Neurological Syndromes Associated with Anti-GAD Antibodies

Abstract
1. Introduction
2. GAD Antibody Titers and Epitope Specificities
3. GAD Ab Detection Strategies
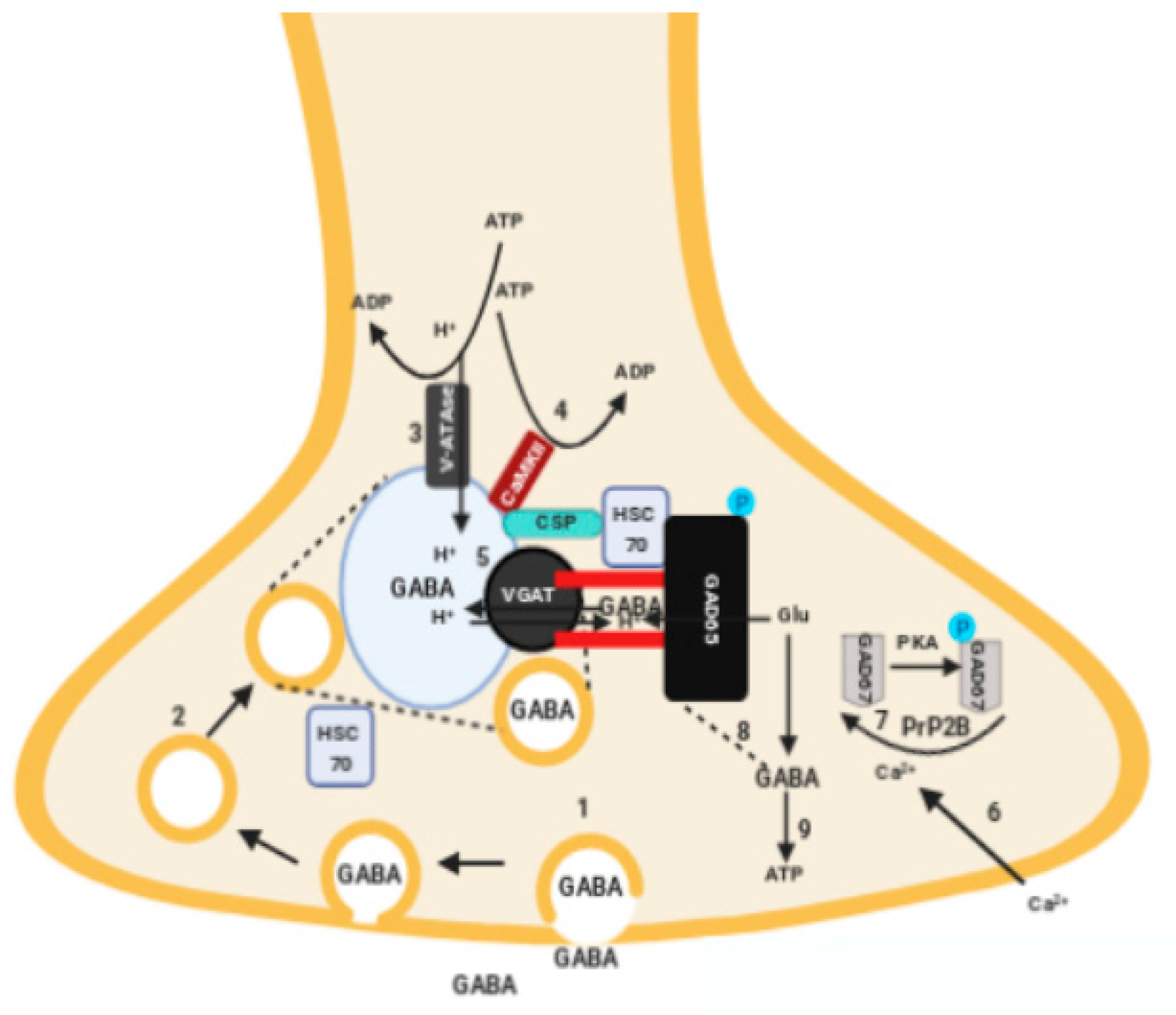
4. Physiopathology: Decreased GABAergic Transmission
5. Immune Effectors and Pathogenetic Mechanisms
6. Epidemiology
7. Genetic Predisposition
8. Coexisting Autoimmune Disorders
9. Association with Cancer
10. Other Triggers of Autoimmunity
11. Neurological Presentation
11.1. Stiff-Person Syndrome
11.2. Cerebellar Ataxia
11.3. Limbic Encephalitis and Autoimmune Epilepsy
11.4. Overlap Syndromes
11.5. Other Neurological Syndromes Associated with GAD Ab
12. Treatment and Outcome
12.1. Stiff-Person Syndrome
12.2. Cerebellar Ataxia
12.3. Limbic Encephalitis and Autoimmune Epilepsy
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GAD | Glutamic acid decarboxylase |
| GABA | Gamma-aminobutyric acid |
| PLP | Pyridoxal 5’-phosphate |
| Ab | Antibodies |
| Svs | Synaptic vesicles |
| HCS70 | Heat Shock Cognate 70 |
| CSP | Cysteine-String Protein |
| VGAT | Vesicular GABA transporter |
| CaMKII | Calcium/calmoduline protein kinase |
| T1DM | Type 1 diabetes mellitus |
| SPS | Stiff-person syndrome |
| CA | Cerebellar ataxia |
| LE | Limbic encephalitis |
| CSF | Cerebrospinal fluid |
| ELISA | Enzyme-linked immunosorbent assay |
| RIA | Radioimmunoassay |
| 125I | Iodine-125 |
| SV | Synaptic vesicles |
| HLA | Human leukocyte antigen |
| TLE | Temporal lobe epilepsy |
| ICI | Immune checkpoint inhibitors |
| EMG | Electromyogram |
| MRI | magnetic resonance imaging |
| MDS | Myelodysplastic syndrome |
| PET | Positron emission tomography |
| PA | Pernicious anemia |
| NSCLC | Non-small cell lung cancer |
| NA | Not available |
| PERM | Progressive encephalomyelitis with rigidity and myoclonus |
| mRS | Modified Rankin Score |
| FLAIR | Fluid-attenuated inversion recovery |
| EEG | Electroencephalogram |
| CS | Corticosteroids |
| FU | Follow-up |
| IV | Intravenous |
| IVIg | Intravenous immunoglobulin |
| MP | Methylprednisolone |
| PE | Plasma exchange |
| ICARS | International Cooperative Ataxia Rating Scale |
| AZA | Azathioprine |
References
- Solimena, M.; De Camilli, P. Autoimmunity to glutamic acid decarboxylase (GAD) in Stiff-Man syndrome and insulin-dependent diabetes mellitus. Trends Neurosci. 1991, 14, 452–457. [Google Scholar] [CrossRef]
- Vincent, S.R.; Hökfelt, T.; Wu, J.Y.; Elde, R.P.; Morgan, L.M.; Kimmel, J.R. Immunohistochemical studies of the GABA system in the pancreas. Neuroendocrinology 1983, 36, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.L.; Rimvall, K. Regulation of Gamma-Aminobutyric Acid Synthesis in the Brain. Available online: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/8419527/?from_term=martin+neurochem+1993&from_pos=1 (accessed on 26 February 2020).
- Kaufman, D.L.; Houser, C.R.; Tobin, A.J. Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J. Neurochem. 1991, 56, 720–723. [Google Scholar] [CrossRef]
- Bu, D.F.; Tobin, A.J. The exon-intron organization of the genes (GAD1 and GAD2) encoding two human glutamate decarboxylases (GAD67 and GAD65) suggests that they derive from a common ancestral GAD. Genomics 1994, 21, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Popp, A.; Urbach, A.; Witte, O.W.; Frahm, C. Adult and embryonic GAD transcripts are spatiotemporally regulated during postnatal development in the rat brain. PLoS ONE 2009, 4, e4371. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyaya, B.; Di Cristo, G.; Wu, C.Z.; Knott, G.; Kuhlman, S.; Fu, Y.; Palmiter, R.D.; Huang, Z.J. GAD67-mediated GABA Synthesis and Signaling Regulate Inhibitory Synaptic Innervation in the Visual Cortex. Neuron 2007, 54, 889–903. [Google Scholar] [CrossRef]
- Magri, C.; Giacopuzzi, E.; La Via, L.; Bonini, D.; Ravasio, V.; Elhussiny, M.E.A.; Orizio, F.; Gangemi, F.; Valsecchi, P.; Bresciani, R.; et al. A novel homozygous mutation in GAD1 gene described in a schizophrenic patient impairs activity and dimerization of GAD67 enzyme. Sci Rep. 2018, 8. [Google Scholar] [CrossRef]
- Kash, S.F.; Johnson, R.S.; Tecott, L.H.; Noebels, J.L.; Mayfield, R.D.; Hanahan, D.; Baekkeskov, S. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc. Natl. Acad. Sci. USA 1997, 94, 14060–14065. [Google Scholar] [CrossRef]
- Erlander, M.G.; Tillakaratne, N.J.; Feldblum, S.; Patel, N.; Tobin, A.J. Two genes encode distinct glutamate decarboxylases. Neuron 1991, 7, 91–100. [Google Scholar] [CrossRef]
- McKeon, A.; Tracy, J.A. GAD65 neurological autoimmunity. Muscle Nerve 2017, 56, 15–27. [Google Scholar] [CrossRef]
- Daif, A.; Lukas, R.V.; Issa, N.P.; Javed, A.; VanHaerents, S.; Reder, A.T.; Tao, J.X.; Warnke, P.; Rose, S.; Towle, V.L.; et al. Antiglutamic acid decarboxylase 65 (GAD65) antibody-associated epilepsy. Epilepsy Behav. 2018, 80, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Wu, H.; Osterhaus, G.; Wei, J.; Davis, K.; Sha, D.; Floor, E.; Hsu, C.-C.; Kopke, R.D.; Wu, J.-Y. Demonstration of functional coupling between gamma -aminobutyric acid (GABA) synthesis and vesicular GABA transport into synaptic vesicles. Proc. Natl. Acad. Sci. USA 2003, 100, 4293–4298. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, E.; Dalmau, J. Neuronal autoantigens—Pathogenesis, associated disorders and antibody testing. Nat. Rev. Neurol. 2012, 8, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Gresa-Arribas, N.; Ariño, H.; Martínez-Hernández, E.; Petit-Pedrol, M.; Sabater, L.; Saiz, A.; Dalmau, J.; Graus, F. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS ONE 2015, 10, e0121364. [Google Scholar] [CrossRef] [PubMed]
- Meinck, H.M.; Faber, L.; Morgenthaler, N.; Seissler, J.; Maile, S.; Butler, M.; Solimena, M.; DeCamilli, P.; Scherbaum, W.A. Antibodies against glutamic acid decarboxylase: Prevalence in neurological diseases. J. Neurol. Neurosurg. Psychiatry 2001, 71, 100–103. [Google Scholar] [CrossRef]
- Guasp, M.; Solà-Valls, N.; Martínez-Hernández, E.; Gil, M.P.; González, C.; Brieva, L.; Saiz, A.; Dalmau, J.; Graus, F.; Ariño, H. Cerebellar ataxia and autoantibodies restricted to glutamic acid decarboxylase 67 (GAD67). J. Neuroimmunol. 2016, 300, 15–17. [Google Scholar] [CrossRef]
- Ariño, H.; Gresa-Arribas, N.; Blanco, Y.; Martínez-Hernández, E.; Sabater, L.; Petit-Pedrol, M.; Rouco, I.; Bataller, L.; Dalmau, J.O.; Saiz, A.; et al. Cerebellar Ataxia and Glutamic Acid Decarboxylase Antibodies: Immunologic Profile and Long-term Effect of Immunotherapy. JAMA Neurol. 2014, 71, 1009. [Google Scholar] [CrossRef]
- Fenalti, G.; Buckle, A.M. Structural biology of the GAD autoantigen. Autoimmun. Rev. 2010, 9, 148–152. [Google Scholar] [CrossRef]
- Reetz, A.; Solimena, M.; Matteoli, M.; Folli, F.; Takei, K.; De Camilli, P. GABA and pancreatic beta-cells: Colocalization of glutamic acid decarboxylase (GAD) and GABA with synaptic-like microvesicles suggests their role in GABA storage and secretion. EMBO J. 1991, 10, 1275–1284. [Google Scholar] [CrossRef]
- Manto, M.; Honnorat, J.; Hampe, C.S.; Guerra-Narbona, R.; López-Ramos, J.C.; Delgado-García, J.M.; Saitow, F.; Suzuki, H.; Yanagawa, Y.; Mizusawa, H.; et al. Disease-specific monoclonal antibodies targeting glutamate decarboxylase impair GABAergic neurotransmission and affect motor learning and behavioral functions. Front. Behav. Neurosci. 2015, 9, 78. [Google Scholar] [CrossRef]
- Raju, R.; Foote, J.; Banga, J.P.; Hall, T.R.; Padoa, C.J.; Dalakas, M.C.; Ortqvist, E.; Hampe, C.S. Analysis of GAD65 autoantibodies in Stiff-Person syndrome patients. J. Immunol. 2005, 175, 7755–7762. [Google Scholar] [CrossRef] [PubMed]
- Fouka, P.; Alexopoulos, H.; Akrivou, S.; Trohatou, O.; Politis, P.K.; Dalakas, M.C. GAD65 epitope mapping and search for novel autoantibodies in GAD-associated neurological disorders. J. Neuroimmunol. 2015, 281, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Burbelo, P.D.; Groot, S.; Dalakas, M.C.; Iadarola, M.J. High Definition Profiling of Autoantibodies to Glutamic Acid Decarboxylases GAD65/GAD67 in Stiff-Person Syndrome. Biochem. Biophys. Res. Commun. 2008, 366, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Baekkeskov, S.; Aanstoot, H.-J.; Christgai, S.; Reetz, A.; Solimena, M.; Cascalho, M.; Folli, F.; Richter-Olesen, H.; Camilli, P.-D. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature 1990, 347, 151–156. [Google Scholar] [CrossRef]
- Muñoz-Lopetegi, A.; de Bruijn, M.A.A.M.; Boukhrissi, S.; Bastiaansen, A.E.M.; Nagtzaam, M.M.P.; Hulsenboom, E.S.P.; Boon, A.J.W.; Neuteboom, R.F.; de Vries, J.M.; Sillevis Smitt, P.A.E.; et al. Neurologic syndromes related to anti-GAD65: Clinical and serologic response to treatment. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef]
- Nakajima, H.; Nakamura, Y.; Inaba, Y.; Tsutsumi, C.; Unoda, K.; Hosokawa, T.; Kimura, F.; Hanafusa, T.; Date, M.; Kitaoka, H. Neurologic disorders associated with anti-glutamic acid decarboxylase antibodies: A comparison of anti-GAD antibody titers and time-dependent changes between neurologic disease and type I diabetes mellitus. J. Neuroimmunol. 2018, 317, 84–89. [Google Scholar] [CrossRef]
- Saiz, A.; Blanco, Y.; Sabater, L.; González, F.; Bataller, L.; Casamitjana, R.; Ramió-Torrentà, L.; Graus, F. Spectrum of neurological syndromes associated with glutamic acid decarboxylase antibodies: Diagnostic clues for this association. Brain 2008, 131, 2553–2563. [Google Scholar] [CrossRef]
- Malter, M.P.; Helmstaedter, C.; Urbach, H.; Vincent, A.; Bien, C.G. Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann. Neurol. 2010, 67, 470–478. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Li, M.; Fujii, M.; Jacobowitz, D.M. Stiff person syndrome: Quantification, specificity, and intrathecal synthesis of GAD65 antibodies. Neurology 2001, 57, 780–784. [Google Scholar] [CrossRef]
- Jarius, S.; Stich, O.; Speck, J.; Rasiah, C.; Wildemann, B.; Meinck, H.M.; Rauer, S. Qualitative and quantitative evidence of anti-glutamic acid decarboxylase-specific intrathecal antibody synthesis in patients with stiff person syndrome. J. Neuroimmunol. 2010, 229, 219–224. [Google Scholar] [CrossRef]
- Rakocevic, G.; Raju, R.; Dalakas, M.C. Anti-glutamic acid decarboxylase antibodies in the serum and cerebrospinal fluid of patients with Stiff-Person syndrome: Correlation with clinical severity. Arch. Neurol. 2004, 61, 902. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C. The role of IVIg in the treatment of patients with stiff person syndrome and other neurological diseases associated with anti-GAD antibodies. J. Neurol. 2005, 252, I19–I25. [Google Scholar] [CrossRef] [PubMed]
- Dalakas, M.C.; Rakocevic, G.; Dambrosia, J.M.; Alexopoulos, H.; McElroy, B. A double-blind, placebo-controlled study of rituximab in patients with stiff person syndrome. Ann. Neurol. 2017, 82, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Di Giacomo, R.; Deleo, F.; Pastori, C.; Didato, G.; Andreetta, F.; Del Sole, A.; de Curtis, M.; Villani, F. Predictive value of high titer of GAD65 antibodies in a case of limbic encephalitis. J. Neuroimmunol. 2019, 337, 577063. [Google Scholar] [CrossRef] [PubMed]
- Saiz, A.; Arpa, J.; Sagasta, A.; Casamitjana, R.; Zarranz, J.J.; Tolosa, E.; Graus, F. Autoantibodies to glutamic acid decarboxylase in three patients with cerebellar ataxia, late-onset insulin-dependent diabetes mellitus, and polyendocrine autoimmunity. Neurology 1997, 49, 1026–1030. [Google Scholar] [CrossRef]
- Höftberger, R. Neuroimmunology: An expanding frontier in autoimmunity. Front. Immunol. 2015, 6, 206. [Google Scholar] [CrossRef]
- Ricken, G.; Schwaiger, C.; De Simoni, D.; Pichler, V.; Lang, J.; Glatter, S.; Macher, S.; Rommer, P.S.; Scholze, P.; Kubista, H.; et al. Detection Methods for Autoantibodies in Suspected Autoimmune Encephalitis. Front. Neurol. 2018, 9, 841. [Google Scholar] [CrossRef]
- Solimena, M.; Folli, F.; Denis-Donini, S.; Comi, G.C.; Pozza, G.; De Camilli, P.; Vicari, A.M. Autoantibodies to glutamic acid decarboxylase in a patient with Stiff-Man syndrome, epilepsy, and type I diabetes mellitus. N. Engl. J. Med. 1988, 318, 1012–1020. [Google Scholar] [CrossRef]
- Pittock, S.J.; Yoshikawa, H.; Ahlskog, J.E.; Tisch, S.H.; Benarroch, E.E.; Kryzer, T.J.; Lennon, V.A. Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction. Mayo Clin. Proc. 2006, 81, 1207–1214. [Google Scholar] [CrossRef]
- Oertel, W.H.; Schmechel, D.E.; Mugnaini, E.; Tappaz, M.L.; Kopin, I.J. Immunocytochemical localization of glutamate decarboxylase in rat cerebellum with a new antiserum. Neuroscience 1981, 6, 2715–2735. [Google Scholar] [CrossRef]
- Solimena, M.; Folli, F.; Aparisi, R.; Pozza, G.; De Camilli, P. Autoantibodies to GABA-ergic neurons and pancreatic beta cells in stiff-man syndrome. N. Engl. J. Med. 1990, 322, 1555–1560. [Google Scholar] [CrossRef] [PubMed]
- Aydin, S. A short history, principles, and types of ELISA, and our laboratory experience with peptide/protein analyses using ELISA. Peptides 2015, 72, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Walikonis, J.E.; Lennon, V.A. Radioimmunoassay for glutamic acid decarboxylase (GAD65) autoantibodies as a diagnostic aid for stiff-man syndrome and a correlate of susceptibility to type 1 diabetes mellitus. Mayo Clin. Proc. 1998, 73, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- McKeon, A.; Robinson, M.T.; McEvoy, K.M.; Matsumoto, J.Y.; Lennon, V.A.; Ahlskog, J.E.; Pittock, S.J. Stiff-man syndrome and variants: Clinical course, treatments, and outcomes. Arch. Neurol. 2012, 69, 230–238. [Google Scholar] [CrossRef]
- Sivilotti, L.; Nistri, A. GABA receptor mechanisms in the central nervous system. Prog. Neurobiol. 1991, 36, 35–92. [Google Scholar] [CrossRef]
- DeFelipe, J.; López-Cruz, P.L.; Benavides-Piccione, R.; Bielza, C.; Larrañaga, P.; Anderson, S.; Burkhalter, A.; Cauli, B.; Fairén, A.; Feldmeyer, D.; et al. New insights into the classification and nomenclature of cortical GABAergic interneurons. Nat. Rev. Neurosci. 2013, 14, 202–216. [Google Scholar] [CrossRef]
- Frangaj, A.; Fan, Q.R. Structural biology of GABAB receptor. Neuropharmacology 2018, 136, 68–79. [Google Scholar] [CrossRef]
- Bowery, N.G.; Bettler, B.; Froestl, W.; Gallagher, J.P.; Marshall, F.; Raiteri, M.; Bonner, T.I.; Enna, S.J. International Union of Pharmacology. XXXIII. Mammalian gamma-aminobutyric acid(B) receptors: Structure and function. Pharmacol. Rev. 2002, 54, 247–264. [Google Scholar] [CrossRef]
- Mitoma, H.; Adhikari, K.; Aeschlimann, D.; Chattopadhyay, P.; Hadjivassiliou, M.; Hampe, C.S.; Honnorat, J.; Joubert, B.; Kakei, S.; Lee, J.; et al. Consensus Paper: Neuroimmune Mechanisms of Cerebellar Ataxias. Cerebellum 2016, 15, 213–232. [Google Scholar] [CrossRef]
- Hansen, N.; Grünewald, B.; Weishaupt, A.; Colaço, M.N.; Toyka, K.V.; Sommer, C.; Geis, C. Human Stiff person syndrome IgG-containing high-titer anti-GAD65 autoantibodies induce motor dysfunction in rats. Exp. Neurol. 2013, 239, 202–209. [Google Scholar] [CrossRef]
- Koerner, C.; Wieland, B.; Richter, W.; Meinck, H.-M. Stiff-person syndromes: Motor cortex hyperexcitability correlates with anti-GAD autoimmunity. Neurology 2004, 62, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- Tian, N.; Petersen, C.; Kash, S.; Baekkeskov, S.; Copenhagen, D.; Nicoll, R. The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc. Natl. Acad. Sci. USA 1999, 96, 12911–12916. [Google Scholar] [CrossRef] [PubMed]
- Mitoma, H.; Song, S.Y.; Ishida, K.; Yamakuni, T.; Kobayashi, T.; Mizusawa, H. Presynaptic impairment of cerebellar inhibitory synapses by an autoantibody to glutamate decarboxylase. J. Neurol. Sci. 2000, 175, 40–44. [Google Scholar] [CrossRef]
- Mitoma, H.; Ishida, K.; Shizuka-Ikeda, M.; Mizusawa, H. Dual impairment of GABAA- and GABAB-receptor-mediated synaptic responses by autoantibodies to glutamic acid decarboxylase. J. Neurol. Sci. 2003, 208, 51–56. [Google Scholar] [CrossRef]
- Bettler, B.; Kaupmann, K.; Mosbacher, J.; Gassmann, M. Molecular structure and physiological functions of GABA(B) receptors. Physiol. Rev. 2004, 84, 835–867. [Google Scholar] [CrossRef] [PubMed]
- Terunuma, M. Diversity of structure and function of GABAB receptors: A complexity of GABAB-mediated signaling. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2018, 94, 390–411. [Google Scholar] [CrossRef]
- Levy, L.M.; Dalakas, M.C.; Floeter, M.K. The stiff-person syndrome: An autoimmune disorder affecting neurotransmission of gamma-aminobutyric acid. Ann. Intern. Med. 1999, 131, 522–530. [Google Scholar] [CrossRef]
- Mitoma, H.; Manto, M.; Hampe, C.S. Pathogenic Roles of Glutamic Acid Decarboxylase 65 Autoantibodies in Cerebellar Ataxias. J. Immunol. Res. 2017, 2017, 2913297. [Google Scholar] [CrossRef]
- Takenoshita, H.; Shizuka-Ikeda, M.; Mitoma, H.; Song, S.; Harigaya, Y.; Igeta, Y.; Yaguchi, M.; Ishida, K.; Shoji, M.; Tanaka, M.; et al. Presynaptic inhibition of cerebellar GABAergic transmission by glutamate decarboxylase autoantibodies in progressive cerebellar ataxia. J. Neurol. Neurosurg. Psychiatry 2001, 70, 386–389. [Google Scholar] [CrossRef]
- Ishida, K.; Mitoma, H.; Song, S.Y.; Uchihara, T.; Inaba, A.; Eguchi, S.; Kobayashi, T.; Mizusawa, H. Selective suppression of cerebellar GABAergic transmission by an autoantibody to glutamic acid decarboxylase. Ann. Neurol. 1999, 46, 263–267. [Google Scholar] [CrossRef]
- Ishida, K.; Mitoma, H.; Mizusawa, H. Reversibility of cerebellar GABAergic synapse impairment induced by anti-glutamic acid decarboxylase autoantibodies. J. Neurol. Sci. 2008, 271, 186–190. [Google Scholar] [CrossRef]
- Manto, M.-U.; Laute, M.-A.; Aguera, M.; Rogemond, V.; Pandolfo, M.; Honnorat, J. Effects of anti-glutamic acid decarboxylase antibodies associated with neurological diseases: GAD-Ab and Cerebellar Function. Ann. Neurol. 2007, 61, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Manto, M.; Mitoma, H.; Hampe, C.S. Anti-GAD Antibodies and the Cerebellum: Where Do We Stand? Cerebellum 2019, 18, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Tomatsu, S.; Tsunoda, Y.; Lee, J.; Hoffman, D.S.; Kakei, S. Releasing dentate nucleus cells from Purkinje cell inhibition generates output from the cerebrocerebellum. PLoS ONE 2014, 9, e108774. [Google Scholar] [CrossRef]
- Vianello, M.; Bisson, G.; Dal Maschio, M.; Vassanelli, S.; Girardi, S.; Mucignat, C.; Fountzoulas, K.; Giometto, B. Increased spontaneous activity of a network of hippocampal neurons in culture caused by suppression of inhibitory potentials mediated by anti-gad antibodies. Autoimmunity 2008, 41, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.-G.; Nepom, G.T. Prediction and Pathogenesis in Type 1 Diabetes. Immunity 2010, 32, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Arneth, B. Activation of CD4+ and CD8+ T-lymphocytes by insulin and GAD in patients with type 1 or 2 diabetes mellitus. Endocr. Connect. 2017, 6, 758–765. [Google Scholar] [CrossRef][Green Version]
- Mallone, R.; Martinuzzi, E.; Blancou, P.; Novelli, G.; Afonso, G.; Dolz, M.; Bruno, G.; Chaillous, L.; Chatenoud, L.; Bach, J.-M.; et al. CD8+ T-cell responses identify beta-cell autoimmunity in human type 1 diabetes. Diabetes 2007, 56, 613–621. [Google Scholar] [CrossRef]
- McKeon, A.; Pittock, S.J. Paraneoplastic encephalomyelopathies: Pathology and mechanisms. Acta Neuropathol. 2011, 122, 381–400. [Google Scholar] [CrossRef]
- Burton, A.R.; Baquet, Z.; Eisenbarth, G.S.; Tisch, R.; Smeyne, R.; Workman, C.J.; Vignali, D.A.A. Central Nervous System Destruction Mediated by Glutamic Acid Decarboxylase-Specific CD4+ T Cells. J. Immunol. 2010, 184, 4863–4870. [Google Scholar] [CrossRef]
- Ishizawa, K.; Komori, T.; Okayama, K.; Qin, X.; Kaneko, K.; Sasaki, S.; Iwata, M. Large motor neuron involvement in Stiff-man syndrome: A qualitative and quantitative study. Acta Neuropathol. 1999, 97, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.D.; Scott, G.; Blumbergs, P.C.; Thompson, P.D. Pathological evidence of encephalomyelitis in the stiff man syndrome with anti-GAD antibodies. J. Clin. Neurosci. 2002, 9, 328–329. [Google Scholar] [CrossRef] [PubMed]
- Holmøy, T.; Skorstad, G.; Røste, L.S.; Scheie, D.; Alvik, K. Stiff person syndrome associated with lower motor neuron disease and infiltration of cytotoxic T cells in the spinal cord. Clin. Neurol. Neurosurg. 2009, 111, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Ishida, K.; Mitoma, H.; Wada, Y.; Oka, T.; Shibahara, J.; Saito, Y.; Murayama, S.; Mizusawa, H. Selective loss of Purkinje cells in a patient with anti-glutamic acid decarboxylase antibody-associated cerebellar ataxia. J. Neurol. Neurosurg. Psychiatry 2007, 78, 190–192. [Google Scholar] [CrossRef]
- Bien, C.G.; Vincent, A.; Barnett, M.H.; Becker, A.J.; Blümcke, I.; Graus, F.; Jellinger, K.A.; Reuss, D.E.; Ribalta, T.; Schlegel, J.; et al. Immunopathology of autoantibody-associated encephalitides: Clues for pathogenesis. Brain 2012, 135, 1622–1638. [Google Scholar] [CrossRef]
- Espay, A.J.; Chen, R. Rigidity and spasms from autoimmune encephalomyelopathies: Stiff-person syndrome. Muscle Nerve 2006, 34, 677–690. [Google Scholar] [CrossRef]
- Dubey, D.; Pittock, S.J.; Kelly, C.R.; McKeon, A.; Lopez-Chiriboga, A.S.; Lennon, V.; Gadoth, A.; Smith, C.Y.; Bryant, S.C.; Klein, C.J.; et al. Autoimmune Encephalitis Epidemiology and a comparison to Infectious Encephalitis. Ann. Neurol. 2018, 83, 166–177. [Google Scholar] [CrossRef]
- Dalakas, M.C.; Fujii, M.; Li, M.; McElroy, B. The clinical spectrum of anti-GAD antibody-positive patients with stiff-person syndrome. Neurology 2000, 55, 1531–1535. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Martindale, J.; Shanmugarajah, P.; Grünewald, R.A.; Sarrigiannis, P.G.; Beauchamp, N.; Garrard, K.; Warburton, R.; Sanders, D.S.; Friend, D.; et al. Causes of progressive cerebellar ataxia: Prospective evaluation of 1500 patients. J. Neurol. Neurosurg. Psychiatry 2017, 88, 301–309. [Google Scholar] [CrossRef]
- Nanri, K.; Okuma, M.; Sato, S.; Yoneda, M.; Taguchi, T.; Mitoma, H.; Yamada, J.; Unezaki, S.; Nagatani, T.; Otsubo, S.; et al. Prevalence of Autoantibodies and the Efficacy of Immunotherapy for Autoimmune Cerebellar Ataxia. Intern. Med. 2016, 55, 449–454. [Google Scholar] [CrossRef]
- Brenner, T.; Sills, G.J.; Hart, Y.; Howell, S.; Waters, P.; Brodie, M.J.; Vincent, A.; Lang, B. Prevalence of neurologic autoantibodies in cohorts of patients with new and established epilepsy. Epilepsia 2013, 54, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Lilleker, J.B.; Biswas, V.; Mohanraj, R. Glutamic acid decarboxylase (GAD) antibodies in epilepsy: Diagnostic yield and therapeutic implications. Seizure 2014, 23, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Errichiello, L.; Perruolo, G.; Pascarella, A.; Formisano, P.; Minetti, C.; Striano, S.; Zara, F.; Striano, P. Autoantibodies to glutamic acid decarboxylase (GAD) in focal and generalized epilepsy: A study on 233 patients. J. Neuroimmunol. 2009, 211, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Bien, C.G.; Scheffer, I.E. Autoantibodies and epilepsy. Epilepsia 2011, 52, 18–22. [Google Scholar] [CrossRef]
- Sørgjerd, E.P.; Thorsby, P.M.; Torjesen, P.A.; Skorpen, F.; Kvaløy, K.; Grill, V. Presence of anti-GAD in a non-diabetic population of adults; time dynamics and clinical influence: Results from the HUNT study. BMJ Open Diabetes Res. Care 2015, 3, e000076. [Google Scholar] [CrossRef]
- Honnorat, J.; Saiz, A.; Giometto, B.; Vincent, A.; Brieva, L.; de Andres, C.; Maestre, J.; Fabien, N.; Vighetto, A.; Casamitjana, R.; et al. Cerebellar ataxia with anti-glutamic acid decarboxylase antibodies: Study of 14 patients. Arch. Neurol. 2001, 58, 225–230. [Google Scholar] [CrossRef]
- Liimatainen, S.; Peltola, M.; Sabater, L.; Fallah, M.; Kharazmi, E.; Haapala, A.-M.; Dastidar, P.; Knip, M.; Saiz, A.; Peltola, J. Clinical significance of glutamic acid decarboxylase antibodies in patients with epilepsy. Epilepsia 2010, 51, 760–767. [Google Scholar] [CrossRef]
- Muñiz-Castrillo, S.; Ambati, A.; Dubois, V.; Vogrig, A.; Joubert, B.; Rogemond, V.; Picard, G.; Lin, L.; Fabien, N.; Mignot, E.; et al. Primary DQ effect in the association between HLA and neurological syndromes with anti-GAD65 antibodies. J. Neurol. 2020. [Google Scholar] [CrossRef]
- Muñiz-Castrillo, S.; Vogrig, A.; Honnorat, J. Associations between HLA and autoimmune neurological diseases with autoantibodies. Autoimmun. Highlights 2020, 11, 2. [Google Scholar] [CrossRef]
- Gough, S.C.L.; Simmonds, M.J. The HLA Region and Autoimmune Disease: Associations and Mechanisms of Action. Curr. Genom. 2007, 8, 453–465. [Google Scholar] [CrossRef]
- Pugliese, A.; Solimena, M.; Awdeh, Z.L.; Alper, C.A.; Bugawan, T.; Erlich, H.A.; De Camilli, P.; Eisenbarth, G.S. Association of HLA-DQB1*0201 with stiff-man syndrome. J. Clin. Endocrinol. Metab. 1993, 77, 1550–1553. [Google Scholar] [CrossRef] [PubMed]
- Joubert, B.; Belbezier, A.; Haesebaert, J.; Rheims, S.; Ducray, F.; Picard, G.; Rogemond, V.; Psimaras, D.; Berzero, G.; Desestret, V.; et al. Long-term outcomes in temporal lobe epilepsy with glutamate decarboxylase antibodies. J. Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Critchley, P.; Lawden, M.; Farooq, S.; Thomas, A.; Proudlock, F.A.; Constantinescu, C.S.; Gottlob, I. Stiff person syndrome with eye movement abnormality, myasthenia gravis, and thymoma. J. Neurol. Neurosurg. Psychiatry 2005, 76, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Matsumura, A.; Okumura, M.; Kitaguchi, M.; Yamamoto, S.; Iuchi, K. Stiff man syndrome with thymoma. Ann. Thorac. Surg. 2005, 80, 739–741. [Google Scholar] [CrossRef]
- Rosin, L.; DeCamilli, P.; Butler, M.; Solimena, M.; Schmitt, H.-P.; Morgenthaler, N.; Meinck, H.-M. Stiff-man syndrome in a woman with breast cancer: An uncommon central nervous system paraneoplastic syndrome. Neurology 1998, 50, 94–98. [Google Scholar] [CrossRef]
- Silverman, I.E. Paraneoplastic stiff limb syndrome. J. Neurol. Neurosurg. Psychiatry 1999, 67, 126–127. [Google Scholar] [CrossRef]
- Ariño, H.; Höftberger, R.; Gresa-Arribas, N.; Martínez-Hernandez, E.; Armangue, T.; Kruer, M.C.; Arpa, J.; Domingo, J.; Rojc, B.; Bataller, L.; et al. Paraneoplastic Neurological Syndromes and Glutamic Acid Decarboxylase Antibodies. JAMA Neurol. 2015, 72, 874–881. [Google Scholar] [CrossRef]
- Bien, C.G. Association of Paraneoplastic Neurological Disorders With Glutamic Acid Decarboxylase Antibodies. JAMA Neurol. 2015, 72, 861–862. [Google Scholar] [CrossRef]
- Hernández-Echebarría, L.; Saiz, A.; Arés, A.; Tejada, J.; García-Tuñón, L.; Nieves, C.; Graus, F. Paraneoplastic encephalomyelitis associated with pancreatic tumor and anti-GAD antibodies. Neurology 2006, 66, 450–451. [Google Scholar] [CrossRef]
- Bataller, L.; Valero, C.; Díaz, R.; Froufe, A.; Garcia-Zarza, A.; Ribalta, T.; Vilchez, J.J.; Saiz, A. Cerebellar ataxia associated with neuroendocrine thymic carcinoma and GAD antibodies. J. Neurol. Neurosurg. Psychiatry 2009, 80, 696–697. [Google Scholar] [CrossRef]
- Graus, F.; Dalmau, J. Paraneoplastic neurological syndromes in the era of immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2019, 16, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Dunn-Pirio, A.; Luedke, M.; Morgenlander, J.; Skeen, M.; Eckstein, C. Nivolumab-Induced Autoimmune Encephalitis in Two Patients with Lung Adenocarcinoma. Case Rep. Neurol. Med. 2018, 2018, 2548528. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.; Jaffer, M.; Verma, N.; Mokhtari, S.; Ramsakal, A.; Peguero, E. Immune checkpoint inhibitor induced anti-glutamic acid decarboxylase 65 (Anti-GAD 65) limbic encephalitis responsive to intravenous immunoglobulin and plasma exchange. J. Neurol. 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Morgan, A.; Shah, S.; Ebeling, P.R. Rapid-onset diabetic ketoacidosis secondary to nivolumab therapy. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- De Filette, J.M.K.; Pen, J.J.; Decoster, L.; Vissers, T.; Bravenboer, B.; Van der Auwera, B.J.; Gorus, F.K.; Roep, B.O.; Aspeslagh, S.; Neyns, B.; et al. Immune checkpoint inhibitors and type 1 diabetes mellitus: A case report and systematic review. Eur. J. Endocrinol. 2019, 181, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Tzoulis, P.; Corbett, R.W.; Ponnampalam, S.; Baker, E.; Heaton, D.; Doulgeraki, T.; Stebbing, J. Nivolumab-induced fulminant diabetic ketoacidosis followed by thyroiditis. Endocrinol. Diabetes Metab. Case Rep. 2018, 2018. [Google Scholar] [CrossRef]
- Moersch, F.P.; Woltman, H.W. Progressive fluctuating muscular rigidity and spasm (“stiff-man” syndrome); report of a case and some observations in 13 other cases. Proc. Staff Meet. Mayo Clin. 1956, 31, 421–427. [Google Scholar]
- Barker, R.; Revesz, T.; Thom, M.; Marsden, C.; Brown, P. Review of 23 patients affected by the stiff man syndrome: Clinical subdivision into stiff trunk (man) syndrome, stiff limb syndrome, and progressive encephalomyelitis with rigidity. J. Neurol. Neurosurg. Psychiatry 1998, 65, 633–640. [Google Scholar] [CrossRef]
- Meinck, H.M.; Ricker, K.; Conrad, B. The stiff-man syndrome: New pathophysiological aspects from abnormal exteroceptive reflexes and the response to clomipramine, clonidine, and tizanidine. J. Neurol. Neurosurg. Psychiatry 1984, 47, 280–287. [Google Scholar] [CrossRef]
- Mitoma, H.; Manto, M.; Hampe, C.S. Immune-mediated Cerebellar Ataxias: Practical Guidelines and Therapeutic Challenges. Curr. Neuropharmacol. 2019, 17, 33–58. [Google Scholar] [CrossRef]
- Aguiar, T.S.; Fragoso, A.; de Albuquerque, C.R.; de Teixeira, P.F.; de Souza, M.V.L.; Zajdenverg, L.; Alves-Leon, S.V.; Rodacki, M.; Lima, M.A.S.D. de Clinical characteristics of patients with cerebellar ataxia associated with anti-GAD antibodies. Arq. Neuropsiquiatr. 2017, 75, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, M.; Tripathi, M.; Roy, S.G.; Parida, G.K.; Ihtisham, K.; Dash, D.; Damle, N.; Shamim, S.A.; Bal, C. Metabolic topography of autoimmune non-paraneoplastic encephalitis. Neuroradiology 2018, 60, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Graus, F.; Delattre, J.Y.; Antoine, J.C.; Dalmau, J.; Giometto, B.; Grisold, W.; Honnorat, J.; Smitt, P.S.; Vedeler, C.; Verschuuren, J.J.G.M.; et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1135–1140. [Google Scholar] [CrossRef] [PubMed]
- Kanter, I.C.; Huttner, H.B.; Staykov, D.; Biermann, T.; Struffert, T.; Kerling, F.; Hilz, M.-J.; Schellinger, P.D.; Schwab, S.; Bardutzky, J. Cyclophosphamide for anti-GAD antibody-positive refractory status epilepticus. Epilepsia 2008, 49, 914–920. [Google Scholar] [CrossRef]
- Triplett, J.; Vijayan, S.; MacDonald, A.; Lawn, N.; McLean-Tooke, A.; Bynevelt, M.; Phatouros, C.; Chemmanam, T. Fulminant Anti-GAD antibody encephalitis presenting with status epilepticus requiring aggressive immunosuppression. J. Neuroimmunol. 2018, 323, 119–124. [Google Scholar] [CrossRef]
- Rakocevic, G.; Raju, R.; Semino-Mora, C.; Dalakas, M.C. Stiff person syndrome with cerebellar disease and high-titer anti-GAD antibodies. Neurology 2006, 67, 1068–1070. [Google Scholar] [CrossRef]
- Jazebi, N.; Rodrigo, S.; Gogia, B.; Shawagfeh, A. Anti-glutamic acid decarboxylase (GAD) positive cerebellar Ataxia with transitioning to progressive encephalomyelitis with rigidity and myoclonus (PERM), responsive to immunotherapy: A case report and review of literature. J. Neuroimmunol. 2019, 332, 135–137. [Google Scholar] [CrossRef]
- Wirth, T.; Kaeuffer, C.; Chanson, J.B.; Echaniz-Laguna, A.; Renaud, M.; Anheim, M.; Schneider, F.; Tranchant, C. Progressive encephalomyelitis with rigidity and myoclonus, a diagnostic challenge. Rev. Neurol. (Paris) 2018, 174, 343–346. [Google Scholar] [CrossRef]
- Joubert, B.; Rostásy, K.; Honnorat, J. Immune-mediated ataxias. Handb. Clin. Neurol. 2018, 155, 313–332. [Google Scholar] [CrossRef]
- Nemni, R.; Braghi, S.; Natali-Sora, M.G.; Lampasona, V.; Bonifacio, E.; Comi, G.; Canal, N. Autoantibodies to glutamic acid decarboxylase in palatal myoclonus and epilepsy. Ann. Neurol. 1994, 36, 665–667. [Google Scholar] [CrossRef]
- Vianello, M.; Morello, F.; Scaravilli, T.; Tavolato, B.; Giometto, B. Tremor of the mouth floor and anti-glutamic acid decarboxylase autoantibodies. Eur. J. Neurol. 2003, 10, 513–514. [Google Scholar] [CrossRef]
- Liu, Z.; Huang, Q.; Li, H.; Qiu, W.; Chen, B.; Luo, J.; Yang, H.; Liu, T.; Liu, S.; Xu, H.; et al. Glutamic Acid Decarboxylase Antibody in a Patient with Myelitis: A Retrospective Study. Neuroimmunomodulation 2018, 25, 68–72. [Google Scholar] [CrossRef]
- Fileccia, E.; Rinaldi, R.; Liguori, R.; Incensi, A.; D’Angelo, R.; Giannoccaro, M.P.; Donadio, V. Post-ganglionic autonomic neuropathy associated with anti-glutamic acid decarboxylase antibodies. Clin. Auton. Res. 2017, 27, 51–55. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Kriscenski-Perry, E.; Wenger, D.A.; Pearce, D.A. An autoantibody to GAD65 in sera of patients with juvenile neuronal ceroid lipofuscinoses. Neurology 2002, 59, 1816–1817. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, G.; Cosi, V.; Zandrini, C.; Moglia, A. Steroid-responsive and dependent stiff-man syndrome: A clinical and electrophysiological study of two cases. Ital. J. Neurol. Sci. 1988, 9, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Brashear, H.R.; Phillips, L.H. Autoantibodies to GABAergic neurons and response to plasmapheresis in stiff-man syndrome. Neurology 1991, 41, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Sevy, A.; Franques, J.; Chiche, L.; Pouget, J.; Attarian, S. Successful treatment with rituximab in a refractory Stiff-person syndrome. Rev. Neurol. (Paris) 2012, 168, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Bacorro, E.; Tehrani, R. Stiff-Person Syndrome Persistent Elevation of Glutamic Acid Decarboxylase Antibodies Despite Successful Treatment with Rituximab. J. Clin. Rheumatol. Pract. Rep. Rheum. Musculoskelet. Dis. 2010, 16, 237–239. [Google Scholar] [CrossRef]
- Pedroso, J.L.; Braga-Neto, P.; Dutra, L.A.; Barsottini, O.G.P. Cerebellar ataxia associated to anti-glutamic acid decarboxylase autoantibody (anti-GAD): Partial improvement with intravenous immunoglobulin therapy. Arq. Neuropsiquiatr. 2011, 69, 993. [Google Scholar] [CrossRef][Green Version]
- Abele, M.; Weller, M.; Mescheriakov, S.; Bürk, K.; Dichgans, J.; Klockgether, T. Cerebellar ataxia with glutamic acid decarboxylase autoantibodies. Neurology 1999, 52, 857–859. [Google Scholar] [CrossRef]
- Lauria, G.; Pareyson, D.; Pitzolu, M.G.; Bazzigaluppi, E. Excellent response to steroid treatment in anti-GAD cerebellar ataxia. Lancet Neurol. 2003, 2, 634–635. [Google Scholar] [CrossRef]
- Virgilio, R.; Corti, S.; Agazzi, P.; Santoro, D.; Lanfranconi, S.; Candelise, L.; Bresolin, N.; Comi, G.P.; Bersano, A. Effect of steroid treatment in cerebellar ataxia associated with anti-glutamic acid decarboxylase antibodies. J. Neurol. Neurosurg. Psychiatry 2009, 80, 95–96. [Google Scholar] [CrossRef] [PubMed]
- Kuchling, J.; Shababi-Klein, J.; Nümann, A.; Gerischer, L.M.; Harms, L.; Prüss, H. GAD Antibody-Associated Late-Onset Cerebellar Ataxia in Two Female Siblings. Case Rep. Neurol. 2014, 6, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Marchiori, G.C.; Vaglia, A.; Vianello, M.; Bardin, P.G.; Giometto, B. Encephalitis associated with glutamic acid decarboxylase autoantibodies. Neurology 2001, 56, 814. [Google Scholar] [CrossRef]
- Mazzi, G.; Roia, D.D.; Cruciatti, B.; Matà, S.; Catapano, R. Plasma exchange for anti-GAD associated non paraneoplastic limbic encephalitis. Transfus. Apher. Sci. 2008, 39, 229–233. [Google Scholar] [CrossRef]
- Malter, M.P.; Frisch, C.; Zeitler, H.; Surges, R.; Urbach, H.; Helmstaedter, C.; Elger, C.E.; Bien, C.G. Treatment of immune-mediated temporal lobe epilepsy with GAD antibodies. Seizure 2015, 30, 57–63. [Google Scholar] [CrossRef]
- Hao, W.; Davis, C.; Hirsch, I.B.; Eng, L.J.; Daniels, T.; Walsh, D.; Lernmark, A. Plasmapheresis and immunosuppression in stiff-man syndrome with type 1 diabetes: A 2-year study. J. Neurol. 1999, 246, 731–735. [Google Scholar] [CrossRef]
- Fekete, R.; Jankovic, J. Childhood Stiff-Person syndrome improved with rituximab. Case Rep. Neurol. 2012, 4, 92–96. [Google Scholar] [CrossRef]
- Mitoma, H.; Hadjivassiliou, M.; Honnorat, J. Guidelines for treatment of immune-mediated cerebellar ataxias. Cerebellum Ataxias 2015, 2, 14. [Google Scholar] [CrossRef]
- Hansen, N.; Widman, G.; Witt, J.-A.; Wagner, J.; Becker, A.J.; Elger, C.E.; Helmstaedter, C. Seizure control and cognitive improvement via immunotherapy in late onset epilepsy patients with paraneoplastic versus GAD65 autoantibody-associated limbic encephalitis. Epilepsy Behav. 2016, 65, 18–24. [Google Scholar] [CrossRef]


{kind=link}
| Reference | Number of Patients | Median Age (Range) | Female Gender | Associated Autoimmune Disorders | Paraneoplastic Cases | Neurological Symptoms/Phenotypes | Intrathecal Synthesis of GAD Ab | Oligoclonal Bands in the CSF |
|---|---|---|---|---|---|---|---|---|
| SPS | ||||||||
| Saiz et al., 2008 [28] | 22 | 56 (14–77) | 19/22 (86%) | T1DM (59%), thyroiditis (18%), vitiligo (9%) | none | classic SPS (82%), focal SPS (18%) | 11/13 (85%) | 5/14 (35%) |
| McKeon et al., 2012 [45] | 79 | NA | NA | T1DM (43%), thyroiditis (35%), vitiligo (9%), PA (8%) | 3/79 (4%); thyroid, kidney, colon | classic SPS (75%), focal SPS (24%), PERM (1%) | NA | NA |
| Arino et al., 2014 [18] | 28 | 56 (19–77) | 26/28 (93%) | T1DM (50%), thyroiditis (25%), PA (11%), vitiligo (11%) | 1/28 (4%) **; breast cancer | NA | 9/11 (82%) | 5/17 (29%) |
| Gresa-Arribas et al., 2015 [15] | 32 | 53 (5–77) | 29/32 (91%) | T1DM (48%), thyroiditis (28%), other (16%) | excluded from the study | NA | NA | 4/15 (27%) |
| Cerebellar Ataxias | ||||||||
| Honnorat et al., 2001 [87] | 14 | 51 (20–74) | 1314 (93%) | T1DM (71%), thyroiditis (57%), PA (14%), myasthenia gravis (7%) | 2/14 (14%); thymomas | gait ataxia (100%), limb ataxia (86%), nystagmus (86%), dysarthria (57%) | 5/6 (83%) | 10/14 (71%) |
| Saiz et al., 2008 [28] | 17 | 59 (39–77) | 16/17 (94%) | T1DM (53%), thyroiditis (41%), PA (12%), vitiligo (6%) | 2/17 (12%); NSCLC, neuroendocrine thymic carcinoma | gait ataxia (100%), limb ataxia (59%), dysarthria (65%), nystagmus (65%) | 12/12 (100%) | 9/13 (69%) |
| Arino et al., 2014 [18] | 34 | 58 (33–80) | 28/34 (82%) | T1DM (38%), thyroiditis (53%), PA (21%), vitiligo (6%) | 4/34 (12%) *; thymoma, endometrial carcinoma, breast cancer, MDS | gait ataxia (91%), limb ataxia (74%), dysarthria (71%), nystagmus (59%) | 13/15 (87%) | 16/22 (73%) |
| Gresa-Arribas et al., 2015 [15] | 39 | 60 (32–79) | 32/39 (82%) | T1DM (38%), thyroiditis (60%), other (23%) | excluded from the study | NA | NA | 18/24 (75%) |
| Limbic Encephalitis | ||||||||
| Malter et al., 2010 [29] | 9 | 23 (17–66) | 7/9 (78%) | T1DM (22%) | none | seizures (100%), overt cognitive impairment or psychiatric disturbances (11%) | 9/9 (100%) | 5/8 (63%) |
| Gresa-Arribas et al., 2015 [15] | 17 | 26 (12–49) | 12/15 (80%) | T1DM (33%), thyroiditis (60%), other (12%) | excluded from the study | NA | NA | 7/7 (100%) |
| Joubert et al., 2020 [93] | 15 | 30 (2–63) | 14/15(93%) | Autoimmune diseases (60%) | none | Seizures (53%), acute amnesia (67%), behavioral disorders (33%) | NA | 12/14 (86%) |
| Number of Patients | Treatment Schedule | Outcome after Treatment | Treatment-Related Complications | Mortality during FU | Reference | |
|---|---|---|---|---|---|---|
| IVIg Placebo controlled study | 16 | 2 g/kg, divided into two daily doses, every month for 3 months | 11 out of 14 patients (79%) improve with regard to muscle rigidity, spasms, and functional ability to walk | None | 12.5% | Dalakas et al., 2005 [33,23] |
| High-dose CS | 2 | Oral or intravenous steroids | - Distal stiffness: 1/5 slight improvement and 1/5 worsening - Axial stiffness: ½ slight improvement | NA | NA | Barker et al., 1998 [109] |
| High-dose CS | 2 | Prednisone 100 mg/d, and decrease | 1. improvement: no symptoms at 10 d 2. improvement | 1. insomnia, increased anxiety, de- pressed mood 2. hypokalemia, cushingoid features | 0% | Piccolo et al., 1988 [126] |
| PE | 1 | No details | Stable: mRS 4 | NA | NA | McKeon et al., 2012 [45] |
| PE | 3 | No details | Stable | NA | NA | Barker et al., 1998 [109] |
| PE | 1 | No details | Marked improvement | NA | NA | Brashear et al., 1991 [127] |
| Rituximab Double blind placebo-controlled study | 14 | 2 biweekly infusions of 1g each | Primary outcome (change in stiffness scores at 6 months): non-significant effect | Some infusion-related reactions | NA | Dalakas et al., 2017 [34] |
| Rituximab | 1 | - 1000 mg at 0 and 14 day | Partial Improvement on scores: -stiff: 4/6 to 1 - sensitivity: 5/7 to 4 | NA | 0% | Sevy et al., 2012 [128] |
| Rituximab | 1 | 1000 mg at 0 and 7 day | Improvement: mRS 4 to 1 | NA | 0% | Bacorro et al. 2010 [129] |
| IVIg | 1 | 2 g/kg over 5 days every month for 3 months | Partial improvement (ICARS 65 to 37) | NA | NA | Pedroso et al., 2011 [130] |
| IVIg | 1 | IVIg every month for 2 months | Partial improvement (ICARS 59 to 48) | NA | 0% | Abele et al., 1999 [131] |
| IVIg | 3 | 0.4 g/kg/day for 5 days, followed by two cycles of single monthly doses 1g/kg | Partial improvement for 1/3 No improvement for 2/3 | NA | NA | Aguiar et al., 2017 [112] |
| High-dose CS | 1 | MP 1000 mg/ day for 5 d | Improvement: ICARS 60 to 36 | NA | 0% | Lauria et al., 2003 [132] |
| High-dose CS | 1 | MP 1000 mg/day for 5 d | Improvement: ICARS 38 to 22 at the beginning and ICARS 7 at 3 months | NA | 0% | Virgilio et al., 2009 [133] |
| PE + Rituximab | 2 | 7–10 cycles of plasmapheresis + 1000 mg rituximab IV | 1. High response during 1 month 2. No change | NA | 0% | Kuchling et al., 2014 [134] |
| High-dose CS | 1 | MP 500 mg/d, 6 days | High improvement | NA | 0% | Marchiori et al., 2001 [135] |
| PE | 1 | Two cycles of 7 PE + 500 mg MPx3/AZA+ oral corticosteroids | Important improvement the first month: decreasing of seizures, low response after | NA | NA | Mazzi et al., 2008 [136] |
| High-dose CS | 11 | median total dose 19 g (3–30 g) | 45% of response | 55%: -Cushing syndrome in three patients, hyperglycaemia, sleep disorders, nervousness and psychosis | NA | Malter et al., 2015 [137] |
| IVIg | 5 | Dose: range 3–4 g for a median of 3 months | 20% (1 patient) with seizure response | 0% | NA | Malter et al., 2015 [137] |
| PE | 8 | 1 (or 2) sequence of 16 sessions in median (range: 11–26) | 13% (1 patient) of response | 0% | NA | Malter et al., 2015 [137] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dade, M.; Berzero, G.; Izquierdo, C.; Giry, M.; Benazra, M.; Delattre, J.-Y.; Psimaras, D.; Alentorn, A. Neurological Syndromes Associated with Anti-GAD Antibodies. Int. J. Mol. Sci. 2020, 21, 3701. https://doi.org/10.3390/ijms21103701
Dade M, Berzero G, Izquierdo C, Giry M, Benazra M, Delattre J-Y, Psimaras D, Alentorn A. Neurological Syndromes Associated with Anti-GAD Antibodies. International Journal of Molecular Sciences. 2020; 21(10):3701. https://doi.org/10.3390/ijms21103701
Chicago/Turabian StyleDade, Maëlle, Giulia Berzero, Cristina Izquierdo, Marine Giry, Marion Benazra, Jean-Yves Delattre, Dimitri Psimaras, and Agusti Alentorn. 2020. "Neurological Syndromes Associated with Anti-GAD Antibodies" International Journal of Molecular Sciences 21, no. 10: 3701. https://doi.org/10.3390/ijms21103701
APA StyleDade, M., Berzero, G., Izquierdo, C., Giry, M., Benazra, M., Delattre, J.-Y., Psimaras, D., & Alentorn, A. (2020). Neurological Syndromes Associated with Anti-GAD Antibodies. International Journal of Molecular Sciences, 21(10), 3701. https://doi.org/10.3390/ijms21103701