Animal Models to Translate Phage Therapy to Human Medicine


Abstract
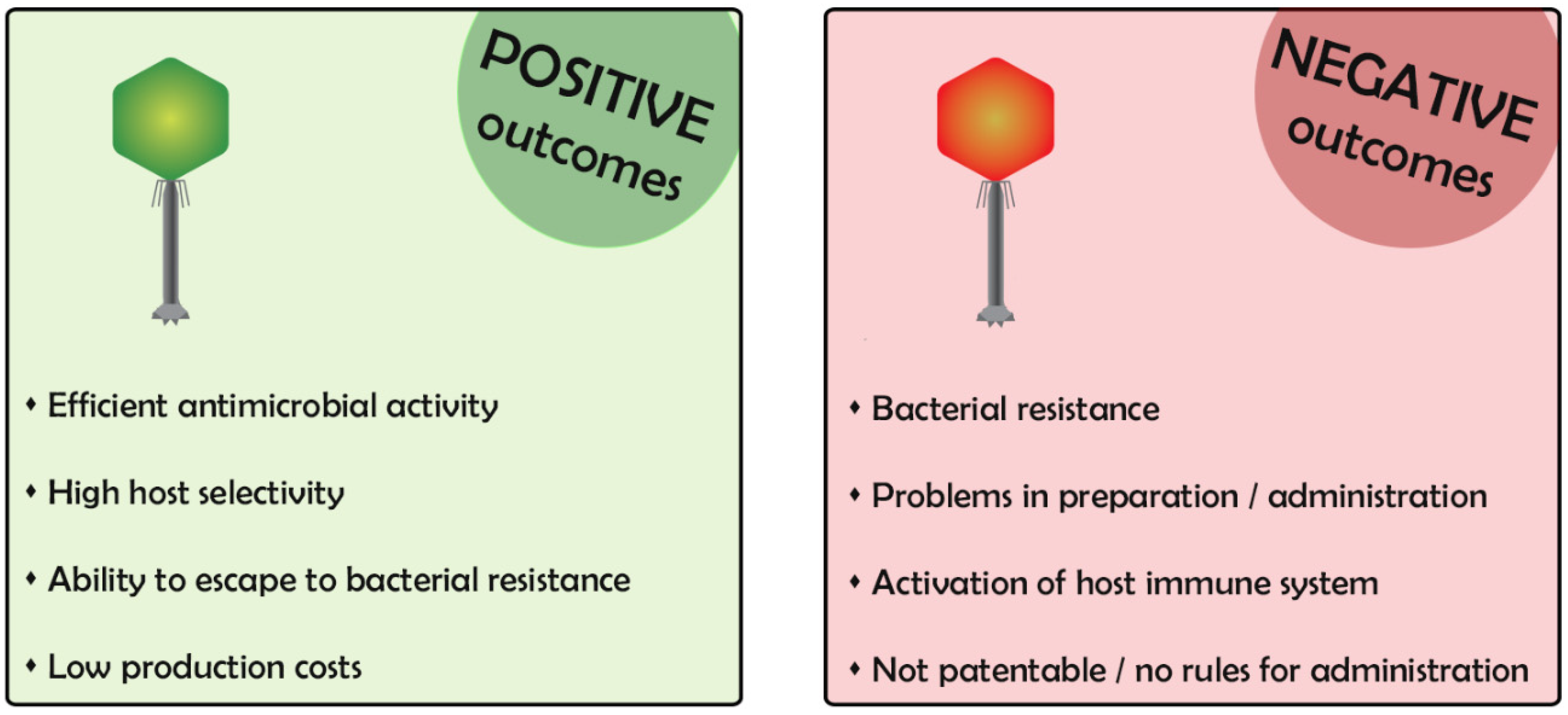
:1. Phages for Therapy: Positive and Negative Outcomes
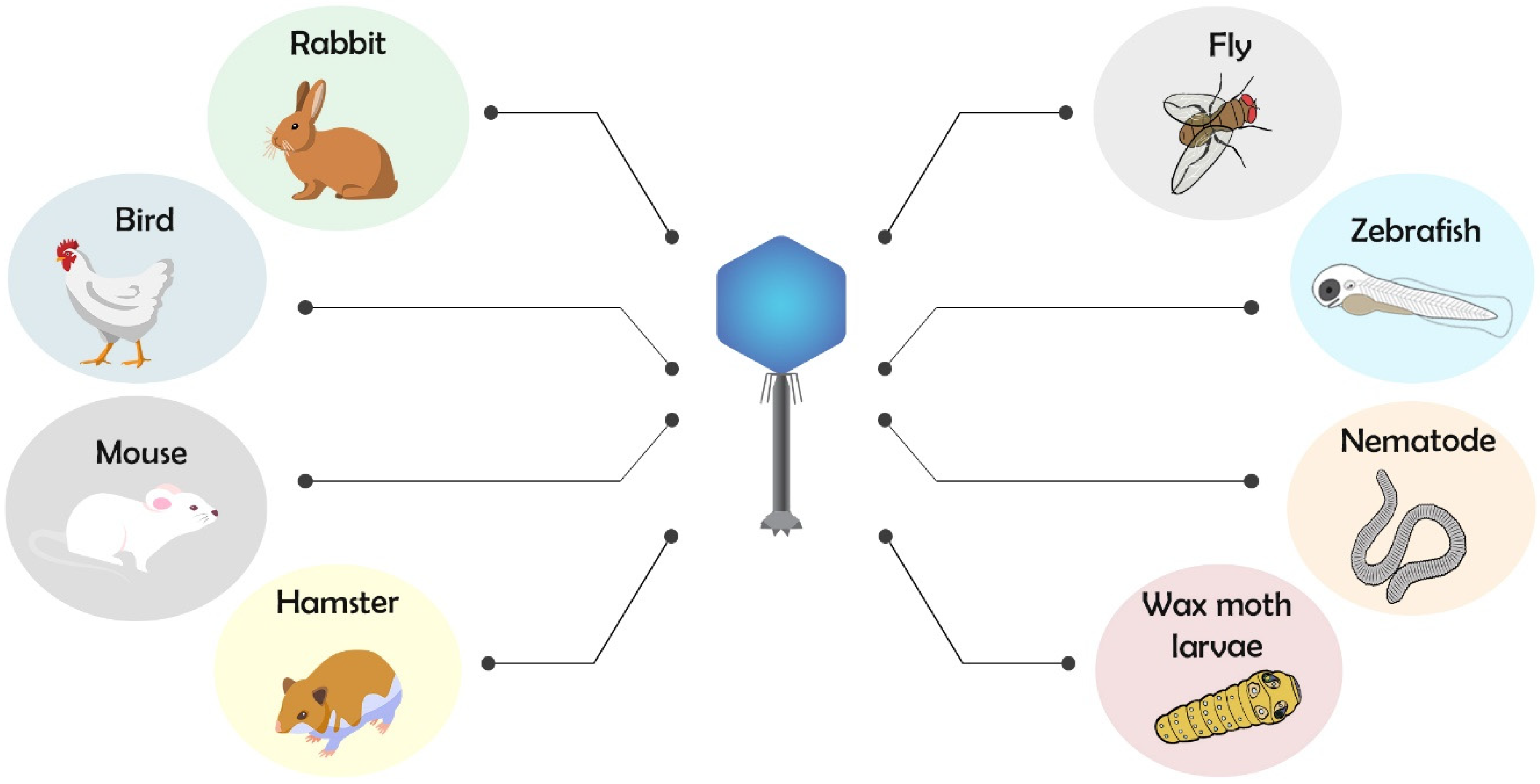
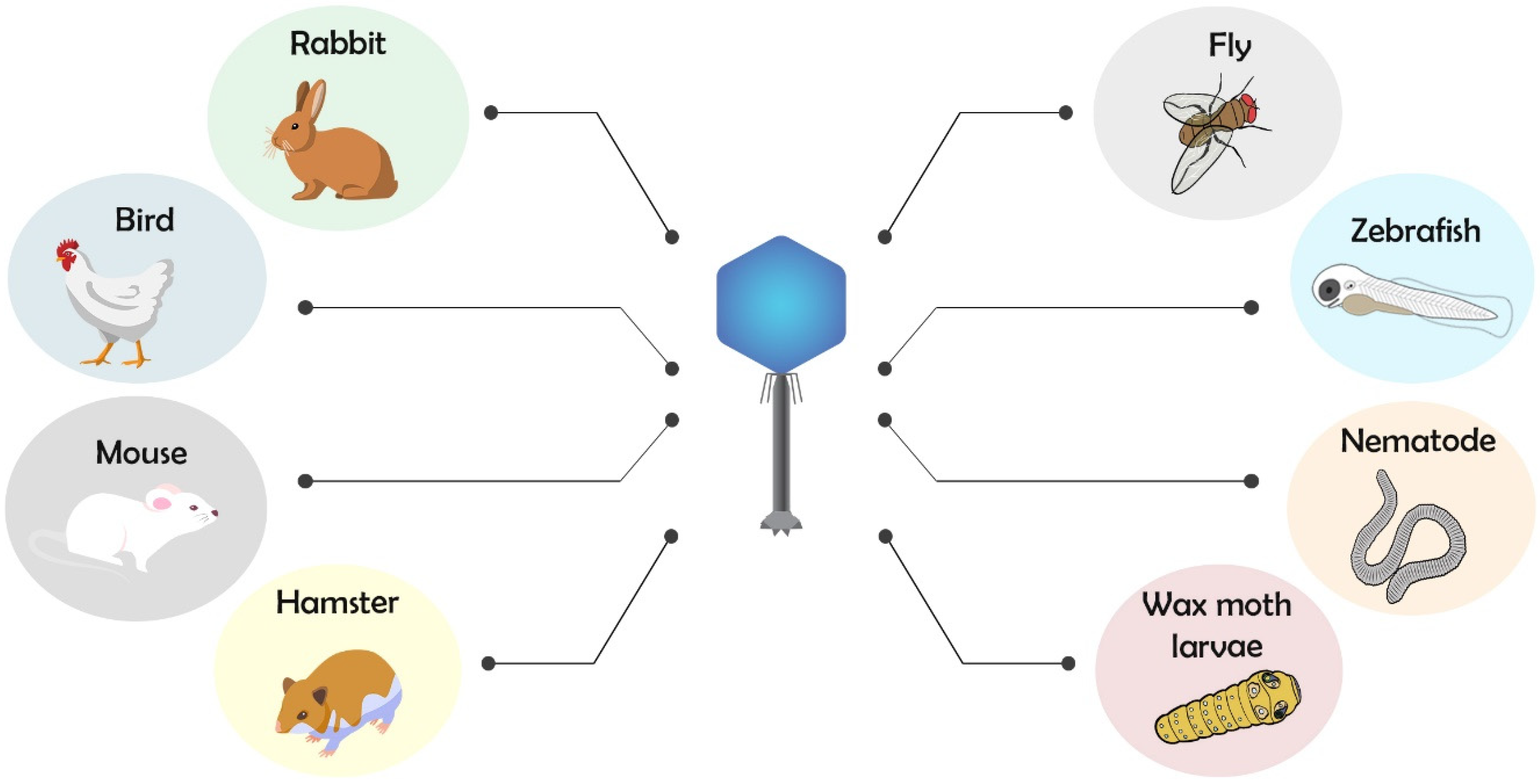
2. Animal Models for Testing Phage Therapy
2.1. Phage Therapy and Antimicrobial Action Using Invertebrate and Vertebrate Animal Models
2.2. Phages and Immune System Interactions Studies Using Animal Models
2.3. Route of Phage Administration in Animal Models
2.4. Methods to Improve Phage Therapy Using Animal Models
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDR | Multi drug resistant |
| CF | Cystic fibrosis |
| EU | Endotoxin units |
| FDA | Food and drug administration |
| CPT | Center for Phage Technology |
| IV | Intravenous |
| PFU | Plaque forming units |
References
- Bordet, J.; Ciuca, M. Remarques sur l’historique de recherches concernant la lyse microbienne transmissible. Compt. Rend. Soc. Biol. 1921, 84, 745–747. [Google Scholar]
- Abedon, S.T. Use of phage therapy to treat long-standing, persistent, or chronic bacterial infections. Adv. Drug Deliv. Rev. 2019, 145, 18–39. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, Z.; Abedon, S.T. Diversity of phage infection types and associated terminology: The problem with ‘Lytic or lysogenic’. FEMS Microbiol. Lett. 2016, 363, fnw047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drulis-Kawa, Z.; Majkowska-Skrobek, G.; Maciejewska, B.; Delattre, A.-S.; Lavigne, R. Learning from bacteriophages—Advantages and limitations of phage and phage-encoded protein applications. Curr. Protein Pept. Sci. 2012, 13, 699–722. [Google Scholar] [CrossRef] [Green Version]
- Goh, S. Phage transduction. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2016; Volume 1476, pp. 177–185. [Google Scholar] [CrossRef]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [Green Version]
- Fauconnier, A. Phage therapy regulation: From night to dawn. Viruses 2019, 11, 352. [Google Scholar] [CrossRef] [Green Version]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [Green Version]
- Law, N.; Logan, C.; Furr, C.; Lehman, S.; Morales, S.; Rosas, F.; Gaidamaka, A.; Bilinsky, I.; Grint, P.; Schooley, R.; et al. Successful bacteriophage therapy for treatment of multidrug-resistant pseudomonas aeruginosa infection in a cystic fibrosis patient. J. Hear. Lung Transplant. 2019, 38, S38. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, M.; Adamia, R. Phage therapy experience at the Eliava Institute. Médecine Mal. Infect. 2008, 38, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, P. Purification of bacteriophages and SDS-PAGE analysis of phage structural proteins from ghost particles. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2009; Volume 502, pp. 227–238. [Google Scholar]
- Gill, J.; Hyman, P. Phage choice, isolation, and preparation for phage therapy. Curr. Pharm. Biotechnol. 2010, 11, 2–14. [Google Scholar] [CrossRef] [PubMed]
- McCallin, S.; Alam Sarker, S.; Barretto, C.; Sultana, S.; Berger, B.; Huq, S.; Krause, L.; Bibiloni, R.; Schmitt, B.; Reuteler, G.; et al. Safety analysis of a Russian phage cocktail: From MetaGenomic analysis to oral application in healthy human subjects. Virology 2013, 443, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Gindin, M.; Febvre, H.P.; Rao, S.; Wallace, T.C.; Weir, T.L. Bacteriophage for gastrointestinal health (PHAGE) study: Evaluating the safety and tolerability of supplemental bacteriophage consumption. J. Am. Coll. Nutr. 2019, 38, 68–75. [Google Scholar] [CrossRef]
- Fish, R.; Kutter, E.; Bryan, D.; Wheat, G.; Kuhl, S. Resolving digital staphylococcal osteomyelitis using bacteriophage—A case report. Antibiotics 2018, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Fish, R.; Kutter, E.; Wheat, G.; Blasdel, B.; Kutateladze, M.; Kuhl, S. Compassionate use of bacteriophage therapy for foot ulcer treatment as an effective step for moving toward clinical trials. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2018; Volume 1693, pp. 159–170. [Google Scholar] [CrossRef]
- Corbellino, M.; Kieffer, N.; Kutateladze, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Tsertsvadze, G.; Rimoldi, S.G.; Nizharadze, D.; Hoyle, N.; et al. Eradication of a multidrug-resistant, carbapenemase-producing Klebsiella pneumoniae isolate following oral and intra-rectal therapy with a custom made, lytic bacteriophage preparation. Clin. Infect. Dis. 2020, 70, 1998–2001. [Google Scholar] [CrossRef]
- Manohar, P.; Ramesh, N. Improved lyophilization conditions for long-term storage of bacteriophages. Sci. Rep. 2019, 9, 15242. [Google Scholar] [CrossRef] [Green Version]
- Melo, L.D.R.; Oliveira, H.; Pires, D.P.; Dabrowska, K.; Azeredo, J. Phage therapy efficacy: A review of the last 10 years of preclinical studies. Crit. Rev. Microbiol. 2020, 46, 78–99. [Google Scholar] [CrossRef]
- Cohen, L.B.; Troemel, E.R. Microbial pathogenesis and host defense in the nematode C. elegans. Curr. Opin. Microbiol. 2015, 23, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Pukkila-Worley, R.; Ausubel, F.M. Immune defense mechanisms in the Caenorhabditis elegans intestinal epithelium. Curr. Opin. Immunol. 2012, 24, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewbank, J.J.; Zugasti, O. C. elegans: Model host and tool for antimicrobial drug discovery. Dis. Model. Mech. 2011, 4, 300–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Augustine, J.; Gopalakrishnan, M.V.; Bhat, S.G. Application of ΦSP-1 and ΦSP-3 as a therapeutic strategy against Salmonella Enteritidis infection using Caenorhabditis elegans as model organism. FEMS Microbiol. Lett. 2014, 356, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Głowacka-Rutkowska, A.; Gozdek, A.; Empel, J.; Gawor, J.; Żuchniewicz, K.; Kozińska, A.; Dębski, J.; Gromadka, R.; Łobocka, M. The ability of lytic staphylococcal podovirus vB_SauP_phiAGO1.3 to coexist in equilibrium with its host facilitates the selection of host mutants of attenuated virulence but does not preclude the phage antistaphylococcal activity in a nematode infection model. Front. Microbiol. 2019, 9, 3227. [Google Scholar] [CrossRef]
- Buchon, N.; Silverman, N.; Cherry, S. Immunity in Drosophila melanogaster—From microbial recognition to whole-organism physiology. Nat. Rev. Immunol. 2014, 14, 796–810. [Google Scholar] [CrossRef]
- Lemaitre, B.; Hoffmann, J. The host defense of Drosophila melanogaster. Annu. Rev. Immunol. 2007, 25, 697–743. [Google Scholar] [CrossRef] [Green Version]
- Hill, L.; Veli, N.; Coote, P.J. Evaluation of Galleria mellonella larvae for measuring the efficacy and pharmacokinetics of antibiotic therapies against Pseudomonas aeruginosa infection. Int. J. Antimicrob. Agents 2014, 43, 254–261. [Google Scholar] [CrossRef]
- Ramarao, N.; Nielsen-Leroux, C.; Lereclus, D. The insect Galleria mellonella as a powerful infection model to investigate bacterial pathogenesis. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, H.M.; McKean, K.A.; Wang, I.-N. Phage fitness may help predict phage therapy efficacy. Bacteriophage 2014, 4, e964081. [Google Scholar] [CrossRef]
- Heo, Y.-J.; Lee, Y.-R.; Jung, H.-H.; Lee, J.; Ko, G.; Cho, Y.-H. Antibacterial efficacy of phages against Pseudomonas aeruginosa infections in mice and Drosophila melanogaster. Antimicrob. Agents Chemother. 2009, 53, 2469–2474. [Google Scholar] [CrossRef] [Green Version]
- Jang, H.-J.; Bae, H.-W.; Cho, Y.-H. Exploitation of Drosophila infection models to evaluate antibacterial efficacy of phages. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2019; Volume 1898, pp. 183–190. [Google Scholar]
- Seed, K.D.; Dennis, J.J. Experimental bacteriophage therapy increases survival of Galleria mellonella larvae infected with clinically relevant strains of the Burkholderia cepacia complex. Antimicrob. Agents Chemother. 2009, 53, 2205–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nale, J.Y.; Chutia, M.; Carr, P.; Hickenbotham, P.T.; Clokie, M.R.J. ‘Get in early’; biofilm and wax moth (Galleria mellonella) models reveal new insights into the therapeutic potential of Clostridium difficile bacteriophages. Front. Microbiol. 2016, 7, 1383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forti, F.; Roach, D.R.; Cafora, M.; Pasini, M.E.; Horner, D.S.; Fiscarelli, E.V.; Rossitto, M.; Cariani, L.; Briani, F.; Debarbieux, L.; et al. Design of a broad-range bacteriophage cocktail that reduces Pseudomonas aeruginosa biofilms and treats acute infections in two animal models. Antimicrob. Agents Chemother. 2018, 62, e02573-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.; Chen, S.; Cao, Z.; Lin, Y.; Mo, D.; Zhang, H.; Gu, J.; Dong, M.; Liu, Z.; Xu, A. Acute phase response in zebrafish upon Aeromonas salmonicida and Staphylococcus aureus infection: Striking similarities and obvious differences with mammals. Mol. Immunol. 2007, 44, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Neely, M.N.; Pfeifer, J.D.; Caparon, M. Streptococcus-zebrafish model of bacterial pathogenesis. Infect. Immun. 2002, 70, 3904–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llamas, M.A.; van der Sar, A.M. Assessing Pseudomonas virulence with nonmammalian host: Zebrafish. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2014; pp. 709–721. [Google Scholar] [CrossRef]
- Al-Zubidi, M.; Widziolek, M.; Court, E.K.; Gains, A.F.; Smith, R.E.; Ansbro, K.; Alrafaie, A.; Evans, C.; Murdoch, C.; Mesnage, S.; et al. Identification of novel bacteriophages with therapeutic potential that target Enterococcus faecalis. Infect. Immun. 2019, 87, e00512-19. [Google Scholar] [CrossRef] [Green Version]
- Cafora, M.; Deflorian, G.; Forti, F.; Ferrari, L.; Binelli, G.; Briani, F.; Ghisotti, D.; Pistocchi, A. Phage therapy against Pseudomonas aeruginosa infections in a cystic fibrosis zebrafish model. Sci. Rep. 2019, 9, 1527. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, M.; Amir Karimi Torshizi, M.; Rahimi, S.; Dennehy, J.J. Prophylactic bacteriophage administration more effective than post-infection administration in reducing Salmonella enterica serovar enteritidis shedding in Quail. Front. Microbiol. 2016, 7, 1253. [Google Scholar] [CrossRef] [Green Version]
- Wernicki, A.; Nowaczek, A.; Urban-Chmiel, R. Bacteriophage therapy to combat bacterial infections in poultry. Virol. J. 2017, 14, 179. [Google Scholar] [CrossRef]
- Colom, J.; Cano-Sarabia, M.; Otero, J.; Aríñez-Soriano, J.; Cortés, P.; Maspoch, D.; Llagostera, M. Microencapsulation with alginate/CaCO3: A strategy for improved phage therapy. Sci. Rep. 2017, 7, 41441. [Google Scholar] [CrossRef]
- Colom, J.; Cano-Sarabia, M.; Otero, J.; Cortés, P.; Maspoch, D.; Llagostera, M. Liposome-encapsulated bacteriophages for enhanced oral phage therapy against Salmonella spp. Appl. Environ. Microbiol. 2015, 81, 4841–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waseh, S.; Hanifi-Moghaddam, P.; Coleman, R.; Masotti, M.; Ryan, S.; Foss, M.; MacKenzie, R.; Henry, M.; Szymanski, C.M.; Tanha, J. Orally administered P22 phage tailspike protein reduces Salmonella colonization in chickens: Prospects of a novel therapy against bacterial infections. PLoS ONE 2010, 5, e13904. [Google Scholar] [CrossRef] [PubMed]
- Trotereau, A.; Schouler, C. Use of a chicken embryo lethality assay to assess the efficacy of phage therapy. In Methods in Molecular Biology; Springer: Cham, Switzerland, 2019; Volume 1898, pp. 199–205. [Google Scholar] [CrossRef]
- Wills, Q.F.; Kerrigan, C.; Soothill, J.S. Experimental bacteriophage protection against Staphylococcus aureus abscesses in a rabbit model. Antimicrob. Agents Chemother. 2005, 49, 1220–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishor, C.; Mishra, R.; Saraf, S.; Kumar, M.; Srivastav, A.; Nath, G. Phage therapy of staphylococcal chronic osteomyelitis in experimental animal model. Indian J. Med. Res. 2016, 143, 87. [Google Scholar] [CrossRef]
- Abedon, S.T. Commentary: Phage therapy of staphylococcal chronic osteomyelitis in experimental animal model. Front. Microbiol. 2016, 7, 1251. [Google Scholar] [CrossRef] [Green Version]
- Yen, M.; Cairns, L.S.; Camilli, A. A cocktail of three virulent bacteriophages prevents Vibrio cholerae infection in animal models. Nat. Commun. 2017, 8, 14187. [Google Scholar] [CrossRef] [Green Version]
- Nale, J.Y.; Spencer, J.; Hargreaves, K.R.; Buckley, A.M.; Trzepiński, P.; Douce, G.R.; Clokie, M.R.J. Bacteriophage combinations significantly reduce Clostridium difficile growth in vitro and proliferation in vivo. Antimicrob. Agents Chemother. 2016, 60, 968–981. [Google Scholar] [CrossRef] [Green Version]
- Pabary, R.; Singh, C.; Morales, S.; Bush, A.; Alshafi, K.; Bilton, D.; Alton, E.W.F.W.; Smithyman, A.; Davies, J.C. Antipseudomonal bacteriophage reduces infective burden and inflammatory response in murine lung. Antimicrob. Agents Chemother. 2016, 60, 744–751. [Google Scholar] [CrossRef] [Green Version]
- Alvi, I.A.; Asif, M.; Tabassum, R.; Aslam, R.; Abbas, Z.; Rehman, S.u. RLP, a bacteriophage of the family Podoviridae, rescues mice from bacteremia caused by multi-drug-resistant Pseudomonas aeruginosa. Arch. Virol. 2020, 165, 1289–1297. [Google Scholar] [CrossRef]
- Debarbieux, L.; Leduc, D.; Maura, D.; Morello, E.; Criscuolo, A.; Grossi, O.; Balloy, V.; Touqui, L. Bacteriophages can treat and prevent Pseudomonas aeruginosa lung infections. J. Infect. Dis. 2010, 201, 1096–1104. [Google Scholar] [CrossRef] [Green Version]
- Leshkasheli, L.; Kutateladze, M.; Balarjishvili, N.; Bolkvadze, D.; Save, J.; Oechslin, F.; Que, Y.-A.; Resch, G. Efficacy of newly isolated and highly potent bacteriophages in a mouse model of extensively drug-resistant Acinetobacter baumannii bacteraemia. J. Glob. Antimicrob. Resist. 2019, 19, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Jamalludeen, N.; Johnson, R.P.; Shewen, P.E.; Gyles, C.L. Evaluation of bacteriophages for prevention and treatment of diarrhea due to experimental enterotoxigenic Escherichia coli O149 infection of pigs. Vet. Microbiol. 2009, 136, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Chadha, P.; Katare, O.P.; Chhibber, S. In vivo efficacy of single phage versus phage cocktail in resolving burn wound infection in BALB/c mice. Microb. Pathog. 2016, 99, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Trigo, G.; Martins, T.G.; Fraga, A.G.; Longatto-Filho, A.; Castro, A.G.; Azeredo, J.; Pedrosa, J. Phage therapy is effective against infection by Mycobacterium ulcerans in a murine footpad model. PLoS Negl. Trop. Dis. 2013, 7, e2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capparelli, R.; Parlato, M.; Borriello, G.; Salvatore, P.; Iannelli, D. Experimental phage therapy against Staphylococcus aureus in mice. Antimicrob. Agents Chemother. 2007, 51, 2765–2773. [Google Scholar] [CrossRef] [Green Version]
- Anand, T.; Virmani, N.; Kumar, S.; Mohanty, A.K.; Pavulraj, S.; Bera, B.C.; Vaid, R.K.; Ahlawat, U.; Tripathi, B.N. Phage therapy for treatment of virulent Klebsiella pneumoniae infection in a mouse model. J. Glob. Antimicrob. Resist. 2020, 21, 34–41. [Google Scholar] [CrossRef]
- Abd El-Aziz, A.M.; Elgaml, A.; Ali, Y.M. Bacteriophage therapy increases complement-mediated lysis of bacteria and enhances bacterial clearance after acute lung infection with multidrug-resistant Pseudomonas aeruginosa. J. Infect. Dis. 2019, 219, 1439–1447. [Google Scholar] [CrossRef]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the host immune system and bacteriophage is essential for successful phage therapy against an acute respiratory pathogen. Cell Host Microbe 2017, 22, 38–47.e4. [Google Scholar] [CrossRef]
- Łusiak-Szelachowska, M.; Żaczek, M.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Letkiewicz, S.; Fortuna, W.; Rogóż, P.; Szufnarowski, K.; Jończyk-Matysiak, E.; Olchawa, E.; et al. Antiphage activity of sera during phage therapy in relation to its outcome. Future Microbiol. 2017, 12, 109–117. [Google Scholar] [CrossRef]
- Żaczek, M.; Łusiak-Szelachowska, M.; Jończyk-Matysiak, E.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Owczarek, B.; Kopciuch, A.; Fortuna, W.; Rogóż, P.; Górski, A. Antibody production in response to staphylococcal MS-1 phage cocktail in patients undergoing phage therapy. Front. Microbiol. 2016, 7, 1681. [Google Scholar] [CrossRef] [Green Version]
- Łusiak-Szelachowska, M.; Żaczek, M.; Weber-Dąbrowska, B.; Międzybrodzki, R.; Kłak, M.; Fortuna, W.; Letkiewicz, S.; Rogóż, P.; Szufnarowski, K.; Jończyk-Matysiak, E.; et al. Phage neutralization by sera of patients receiving phage therapy. Viral Immunol. 2014, 27, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruttin, A.; Brüssow, H. Human volunteers receiving Escherichia coli phage T4 orally: A safety test of phage therapy. Antimicrob. Agents Chemother. 2005, 49, 2874–2878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majewska, J.; Beta, W.; Lecion, D.; Hodyra-Stefaniak, K.; Kłopot, A.; Kaźmierczak, Z.; Miernikiewicz, P.; Piotrowicz, A.; Ciekot, J.; Owczarek, B.; et al. Oral application of T4 phage induces weak antibody production in the gut and in the blood. Viruses 2015, 7, 4783–4799. [Google Scholar] [CrossRef]
- Kim, K.-P.; Cha, J.-D.; Jang, E.-H.; Klumpp, J.; Hagens, S.; Hardt, W.-D.; Lee, K.-Y.; Loessner, M.J. PEGylation of bacteriophages increases blood circulation time and reduces T-helper type 1 immune response. Microb. Biotechnol. 2008, 1, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dąbrowska, K.; Miernikiewicz, P.; Piotrowicz, A.; Hodyra, K.; Owczarek, B.; Lecion, D.; Kaźmierczak, Z.; Letarov, A.; Górski, A. Immunogenicity studies of proteins forming the T4 phage head surface. J. Virol. 2014, 88, 12551–12557. [Google Scholar] [CrossRef] [Green Version]
- Dąbrowska, K.; Abedon, S.T. Pharmacologically aware phage therapy: Pharmacodynamic and pharmacokinetic obstacles to phage antibacterial action in animal and human bodies. Microbiol. Mol. Biol. Rev. 2019, 83. [Google Scholar] [CrossRef]
- Morozova, V.V.; Vlassov, V.V.; Tikunova, N.V. Applications of bacteriophages in the treatment of localized infections in humans. Front. Microbiol. 2018, 9, 1696. [Google Scholar] [CrossRef] [Green Version]
- Kumari, S. Evidence to support the therapeutic potential of bacteriophage Kpn5 in burn wound infection caused by Klebsiella pneumoniae in BALB/c mice. J. Microbiol. Biotechnol. 2010, 20, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Rose, T.; Verbeken, G.; De Vos, D.; Merabishvili, M.; Vaneechoutte, M.; Lavigne, R.; Jennes, S.; Zizi, M.; Pirnay, J.-P. Experimental phage therapy of burn wound infection: Difficult first steps. Int. J. Burns Trauma 2014, 4, 66–73. [Google Scholar]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Hoyle, N.; Zhvaniya, P.; Balarjishvili, N.; Bolkvadze, D.; Nadareishvili, L.; Nizharadze, D.; Wittmann, J.; Rohde, C.; Kutateladze, M. Phage therapy against Achromobacter xylosoxidans lung infection in a patient with cystic fibrosis: A case report. Res. Microbiol. 2018, 169, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Waters, E.M.; Neill, D.R.; Kaman, B.; Sahota, J.S.; Clokie, M.R.J.; Winstanley, C.; Kadioglu, A. Phage therapy is highly effective against chronic lung infections with Pseudomonas aeruginosa. Thorax 2017, 72, 666–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Speck, P.; Smithyman, A. Safety and efficacy of phage therapy via the intravenous route. FEMS Microbiol. Lett. 2016, 363, 242. [Google Scholar] [CrossRef] [PubMed]
- Duplessis, C.; Biswas, B.; Hanisch, B.; Perkins, M.; Henry, M.; Quinones, J.; Wolfe, D.; Estrella, L.; Hamilton, T. Refractory pseudomonas bacteremia in a 2-year-old sterilized by bacteriophage therapy. J. Pediatric Infect. Dis. Soc. 2018, 7, 253–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dąbrowska, K. Phage therapy: What factors shape phage pharmacokinetics and bioavailability? Systematic and critical review. Med. Res. Rev. 2019, 39, 2000–2025. [Google Scholar] [CrossRef] [Green Version]
- Sarker, S.A.; McCallin, S.; Barretto, C.; Berger, B.; Pittet, A.-C.; Sultana, S.; Krause, L.; Huq, S.; Bibiloni, R.; Bruttin, A.; et al. Oral T4-like phage cocktail application to healthy adult volunteers from Bangladesh. Virology 2012, 434, 222–232. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, W.N.; Concepción-Acevedo, J.; Park, T.; Andleeb, S.; Bull, J.J.; Levin, B.R. Synergy and order effects of antibiotics and phages in killing Pseudomonas aeruginosa biofilms. PLoS ONE 2017, 12, e0168615. [Google Scholar] [CrossRef]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef] [Green Version]
- Abu Lila, A.S.; Ishida, T. Liposomal delivery systems: Design optimization and current applications. Biol. Pharm. Bull. 2017, 40, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Singh, D.; Pradhan, M.; Nag, M.; Singh, M.R. Vesicular system: Versatile carrier for transdermal delivery of bioactives. Artif. Cells Nanomed. Biotechnol. 2015, 43, 282–290. [Google Scholar] [CrossRef]
- Chhibber, S.; Shukla, A.; Kaur, S. Transfersomal phage cocktail is an effective treatment against methicillin-resistant Staphylococcus aureus-mediated skin and soft tissue infections. Antimicrob. Agents Chemother. 2017, 61, e02146-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.; Abedon, S. Bacteriophages and their enzymes in biofilm control. Curr. Pharm. Des. 2014, 21, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Fischetti, V. Development of phage lysins as novel therapeutics: A historical perspective. Viruses 2018, 10, 310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, S.Y.; Jang, I.J.; Yoon, S.; Jang, K.; Yu, K.-S.; Cho, J.Y.; Seong, M.-W.; Jung, G.M.; Yoon, S.J.; Kang, S.H. Pharmacokinetics and tolerance of the phage endolysin-based candidate drug SAL200 after a single intravenous administration among healthy volunteers. Antimicrob. Agents Chemother. 2017, 61, e02629-16. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Paff, M.L.; Molineux, I.J.; Bull, J.J. Therapeutic application of phage capsule depolymerases against K1, K5, and K30 capsulated E. coli in mice. Front. Microbiol. 2017, 8, 2257. [Google Scholar] [CrossRef]
- Lin, H.; Paff, M.; Molineux, I.; Bull, J. Antibiotic therapy using phage depolymerases: Robustness across a range of conditions. Viruses 2018, 10, 622. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Animal Model | Challenge (Pathogen) | Condition | Phage Treatment | Route of Administration | Results Summary | Reference |
|---|---|---|---|---|---|---|
| C. elegans | Salmonella enterica; spread on agar plate | lethal systemic infection | mono-phage, delay (24 hpi); 5 × 109–1× 1010 pfu | in growth medium | >survival rate | Augustine et al., 2014 [26] |
| C. elegans | Staphylococcus aureus; spread on agar plate | lethal systemic infection | mono-phage, delay (24 hpi); 109 pfu/ml | in growth medium | >survival rate | Glowacka-Rutkowska et al., 2019 [27] |
| D. melanogaster | Pseudomonas aeruginosa; intrathorax injection of 103 cfu/fly | lethal systemic infection | mono-phage, delay (6 hpi); 104 pfu/fly | intrathorax injection | >survival rate | Lindberg et al., 2014 [32] |
| D. melanogaster | Pseudomonas aeruginosa; intrathorax injection of 50–200 cfu/fly | lethal systemic infection | mono-phage, co-adm; 106 pfu/fly | oral (force feed) | >survival rate; <BB | Heo et al., 2009 [33] |
| G. mellonella | Clostridium difficile; oral administration 105 cfu/larva | lethal systemic infection | 4-phage cocktail: proph (2 hbi), delay (2 hpi) or co-adm; 1 to 4 doses of 106 pfu/larva | oral | reduced mortality (100% in proph); dose-dependence | Nale et al., 2016 [36] |
| G. mellonella | Burkholderia cepacia; injection of 2.5 × 103 cfu/larva | lethal bacteremia | mono-phage, delay (6 or 12 hpi); 2.5 × 103 pfu/larva | injection | >survival rate; <BB | Seed et al., 2009 [35] |
| G. mellonella | Pseudomonas aeruginosa (lab and clinical strains); injection of 30 cfu/larva | lethal bacteremia | 6-phage cocktail: proph (1 hbi) or delay (1 hpi); 1.5 to 4.5 × 103 pfu/larva | injection | prolonged survival time after infection | Forti et al., 2018 [37] |
| G. mellonella | Acinetobacter baumanii (XDR); injection of 5 × 105 cfu/larva | lethal bacteremia | 2-phage cocktail or mono-phage, delay (0.5 hpi); 5 × 107 pfu/larva | injection | >survival rate (≥80%) | Leshkasheli et al., 2019 [57] |
| Zebrafish | Enterococcus faecalis (clinical strain); injection in circulation of 3 × 104 cfu/embryo | lethal systemic infection | mono-phage, delay (2 hpi); 6 × 105 pfu/embryo in 2 nL | injection in circulation | >survival rate (of 57%); >healthy state | Al-Zubidi et al., 2019 [41] |
| Zebrafish | Pseudomonas aeruginosa; injection in circulation of 30 cfu/embryo | lethal systemic infection | 4-phage cocktail, delay (0.5 or 7 hpi); 500–1000 pfu/embryo in 2 nL | injection in circulation | >survival rate (of about 30%); <BB; reduced inflammatory response | Cafora et al., 2019 [42] |
| Quail | Salmonella enterica (Enteriditis); oral administration of 1.2 × 108 cfu/quail | gastrointestinal infection | mono-phage, proph or delay *; 105 pfu/mL, 3 doses daily | oral (oral gavage or vent lip) | <BB in cecal tonsils | Ahmadi et al., 2016 [43] |
| Chicken | Salmonella enterica (Typhimurium); oral administration of 107 cfu/chicken | gastrointestinal infection | 3-phage cocktail (liposome/alginate encapsulated), delay (24 hpi); 109/1010 pfu/chicken, 8 doses daily | oral | <BB in cecum (of 1.5–3.9 Log10 cfu) | Colom et al., 2015, 2017 [45,46] |
| Rabbit | Staphylococcus aureus; subcutaneous injection of 8 × 107 cfu/rabbit | local infection (abscess) | mono-phage, co-adm or delay (6, 12 or 24 hpi); 2 × 109 pfu/rabbit | subcutaneous injection | <BB of infected area and abscesses prevention in co-adm (no effect in delay) | Wills et al., 2005 [49] |
| Rabbit | Staphylococcus aureus (MRSA); Intramedullary injection of ≤5 × 106 cfu/rabbit (*) | chronic osteomyelitis | 7-phage cocktail, delay (21, or 42 dpi); 5 × 1011 pfu/rabbit, 4 doses total every 2 days | Intralesional injection | cure of infection in 21 dpf treatment | Kishor et al, 2016 [50] |
| Rabbit | Vibrio cholerae; oral administration of 5 × 108 cfu/rabbit | gastrointestinal infection | 3-phage cocktail: proph (3 or 24 hbi); 4–8 × 109 pfu/rabbit | oral | prevention of diarrheal symptoms; <BB in intestine (of 1–4 Log10 cfu) | Yen et al., 2017 [52] |
| Hamster | Clostridium difficile; oral administration of 2 × 103 spores/hamster | gastrointestinal infection | 2,3,4-phage cocktails or mono-phage, delay *; 8 × 107 pfu/mL, every 8 h × 36 hpi | oral | <BB in cecum and colon (of 2 Log10 cfu) | Nale et al., 2017 [53] |
| Pig | Escherichia coli (ETEC); oral administration of 1010 cfu/pig | gastrointestinal infection | 2,3-phage cocktail or mono-phage, proph (0.25 hbi, 3 × 109–1010 pfu/pig) or delay (24 hpi, 6 doses every 3 h, 108 pfu/pig) | oral | diarrhea symptoms ameliorate | Jamalludeen et al., 2009 [58] |
| Murine | Pseudomonas aeruginosa, intranasal injection of 1 × 107 cfu/mouse | lethal respiratory infection | 6-phage cocktail, delay (2 hpi); 107 pfu/mouse | intranasal injection | 100% reduced mortality; <BB (about 3 Log10 times) | Forti et al., 2018 [37] |
| Murine | Pseudomonas aeruginosa, intranasal injection of 2.5 × 107 cfu/mouse | respiratory infection | 3-phage cocktail: proph (48 hbi), co-adm or delay (24 hpi); 1.24 × 109 pfu/mouse | intranasal injection | >survival rate; bacterial clearance in BALs (proph 71%, co-adm 100% and delay 86%); reduced inflammatory response | Pabary et al., 2016 [54] |
| Murine | Pseudomonas aeruginosa (MDR), intraperitoneal injection of 107 cfu/mouse | lethal bacteremia | mono-phage, co-adm; 1 ×109 pfu/mouse | intraperitoneal injection | 85% reduced mortality; bacterial clearance in blood; reduced inflammatory response | Alvi et al., 2020 [55] |
| Murine | Pseudomonas aeruginosa (clinical strain), intranasal injection of 107 cfu/mouse | lethal lung infection | mono-phage, proph (24 hbi) or delay (2, 4, 6 hpi); 108 pfu/mouse | intranasal injection | >survival rate: delay-dependent (from 100% in 2 hpi to 25% in 6 hpi) and 100% in proph; reduced inflammatory response | Debarbieux et al., 2010 [56] |
| Murine | Acinetobacter baumanii (XDR), intraperitoneal injection of 6 × 107 cfu/mouse | lethal bacteremia | 2-phage cocktail or mono-phage, delay (2 hpi); 6 × 109 pfu/mouse | intraperitoneal injection | >survival rate (≥80%) | Leshkasheli et al., 2019 [57] |
| Murine | Klebsiella pneumoniae, topical administration 50 ul of 108 cfu/mL | burn wound infection | 5-phage cocktail or mono-phage, delay (6 hpi); 50 uL of 108 pfu/ml | topical | <BB in skin tissue; faster wound healing; reduced inflammatory response | Chadha et al., 2016 [59] |
| Murine | Mycobacterium ulcerans, subcutaneous injection of 105.5 afb | local infection (ulceration) | mono-phage, delay (33 dpi); 108 pfu/mouse | subcutaneous injection | <BB in skin tissue; prevent ulceration | Trigo et al., 2013 [60] |
| Murine | Staphylococcus aureus (MRSA), subcutaneous injection of 107 cfu/mouse | local infection (abscess) | mono-phage, co-adm or delay (4 dpi); 109 pfu/mouse | subcutaneous injection | prevent/ameliorate abscess development | Capparelli et al., 2007 [61] |
| Murine | Staphylococcus aureus (MRSA), intravenous injection of 108 cfu/mouse | systemic infection | mono-phage, co-adm; 109 pfu/mouse | intravenous injection | 97% reduced mortality; bacterial clearance in blood | Capparelli et al., 2007 [61] |
| Murine | Klebsiella pneumoniae, intranasal instillation of 109 cfu/mL | Lung infection | mono-phage, delay (2 hpi); 109 pfu/mouse | intranasal instillation | <BB in lung and serum; prevent severe lung lesions | Anand et al., 2019 [62] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brix, A.; Cafora, M.; Aureli, M.; Pistocchi, A. Animal Models to Translate Phage Therapy to Human Medicine. Int. J. Mol. Sci. 2020, 21, 3715. https://doi.org/10.3390/ijms21103715
Brix A, Cafora M, Aureli M, Pistocchi A. Animal Models to Translate Phage Therapy to Human Medicine. International Journal of Molecular Sciences. 2020; 21(10):3715. https://doi.org/10.3390/ijms21103715
Chicago/Turabian StyleBrix, Alessia, Marco Cafora, Massimo Aureli, and Anna Pistocchi. 2020. "Animal Models to Translate Phage Therapy to Human Medicine" International Journal of Molecular Sciences 21, no. 10: 3715. https://doi.org/10.3390/ijms21103715
APA StyleBrix, A., Cafora, M., Aureli, M., & Pistocchi, A. (2020). Animal Models to Translate Phage Therapy to Human Medicine. International Journal of Molecular Sciences, 21(10), 3715. https://doi.org/10.3390/ijms21103715