. [Google Scholar] [CrossRef] [Green Version]
- Malliri, A.; van der Kammen, R.A.; Clark, K.; van der Valk, M.; Michiels, F.; Collard, J.G. Mice deficient in the Rac activator Tiam1 are resistant to Ras-induced skin tumours. Nature 2002, 417, 867–871. [Google Scholar] [CrossRef]
- Wang, Z.; Pedersen, E.; Basse, A.; Lefever, T.; Peyrollier, K.; Kapoor, S.; Mei, Q.; Karlsson, R.; Chrostek-Grashoff, A.; Brakebusch, C. Rac1 is crucial for Ras-dependent skin tumor formation by controlling Pak1-Mek-Erk hyperactivation and hyperproliferation in vivo. Oncogene 2010, 29, 3362–3373. [Google Scholar] [CrossRef] [Green Version]
- Kissil, J.L.; Walmsley, M.J.; Hanlon, L.; Haigis, K.M.; Bender Kim, C.F.; Sweet-Cordero, A.; Eckman, M.S.; Tuveson, D.A.; Capobianco, A.J.; Tybulewicz, V.L.; et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007, 67, 8089–8094. [Google Scholar] [CrossRef] [Green Version]
- Heid, I.; Lubeseder-Martellato, C.; Sipos, B.; Mazur, P.K.; Lesina, M.; Schmid, R.M.; Siveke, J.T. Early requirement of Rac1 in a mouse model of pancreatic cancer. Gastroenterology 2011, 141, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Joneson, T.; Bar-Sagi, D. Suppression of Ras-induced apoptosis by the Rac GTPase. Mol. Cell. Biol. 1999, 19, 5892–5901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, H.Y.; Jubb, A.M.; Koch, J.N.; Jaffer, Z.M.; Stepanova, D.; Campbell, D.A.; Duron, S.G.; O‘Farrell, M.; Cai, K.Q.; Klein-Szanto, A.J.; et al. P21-Activated kinase 1 is required for efficient tumor formation and progression in a Ras-mediated skin cancer model. Cancer Res. 2012, 72, 5966–5975. [Google Scholar] [CrossRef] [Green Version]
- Baker, N.M.; Yee Chow, H.; Chernoff, J.; Der, C.J. Molecular pathways: Targeting RAC-p21-activated serine-threonine kinase signaling in RAS-driven cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 4740–4746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.W.; Shin, M.G.; Lee, S.; Kim, J.R.; Park, W.S.; Cho, K.H.; Meyer, T.; Heo, W.D. Cooperative activation of PI3K by Ras and Rho family small GTPases. Mol. Cell 2012, 47, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Nimnual, A.; Bar-Sagi, D. The two hats of SOS. Sci. Stke Signal Transduct. Knowl. Environ. 2002, 2002, 36. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GTPase | Function | Interaction method | Species | Calmodulin (CaM) action | Refs |
|---|---|---|---|---|---|
| Cdc42 | Filopodia formation and cell-cycle progression | CoIP, AC, RC | Human (platelets) | Cdc42 inhibition CBR:151AVKYVECSALTQK164 (hypothetical amphipathic α-helix) | [23] |
| AC | Human (MCF-7) | Regulation of Cdc42 signalling through IQGAP1 | [48] | ||
| Kir/Gem | Cell proliferation, Neurite extension and flattening (Gem); invasion and metastasis (Kir). Inhibition of Ca2+ channels (Kir/Gem) | FS, RC | Synthetic peptides, in vitro | Kir/Gem inactivation (inhibits GTP binding), regulates intracellular localization CBR:264/265KARRFWGKIVAKNNKNMAFKLKSKS288/289 (hypothetical amphipathic α-helix) | [31] |
| Xenopus oocytes | Inhibition of high voltage-activated calcium channels and regulation of intracellular distribution | [63] | |||
| Monkey (COS-1) | Regulate intracellular localization between cytoplasm and nucleus, in a complex with 14-3-3 | [61] | |||
| CoIP, RC | Monkey (COS-1) | Inhibits association with importin α5 and nuclear translocation | [66] | ||
| KRas-4B | Cell proliferation, differentiation, survival and apoptosis | CoIP, AC, RC | Mouse (NIH3T3) | KRas inactivation | [19] |
| RC, YTH. | Human (platelets) | Down-regulation of KRas signalling and KRas membrane transport | [20] | ||
| AC | Mouse (NIH3T3) | CBR:151GVDDAFYTLVREIRKH166 (amphipathic α-helix5) CBR: C-terminal prenyl group | [16] | ||
| RC | CBR:170KMSKDGKKKKKSKTKC185+ prenyl CBR:180KSKTKC185+prenyl group | [67,68] | |||
| AC | Mouse (NIH3T3) | Inhibits KRas Ser181 phosphorylation | [29] | ||
| Rab3A | Synaptic and axonal transport. Regulated exocytosis in neuronal and endocrine cells | PXL | Rat brain synaptosomes |
Dissociation of Rab3A from synaptic membranes CBR:62KTIYRNDKRIKLQIWDTA GQERYR85 | [36,69] |
| Rat (PC12) | Inhibition of exocytosis | [30] | |||
| CoIP, PXL | Rat (pancreatic islets) | Inhibits insulin secretion | [42] | ||
| Rat brain synaptosomes | Promotes GTP binding | [70] | |||
| Human (spermatozoa from sperm donors) | Acrosomal exocytosis | [71] | |||
| Rab3B | Calcium-dependent exocytosis | AC | Mouse brain | [10] | |
| RC | Human (platelets) | Ca2+-dependent secretion in platelets | [33] | ||
| Rab3D | Maintenance of osteoclastic border membrane | AC | Mouse brain | [10] | |
| YTH, BRET, RC | Monkey (COS-1, reconstituted complex) | Promotes osteoclastic bone resorption | [46] | ||
| Rabs: 6B, 8B,10, 15, 33B, 37. | AC | Mouse brain | [10] | ||
| Rac1 | Cytoskeletal organization, migration, adhesion, proliferation, endocytosis, vesicular trafficking | CoIP, AC, RC | Human (platelets) | Rac1 activation CBR:151AVKYLECSALTQRG164 (hypothetical amphipathic α-helix) | [23] |
| AC, RC, CompMod | Human (HeLa) | Rac1 activation | [21] | ||
| AC | Monkey (COS-1) | Rac1 activation Impairs binding to PIP5K CBR:171EAIRAVLCPPPVKKRKRK188 and the C-terminal prenyl group | [22] | ||
| SF-TAP | Human (HEK293T) | [72] | |||
| Rad | Skeletal muscle motor function, cytoskeletal organization and glucose transport | RC | GST-Rad incubated with CaM-sepharose in vitro | Regulates intracellular localization CBR:278AKRFLGRIVARNSRKMA FRA297 (hypothetical amphipathic α-helix) | [31] |
| [65] | |||||
| RalA | Cell proliferation, migration, filopodia formation, differentiation, cytoskeletal organization, vesicular transport, exocytosis and receptor endocytosis. Synaptic and axonal transport | AC, RC, YTH | Human (platelets) | RalA activation | [38] |
| RC | Human (HeLa) | RalA activation CBR: C-terminal prenyl group | [39] | ||
| Rat brain synaptosomes | Dissociation of RalA from synaptic membranes | [69] | |||
| Purified RalA, in vitro | Regulates GTP binding CBR:183SKEKNGKKKRKSLAKRIR200 (hypothetical amphipathic α-helix) | [34,40] | |||
| Rat brain synaptosomes | Regulates GTP binding | [73] | |||
| RalB | Cell proliferation, oncogenic transformation | CoIP, AC, RC, YTH | Human (platelets) | RalB activation CBR: C-terminal prenyl group | [38,39] |
| Rat brain synaptosomes | Regulates GTP binding | [73] | |||
| Rem | Ca2+channel regulation | CoIP, AC | Human (HEK293, TSA20) | No Ca2+ channel inhibition CBR:165QRARRFLARLTARSARRR282 | [74] |
| Ric | Dot-blot, AC, CoIP | Drosophila | Regulation of Ca2+-mediated neuronal signal transduction CBR:242RRSRWWRIRSIFALVFRR RR261 | [35] | |
| Drosophila (cross of Ric mutants with CaM mutants) | Ric inhibition | [56] | |||
| Rin | Ca2+ signalling in neurons | Dot-blot | Mouse |
Regulation of calcium-mediated neuronal signal transduction CBR:194RKLKRKDSLWKKIKASLKKKRENML218 | [32] |
| CoIP | Monkey (COS-7) | Regulation of Rin activation | [54] | ||
| Rat (PC12) | Induce neurite outgrowth | [55] |
| Cell Type (Species) | Ca2+/Calmodulin Effects | |
|---|---|---|
| MAPK Pathway (Refs) | Rac1 Activity (Refs) | |
| NIH3T3 fibroblasts (mouse) | - [144] | + [157] |
| Swiss3T3 fibroblasts (mouse) | - [19] | + [158] |
| HeLa (cervix carcinoma, human) | - [145] | + [21] |
| NRK (kidney epithelial, rat) | - [144] | ND |
| A431 (epidermoid carcinoma, human) | - [142] | ND |
| COS1 fibroblasts (kidney, monkey) | + [143] | + [22] |
| CHO (epithelial ovary, hamster) | + [143] | ND |
| HEK293 (embryonic kidney, human) | + [142] | ND |
| PC12 (adrenal phaeochromocytoma, rat) | + [156] | ND |
| Primary hepatocytes (rat) | + [155] | ND |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tebar, F.; Chavero, A.; Agell, N.; Lu, A.; Rentero, C.; Enrich, C.; Grewal, T. Pleiotropic Roles of Calmodulin in the Regulation of KRas and Rac1 GTPases: Functional Diversity in Health and Disease. Int. J. Mol. Sci. 2020, 21, 3680. https://doi.org/10.3390/ijms21103680
Tebar F, Chavero A, Agell N, Lu A, Rentero C, Enrich C, Grewal T. Pleiotropic Roles of Calmodulin in the Regulation of KRas and Rac1 GTPases: Functional Diversity in Health and Disease. International Journal of Molecular Sciences. 2020; 21(10):3680. https://doi.org/10.3390/ijms21103680
Chicago/Turabian StyleTebar, Francesc, Albert Chavero, Neus Agell, Albert Lu, Carles Rentero, Carlos Enrich, and Thomas Grewal. 2020. "Pleiotropic Roles of Calmodulin in the Regulation of KRas and Rac1 GTPases: Functional Diversity in Health and Disease" International Journal of Molecular Sciences 21, no. 10: 3680. https://doi.org/10.3390/ijms21103680