Phosphorylation Sites in Protein Kinases and Phosphatases Regulated by Formyl Peptide Receptor 2 Signaling

 ,
, 
 and
and 
Abstract

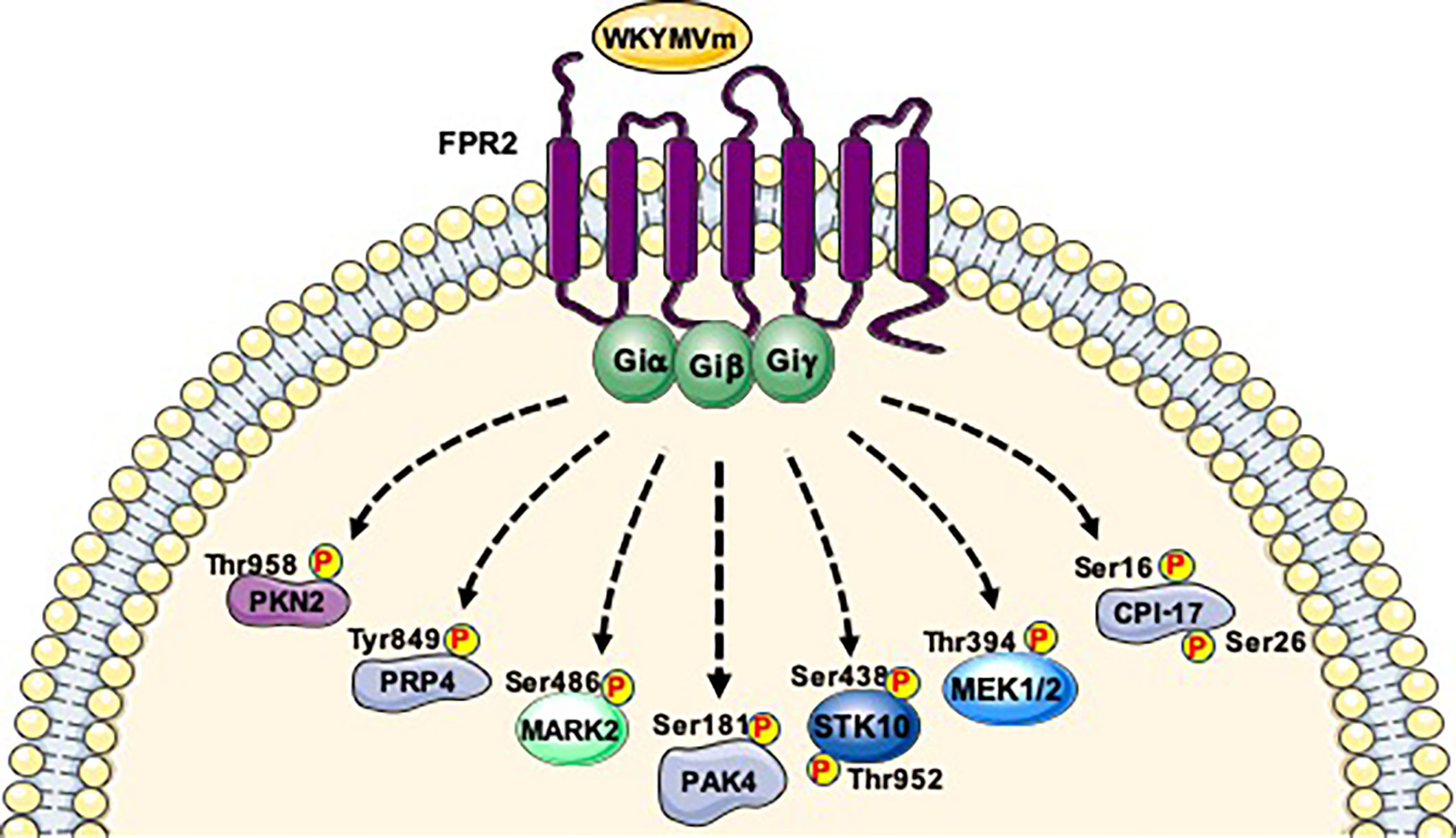{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Serine/Threonine-Protein Kinase N2 (PKN2; UniProt: Q16513)
3. Serine/Threonine-Protein Kinase PRP4 Homolog (PRP4; UniProt: Q13523)
4. Serine/Threonine-Protein Kinase MARK2 (MARK2; Uniprot: Q7KZI7)
5. Serine/Threonine-Protein Kinase PAK4 (PAK4; Uniprot: O96013)
6. Serine/Threonine-Protein Kinase 10 (STK10; UniProt: O94804)
7. Dual Specificity Mitogen-Activated Protein Kinase, Kinase 2 (MAP2K2; UniProt: P36507)
8. Protein Phosphatase 1 Regulatory Subunit 14A (PPP1R14A; UniProt: Q96A00)
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Bononi, A.; Agnoletto, C.; De Marchi, E.; Marchi, S.; Patergnani, S.; Bonora, M.; Giorgi, C.; Missiroli, S.; Poletti, F.; Rimessi, A.; et al. Protein kinases and phosphatases in the control of cell fate. Enzyme Res. 2011, 2011, 329098. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; MacDonald, G. ErbB receptors and signaling pathways in cancer. Curr. Opin. Cell. Biol. 2009, 21, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Levitzki, A. Targeting the EGFR and the PKB pathway in cancer. Curr. Opin. Cell. Biol. 2009, 21, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Freschi, L.; Osseni, M.; Landry, C.R. Functional divergence and evolutionary turnover in mammalian phosphoproteomes. PLoS Genet. 2014, 10, e1004062. [Google Scholar] [CrossRef]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef]
- Amano, M.; Nishioka, T.; Tsuboi, D.; Kuroda, K.; Funahashi, Y.; Yamahashi, Y.; Kaibuchi, K. Comprehensive analysis of kinase-oriented phospho-signalling pathways. J. Biochem. 2019, 165, 301–307. [Google Scholar] [CrossRef]
- Macho, A.P.; Lozano-Duran, R.; Zipfel, C. Importance of tyrosine phosphorylation in receptor kinase complexes. Trends Plant Sci. 2015, 20, 269–272. [Google Scholar] [CrossRef]
- Jung, K.J.; Lee, E.K.; Yu, B.P.; Chung, H.Y. Significance of protein tyrosine kinase/protein tyrosine phosphatase balance in the regulation of NF-kappaB signaling in the inflammatory process and aging. Free Radic. Biol. Med. 2009, 47, 983–991. [Google Scholar] [CrossRef]
- Yang, X.J. Multisite protein modification and intramolecular signaling. Oncogene 2005, 24, 1653–1662. [Google Scholar] [CrossRef]
- Holmberg, C.I.; Tran, S.E.; Eriksson, J.E.; Sistonen, L. Multisite phosphorylation provides sophisticated regulation of transcription factors. Trends Biochem. Sci. 2002, 27, 619–627. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Ammendola, R. Distinct signaling cascades elicited by different formyl peptide receptor 2 (FPR2) agonists. Int. J. Mol. Sci 2013, 14, 7193–7230. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Liu, H.; Edward Zhou, X.; Kumar Verma, R.; de Waal, P.W.; Jang, W.; Xu, T.H.; Wang, L.; Meng, X.; Zhao, G.; et al. Structure of formylpeptide receptor 2-Gi complex reveals insights into ligand recognition and signaling. Nat. Commun. 2020, 11, 885. [Google Scholar] [CrossRef] [PubMed]
- He, H.Q.; Ye, R.D. The Formyl Peptide Receptors: Diversity of Ligands and Mechanism for Recognition. Molecules 2017, 22, 455. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Fioretti, T.; Sarnataro, D.; Esposito, G.; Ammendola, R. Nuclear localization of Formyl-Peptide Receptor 2 in human cancer cells. Arch. Biochem. Biophys. 2016, 603, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.; Kretschmer, D. Formyl-Peptide Receptors in Infection, Inflammation, and Cancer. Trends Immunol. 2018, 39, 815–829. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Ammendola, R. Expression and signaling of formyl-peptide receptors in the brain. Neurochem. Res. 2010, 35, 2018–2026. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Parisi, M.; de Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-surface receptors transactivation mediated by g protein-coupled receptors. Int. J. Mol. Sci. 2014, 15, 19700–19728. [Google Scholar] [CrossRef]
- Leoni, G.; Alam, A.; Neumann, P.A.; Lambeth, J.D.; Cheng, G.; McCoy, J.; Hilgarth, R.S.; Kundu, K.; Murthy, N.; Kusters, D.; et al. Annexin A1, formyl peptide receptor, and NOX1 orchestrate epithelial repair. J. Clin. Investig. 2013, 123, 443–454. [Google Scholar] [CrossRef]
- Wentworth, C.C.; Alam, A.; Jones, R.M.; Nusrat, A.; Neish, A.S. Enteric commensal bacteria induce extracellular signal-regulated kinase pathway signaling via formyl peptide receptor-dependent redox modulation of dual specific phosphatase 3. J. Biol. Chem. 2011, 286, 38448–38455. [Google Scholar] [CrossRef]
- Ammendola, R.; Russo, L.; de Felice, C.; Esposito, F.; Russo, T.; Cimino, F. Low-affinity receptor-mediated induction of superoxide by N-formyl-methionyl-leucyl-phenylalanine and WKYMVm in IMR90 human fibroblasts. Free Radic. Biol. Med. 2004, 36, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Iaccio, A.; Cattaneo, F.; Mauro, M.; Ammendola, R. FPRL1-mediated induction of superoxide in LL-37-stimulated IMR90 human fibroblast. Arch. Biochem. Biophys. 2009, 481, 94–100. [Google Scholar] [CrossRef]
- Cattaneo, F.; Castaldo, M.; Parisi, M.; Faraonio, R.; Esposito, G.; Ammendola, R. Formyl Peptide Receptor 1 Modulates Endothelial Cell Functions by NADPH Oxidase-Dependent VEGFR2 Transactivation. Oxid. Med. Cell Longev. 2018, 2018, 2609847. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, M.; Zollo, C.; Esposito, G.; Ammendola, R.; Cattaneo, F. NOX2-Dependent Reactive Oxygen Species Regulate Formyl-Peptide Receptor 1-Mediated TrkA Transactivation in SH-SY5Y Cells. Oxid. Med. Cell. Longev. 2019, 2019, 2051235. [Google Scholar] [CrossRef]
- Cattaneo, F.; Parisi, M.; Ammendola, R. WKYMVm-induced cross-talk between FPR2 and HGF receptor in human prostate epithelial cell line PNT1A. FEBS Lett. 2013, 587, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Iaccio, A.; Guerra, G.; Montagnani, S.; Ammendola, R. NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic. Biol. Med. 2011, 51, 1126–1136. [Google Scholar] [CrossRef] [PubMed]
- Cussell, P.J.G.; Gomez Escalada, M.; Milton, N.G.N.; Paterson, A.W.J. The N-formyl peptide receptors: Contemporary roles in neuronal function and dysfunction. Neural. Regen. Res. 2020, 15, 1191–1198. [Google Scholar] [CrossRef]
- Russo, R.; Cattaneo, F.; Lippiello, P.; Cristiano, C.; Zurlo, F.; Castaldo, M.; Irace, C.; Borsello, T.; Santamaria, R.; Ammendola, R.; et al. Motor coordination and synaptic plasticity deficits are associated with increased cerebellar activity of NADPH oxidase, CAMKII, and PKC at preplaque stage in the TgCRND8 mouse model of Alzheimer’s disease. Neurobiol. Aging 2018, 68, 123–133. [Google Scholar] [CrossRef]
- Mollica, A.; Stefanucci, A.; Costante, R.; Pinnen, F. Role of formyl peptide receptors (FPR) in abnormal inflammation responses involved in neurodegenerative diseases. Antiinflamm. Antiallergy Agents Med. Chem. 2012, 11, 20–36. [Google Scholar] [CrossRef]
- Li, Y.; Ye, D. Molecular biology for formyl peptide receptors in human diseases. J. Mol. Med. (Berl). 2013, 91, 781–789. [Google Scholar] [CrossRef]
- Ansari, J.; Kaur, G.; Gavins, F.N.E. Therapeutic Potential of Annexin A1 in Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2018, 19, 1211. [Google Scholar] [CrossRef] [PubMed]
- Snapkov, I.; Oqvist, C.O.; Figenschau, Y.; Kogner, P.; Johnsen, J.I.; Sveinbjornsson, B. The role of formyl peptide receptor 1 (FPR1) in neuroblastoma tumorigenesis. BMC Cancer 2016, 16, 490. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Yang, M.; Ding, Y.; Yu, L.; Chen, J. Formyl peptide receptor 2 expression predicts poor prognosis and promotes invasion and metastasis in epithelial ovarian cancer. Oncol. Rep. 2017, 38, 3297–3308. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, F.; Guerra, G.; Parisi, M.; Lucariello, A.; de Luca, A.; de Rosa, N.; Mazzarella, G.; Bianco, A.; Ammendola, R. Expression of Formyl-peptide Receptors in Human Lung Carcinoma. Anticancer Res. 2015, 35, 2769–2774. [Google Scholar]
- Belvedere, R.; Bizzarro, V.; Popolo, A.; Dal Piaz, F.; Vasaturo, M.; Picardi, P.; Parente, L.; Petrella, A. Role of intracellular and extracellular annexin A1 in migration and invasion of human pancreatic carcinoma cells. BMC Cancer 2014, 14, 961. [Google Scholar] [CrossRef]
- Vacchelli, E.; Enot, D.P.; Pietrocola, F.; Zitvogel, L.; Kroemer, G. Impact of Pattern Recognition Receptors on the Prognosis of Breast Cancer Patients Undergoing Adjuvant Chemotherapy. Cancer Res. 2016, 76, 3122–3126. [Google Scholar] [CrossRef]
- Vacchelli, E.; Ma, Y.; Baracco, E.E.; Sistigu, A.; Enot, D.P.; Pietrocola, F.; Yang, H.; Adjemian, S.; Chaba, K.; Semeraro, M.; et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science 2015, 350, 972–978. [Google Scholar] [CrossRef]
- Cattaneo, F.; Russo, R.; Castaldo, M.; Chambery, A.; Zollo, C.; Esposito, G.; Pedone, P.V.; Ammendola, R. Phosphoproteomic analysis sheds light on intracellular signaling cascades triggered by Formyl-Peptide Receptor 2. Sci. Rep. 2019, 9, 17894. [Google Scholar] [CrossRef]
- Mukai, H.; Ono, Y. A novel protein kinase with leucine zipper-like sequences: Its catalytic domain is highly homologous to that of protein kinase C. Biochem. Biophys. Res. Commun. 1994, 199, 897–904. [Google Scholar] [CrossRef]
- Palmer, R.H.; Ridden, J.; Parker, P.J. Cloning and expression patterns of two members of a novel protein-kinase-C-related kinase family. Eur. J. Biochem. 1995, 227, 344–351. [Google Scholar] [CrossRef]
- Taylor, S.S.; Radzio-Andzelm, E. Three protein kinase structures define a common motif. Structure 1994, 2, 345–355. [Google Scholar] [CrossRef]
- Thauerer, B.; Zur Nedden, S.; Baier-Bitterlich, G. Protein Kinase C-Related Kinase (PKN/PRK). Potential Key-Role for PKN1 in Protection of Hypoxic Neurons. Curr. Neuropharmacol. 2014, 12, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaga, C.; Mukai, H.; Toshimori, M.; Miyamoto, M.; Ono, Y. Mutational analysis of the regulatory mechanism of PKN: The regulatory region of PKN contains an arachidonic acid-sensitive autoinhibitory domain. J. Biochem. 1999, 126, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Lachmann, S.; Jevons, A.; de Rycker, M.; Casamassima, A.; Radtke, S.; Collazos, A.; Parker, P.J. Regulatory domain selectivity in the cell-type specific PKN-dependence of cell migration. PLoS ONE 2011, 6, e21732. [Google Scholar] [CrossRef]
- Wallace, S.W.; Magalhaes, A.; Hall, A. The Rho target PRK2 regulates apical junction formation in human bronchial epithelial cells. Mol. Cell. Biol. 2011, 31, 81–91. [Google Scholar] [CrossRef]
- Collazos, A.; Michael, N.; Whelan, R.D.; Kelly, G.; Mellor, H.; Pang, L.C.; Totty, N.; Parker, P.J. Site recognition and substrate screens for PKN family proteins. Biochem. J. 2011, 438, 535–543. [Google Scholar] [CrossRef]
- Bauer, A.F.; Sonzogni, S.; Meyer, L.; Zeuzem, S.; Piiper, A.; Biondi, R.M.; Neimanis, S. Regulation of protein kinase C-related protein kinase 2 (PRK2) by an intermolecular PRK2-PRK2 interaction mediated by Its N-terminal domain. J. Biol. Chem. 2012, 287, 20590–20602. [Google Scholar] [CrossRef]
- Unsal-Kacmaz, K.; Ragunathan, S.; Rosfjord, E.; Dann, S.; Upeslacis, E.; Grillo, M.; Hernandez, R.; Mack, F.; Klippel, A. The interaction of PKN3 with RhoC promotes malignant growth. Mol. Oncol. 2012, 6, 284–298. [Google Scholar] [CrossRef]
- Owen, D.; Lowe, P.N.; Nietlispach, D.; Brosnan, C.E.; Chirgadze, D.Y.; Parker, P.J.; Blundell, T.L.; Mott, H.R. Molecular dissection of the interaction between the small G proteins Rac1 and RhoA and protein kinase C-related kinase 1 (PRK1). J. Biol. Chem. 2003, 278, 50578–50587. [Google Scholar] [CrossRef]
- Mukai, H. The structure and function of PKN, a protein kinase having a catalytic domain homologous to that of PKC. J. Biochem. 2003, 133, 17–27. [Google Scholar] [CrossRef]
- Flynn, P.; Mellor, H.; Casamassima, A.; Parker, P.J. Rho GTPase control of protein kinase C-related protein kinase activation by 3-phosphoinositide-dependent protein kinase. J. Biol. Chem. 2000, 275, 11064–11070. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.G.; Tan, B.J.; Zhu, Y.; Zhou, S.; Armstrong, J.S.; Li, Q.T.; Dong, Q.; Chan, E.; Smith, D.; Verma, C.; et al. The very C-terminus of PRK1/PKN is essential for its activation by RhoA and downstream signaling. Cell Signal 2006, 18, 1473–1481. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.G.; Chen, X.; Liu, J.P.; Tan, B.J.; Zhou, S.; Smith, A.; Lees, N.; Hou, L.; Gu, F.; Yu, X.Y.; et al. The C-terminus of PRK2/PKNgamma is required for optimal activation by RhoA in a GTP-dependent manner. Arch. Biochem. Biophys. 2008, 479, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Parekh, D.B.; Ziegler, W.; Parker, P.J. Multiple pathways control protein kinase C phosphorylation. EMBO J. 2000, 19, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Newton, A.C. Protein kinase C: Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 2001, 101, 2353–2364. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Toker, A.; Newton, A.C. The carboxyl terminus of protein kinase c provides a switch to regulate its interaction with the phosphoinositide-dependent kinase, PDK-1. J. Biol. Chem. 2001, 276, 19588–19596. [Google Scholar] [CrossRef]
- Hauge, C.; Antal, T.L.; Hirschberg, D.; Doehn, U.; Thorup, K.; Idrissova, L.; Hansen, K.; Jensen, O.N.; Jorgensen, T.J.; Biondi, R.M.; et al. Mechanism for activation of the growth factor-activated AGC kinases by turn motif phosphorylation. EMBO J. 2007, 26, 2251–2261. [Google Scholar] [CrossRef]
- Yang, C.S.; Melhuish, T.A.; Spencer, A.; Ni, L.; Hao, Y.; Jividen, K.; Harris, T.E.; Snow, C.; Frierson, H.F., Jr.; Wotton, D.; et al. The protein kinase C super-family member PKN is regulated by mTOR and influences differentiation during prostate cancer progression. Prostate 2017, 77, 1452–1467. [Google Scholar] [CrossRef]
- Gan, X.; Wang, J.; Su, B.; Wu, D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 2011, 286, 10998–11002. [Google Scholar] [CrossRef]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef]
- Albert, V.; Svensson, K.; Shimobayashi, M.; Colombi, M.; Munoz, S.; Jimenez, V.; Handschin, C.; Bosch, F.; Hall, M.N. mTORC2 sustains thermogenesis via Akt-induced glucose uptake and glycolysis in brown adipose tissue. EMBO Mol. Med. 2016, 8, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Evans, B.A.; Sandstrom, A.L.; Chia, L.Y.; Mukaida, S.; Thai, B.S.; Nguyen, A.; Lim, L.; Tan, C.Y.R.; Baltos, J.A.; et al. alpha1A-Adrenoceptors activate mTOR signalling and glucose uptake in cardiomyocytes. Biochem. Pharmacol. 2018, 148, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Comer, F.I.; Sasaki, A.; McLeod, I.X.; Duong, Y.; Okumura, K.; Yates, J.R., III; Parent, C.A.; Firtel, R.A. TOR complex 2 integrates cell movement during chemotaxis and signal relay in Dictyostelium. Mol. Biol. Cell. 2005, 16, 4572–4583. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Das, S.; Losert, W.; Parent, C.A. mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev. Cell 2010, 19, 845–857. [Google Scholar] [CrossRef]
- Petri, B.; Sanz, M.J. Neutrophil chemotaxis. Cell Tissue Res. 2018, 371, 425–436. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, G.; Chen, X.; Xue, X.; Guo, Q.; Liu, M.; Zhao, J. Formyl peptide receptors promotes neural differentiation in mouse neural stem cells by ROS generation and regulation of PI3K-AKT signaling. Sci. Rep. 2017, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Faour, W.H.; Fayyad-Kazan, H.; el Zein, N. fMLP-dependent activation of Akt and ERK1/2 through ROS/Rho A pathways is mediated through restricted activation of the FPRL1 (FPR2) receptor. Inflamm. Res. 2018, 67, 711–722. [Google Scholar] [CrossRef]
- Vlahos, C.J.; Matter, W.F.; Brown, R.F.; Traynor-Kaplan, A.E.; Heyworth, P.G.; Prossnitz, E.R.; Ye, R.D.; Marder, P.; Schelm, J.A.; Rothfuss, K.J.; et al. Investigation of neutrophil signal transduction using a specific inhibitor of phosphatidylinositol 3-kinase. J. Immunol. 1995, 154, 2413–2422. [Google Scholar]
- Quilliam, L.A.; Lambert, Q.T.; Mickelson-Young, L.A.; Westwick, J.K.; Sparks, A.B.; Kay, B.K.; Jenkins, N.A.; Gilbert, D.J.; Copeland, N.G.; Der, C.J. Isolation of a NCK-associated kinase, PRK2, an SH3-binding protein and potential effector of Rho protein signaling. J. Biol. Chem. 1996, 271, 28772–28776. [Google Scholar] [CrossRef]
- Li, W.; Hu, P.; Skolnik, E.Y.; Ullrich, A.; Schlessinger, J. The SH2 and SH3 domain-containing Nck protein is oncogenic and a common target for phosphorylation by different surface receptors. Mol. Cell. Biol. 1992, 12, 5824–5833. [Google Scholar] [CrossRef][Green Version]
- Guo, D.; Jia, Q.; Song, H.Y.; Warren, R.S.; Donner, D.B. Vascular endothelial cell growth factor promotes tyrosine phosphorylation of mediators of signal transduction that contain SH2 domains. Association with endothelial cell proliferation. J. Biol. Chem. 1995, 270, 6729–6733. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Zhang, S.; Wang, J.; Xia, F.; Wan, J.B.; Lu, J.; Ye, R.D. Dual modulation of formyl peptide receptor 2 by aspirin-triggered lipoxin contributes to its anti-inflammatory activity. FASEB J. 2020, 34, 6920–6933. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Yacoubian, S.; Lee, C.H.; Yang, R.; Petasis, N.A.; Serhan, C.N. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc. Natl. Acad. Sci. USA 2010, 107, 1660–1665. [Google Scholar] [CrossRef] [PubMed]
- Huet, E.; Boulay, F.; Barral, S.; Rabiet, M.J. The role of beta-arrestins in the formyl peptide receptor-like 1 internalization and signaling. Cell. Signal 2007, 19, 1939–1948. [Google Scholar] [CrossRef]
- Jean-Charles, P.Y.; Kaur, S.; Shenoy, S.K. G Protein-Coupled Receptor Signaling Through beta-Arrestin-Dependent Mechanisms. J. Cardiovasc. Pharmacol. 2017, 70, 142–158. [Google Scholar] [CrossRef]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef]
- Daub, H.; Olsen, J.V.; Bairlein, M.; Gnad, F.; Oppermann, F.S.; Korner, R.; Greff, Z.; Keri, G.; Stemmann, O.; Mann, M. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol. Cell 2008, 31, 438–448. [Google Scholar] [CrossRef]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal 2010, 3, ra3. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Di Palma, S.; Preisinger, C.; Peng, M.; Polat, A.N.; Heck, A.J.; Mohammed, S. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J. Proteome Res. 2013, 12, 260–271. [Google Scholar] [CrossRef]
- Vincent, S.; Settleman, J. The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Mol. Cell. Biol. 1997, 17, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Calautti, E.; Grossi, M.; Mammucari, C.; Aoyama, Y.; Pirro, M.; Ono, Y.; Li, J.; Dotto, G.P. Fyn tyrosine kinase is a downstream mediator of Rho/PRK2 function in keratinocyte cell-cell adhesion. J. Cell. Biol. 2002, 156, 137–148. [Google Scholar] [CrossRef]
- Cryns, V.L.; Byun, Y.; Rana, A.; Mellor, H.; Lustig, K.D.; Ghanem, L.; Parker, P.J.; Kirschner, M.W.; Yuan, J. Specific proteolysis of the kinase protein kinase C-related kinase 2 by caspase-3 during apoptosis. Identification by a novel, small pool expression cloning strategy. J. Biol. Chem. 1997, 272, 29449–29453. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.; Lee, K.H.; Kim, D.; Kim, S.; Kim, J.W.; Chung, J. Inhibition of Akt and its anti-apoptotic activities by tumor necrosis factor-induced protein kinase C-related kinase 2 (PRK2) cleavage. J. Biol. Chem. 2000, 275, 34451–34458. [Google Scholar] [CrossRef] [PubMed]
- Ammer, A.G.; Weed, S.A. Cortactin branches out: Roles in regulating protrusive actin dynamics. Cell Motil. Cytoskeleton 2008, 65, 687–707. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Zama, T.; Wada, K.; Onogi, H.; Hagiwara, M. Cloning of human PRP4 reveals interaction with Clk1. J. Biol Chem. 2001, 276, 32247–32256. [Google Scholar] [CrossRef]
- Gross, T.; Lutzelberger, M.; Weigmann, H.; Klingenhoff, A.; Shenoy, S.; Kaufer, N.F. Functional analysis of the fission yeast Prp4 protein kinase involved in pre-mRNA splicing and isolation of a putative mammalian homologue. Nucleic Acids Res. 1997, 25, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Hanks, S.K.; Hunter, T. Protein kinases 6. The eukaryotic protein kinase superfamily: Kinase (catalytic) domain structure and classification. FASEB J. 1995, 9, 576–596. [Google Scholar] [CrossRef]
- Davis, R.J. Transcriptional regulation by MAP kinases. Mol. Reprod Dev. 1995, 42, 459–467. [Google Scholar] [CrossRef]
- Huang, Y.; Deng, T.; Winston, B.W. Characterization of hPRP4 kinase activation: Potential role in signaling. Biochem Biophys Res. Commun 2000, 271, 456–463. [Google Scholar] [CrossRef]
- Miyata, Y.; Nishida, E. Distantly related cousins of MAP kinase: Biochemical properties and possible physiological functions. Biochem Biophys Res. Commun 1999, 266, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.Y.; Wang, P.; Han, J.; Rosenfeld, M.G.; Fu, X.D. SR proteins in vertical integration of gene expression from transcription to RNA processing to translation. Mol. Cell 2009, 35, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Hsiao, H.H.; Will, C.L.; Giet, R.; Urlaub, H.; Luhrmann, R. Human PRP4 kinase is required for stable tri-snRNP association during spliceosomal B complex formation. Nat. Struct. Mol. Biol. 2010, 17, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Shehzad, A.; Lee, J.; Huh, T.L.; Lee, Y.S. Curcumin induces apoptosis in human colorectal carcinoma (HCT-15) cells by regulating expression of Prp4 and p53. Mol. Cells 2013, 35, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Rigbolt, K.T.; Prokhorova, T.A.; Akimov, V.; Henningsen, J.; Johansen, P.T.; Kratchmarova, I.; Kassem, M.; Mann, M.; Olsen, J.V.; Blagoev, B. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal 2011, 4, rs3. [Google Scholar] [CrossRef]
- Johnson, H.; Lescarbeau, R.S.; Gutierrez, J.A.; White, F.M. Phosphotyrosine profiling of NSCLC cells in response to EGF and HGF reveals network specific mediators of invasion. J. Proteome Res. 2013, 12, 1856–1867. [Google Scholar] [CrossRef][Green Version]
- Corkery, D.P.; Le Page, C.; Meunier, L.; Provencher, D.; Mes-Masson, A.M.; Dellaire, G. PRP4K is a HER2-regulated modifier of taxane sensitivity. Cell Cycle 2015, 14, 1059–1069. [Google Scholar] [CrossRef]
- Drewes, G.; Ebneth, A.; Preuss, U.; Mandelkow, E.M.; Mandelkow, E. MARK, a novel family of protein kinases that phosphorylate microtubule-associated proteins and trigger microtubule disruption. Cell 1997, 89, 297–308. [Google Scholar] [CrossRef]
- Schwalbe, M.; Biernat, J.; Bibow, S.; Ozenne, V.; Jensen, M.R.; Kadavath, H.; Blackledge, M.; Mandelkow, E.; Zweckstetter, M. Phosphorylation of human Tau protein by microtubule affinity-regulating kinase 2. Biochemistry 2013, 52, 9068–9079. [Google Scholar] [CrossRef] [PubMed]
- Kemphues, K. PARsing embryonic polarity. Cell 2000, 101, 345–348. [Google Scholar] [CrossRef]
- Hurov, J.; Piwnica-Worms, H. The Par-1/MARK family of protein kinases: From polarity to metabolism. Cell Cycle 2007, 6, 1966–1969. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Funahashi, Y.; Nakamuta, S.; Xu, C.; Takano, T.; Kaibuchi, K. Extracellular and Intracellular Signaling for Neuronal Polarity. Physiol Rev. 2015, 95, 995–1024. [Google Scholar] [CrossRef] [PubMed]
- Krummel, M.F.; Macara, I. Maintenance and modulation of T cell polarity. Nat. Immunol 2006, 7, 1143–1149. [Google Scholar] [CrossRef]
- Monteverde, T.; Muthalagu, N.; Port, J.; Murphy, D.J. Evidence of cancer-promoting roles for AMPK and related kinases. FEBS J. 2015, 282, 4658–4671. [Google Scholar] [CrossRef]
- Marx, A.; Nugoor, C.; Panneerselvam, S.; Mandelkow, E. Structure and function of polarity-inducing kinase family MARK/Par-1 within the branch of AMPK/Snf1-related kinases. FASEB J. 2010, 24, 1637–1648. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Hennemann, H.; Xing, P.X.; Hoffmann, I.; Moroy, T. The oncogenic serine/threonine kinase Pim-1 phosphorylates and inhibits the activity of Cdc25C-associated kinase 1 (C-TAK1): A novel role for Pim-1 at the G2/M cell cycle checkpoint. J. Biol. Chem. 2004, 279, 48319–48328. [Google Scholar] [CrossRef] [PubMed]
- Long, Y.C.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Investig. 2006, 116, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Hurov, J.B.; Huang, M.; White, L.S.; Lennerz, J.; Choi, C.S.; Cho, Y.R.; Kim, H.J.; Prior, J.L.; Piwnica-Worms, D.; Cantley, L.C.; et al. Loss of the Par-1b/MARK2 polarity kinase leads to increased metabolic rate, decreased adiposity, and insulin hypersensitivity in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 5680–5685. [Google Scholar] [CrossRef] [PubMed]
- Lennerz, J.K.; Hurov, J.B.; White, L.S.; Lewandowski, K.T.; Prior, J.L.; Planer, G.J.; Gereau, R.W.t.; Piwnica-Worms, D.; Schmidt, R.E.; Piwnica-Worms, H. Loss of Par-1a/MARK3/C-TAK1 kinase leads to reduced adiposity, resistance to hepatic steatosis, and defective gluconeogenesis. Mol. Cell Biol. 2010, 30, 5043–5056. [Google Scholar] [CrossRef] [PubMed]
- Bessone, S.; Vidal, F.; Le Bouc, Y.; Epelbaum, J.; Bluet-Pajot, M.T.; Darmon, M. EMK protein kinase-null mice: Dwarfism and hypofertility associated with alterations in the somatotrope and prolactin pathways. Dev. Biol. 1999, 214, 87–101. [Google Scholar] [CrossRef]
- Hurov, J.B.; Stappenbeck, T.S.; Zmasek, C.M.; White, L.S.; Ranganath, S.H.; Russell, J.H.; Chan, A.C.; Murphy, K.M.; Piwnica-Worms, H. Immune system dysfunction and autoimmune disease in mice lacking Emk (Par-1) protein kinase. Mol. Cell Biol. 2001, 21, 3206–3219. [Google Scholar] [CrossRef] [PubMed]
- Segu, L.; Pascaud, A.; Costet, P.; Darmon, M.; Buhot, M.C. Impairment of spatial learning and memory in ELKL Motif Kinase1 (EMK1/MARK2) knockout mice. Neurobiol. Aging 2008, 29, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Dequiedt, F.; Martin, M.; Von Blume, J.; Vertommen, D.; Lecomte, E.; Mari, N.; Heinen, M.F.; Bachmann, M.; Twizere, J.C.; Huang, M.C.; et al. New role for hPar-1 kinases EMK and C-TAK1 in regulating localization and activity of class IIa histone deacetylases. Mol. Cell Biol. 2006, 26, 7086–7102. [Google Scholar] [CrossRef] [PubMed]
- Muller, J.; Ritt, D.A.; Copeland, T.D.; Morrison, D.K. Functional analysis of C-TAK1 substrate binding and identification of PKP2 as a new C-TAK1 substrate. EMBO J. 2003, 22, 4431–4442. [Google Scholar] [CrossRef]
- Timm, T.; Li, X.Y.; Biernat, J.; Jiao, J.; Mandelkow, E.; Vandekerckhove, J.; Mandelkow, E.M. MARKK, a Ste20-like kinase, activates the polarity-inducing kinase MARK/PAR-1. EMBO J. 2003, 22, 5090–5101. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Goransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Makela, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef]
- Timm, T.; Balusamy, K.; Li, X.; Biernat, J.; Mandelkow, E.; Mandelkow, E.M. Glycogen synthase kinase (GSK) 3beta directly phosphorylates Serine 212 in the regulatory loop and inhibits microtubule affinity-regulating kinase (MARK) 2. J. Biol. Chem. 2008, 283, 18873–18882. [Google Scholar] [CrossRef]
- Uboha, N.V.; Flajolet, M.; Nairn, A.C.; Picciotto, M.R. A calcium- and calmodulin-dependent kinase Ialpha/microtubule affinity regulating kinase 2 signaling cascade mediates calcium-dependent neurite outgrowth. J. Neurosci 2007, 27, 4413–4423. [Google Scholar] [CrossRef]
- Hurov, J.B.; Watkins, J.L.; Piwnica-Worms, H. Atypical PKC phosphorylates PAR-1 kinases to regulate localization and activity. Curr. Biol. 2004, 14, 736–741. [Google Scholar] [CrossRef]
- Suzuki, A.; Hirata, M.; Kamimura, K.; Maniwa, R.; Yamanaka, T.; Mizuno, K.; Kishikawa, M.; Hirose, H.; Amano, Y.; Izumi, N.; et al. aPKC acts upstream of PAR-1b in both the establishment and maintenance of mammalian epithelial polarity. Curr. Biol. 2004, 14, 1425–1435. [Google Scholar] [CrossRef]
- Watkins, J.L.; Lewandowski, K.T.; Meek, S.E.; Storz, P.; Toker, A.; Piwnica-Worms, H. Phosphorylation of the Par-1 polarity kinase by protein kinase D regulates 14-3-3 binding and membrane association. Proc. Natl. Acad. Sci. USA 2008, 105, 18378–18383. [Google Scholar] [CrossRef]
- Matenia, D.; Griesshaber, B.; Li, X.Y.; Thiessen, A.; Johne, C.; Jiao, J.; Mandelkow, E.; Mandelkow, E.M. PAK5 kinase is an inhibitor of MARK/Par-1, which leads to stable microtubules and dynamic actin. Mol. Biol. Cell 2005, 16, 4410–4422. [Google Scholar] [CrossRef] [PubMed]
- Dan, I.; Watanabe, N.M.; Kusumi, A. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 2001, 11, 220–230. [Google Scholar] [CrossRef]
- Zheng, B.; Jeong, J.H.; Asara, J.M.; Yuan, Y.Y.; Granter, S.R.; Chin, L.; Cantley, L.C. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol. Cell 2009, 33, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Oppermann, F.S.; Gnad, F.; Olsen, J.V.; Hornberger, R.; Greff, Z.; Keri, G.; Mann, M.; Daub, H. Large-scale proteomics analysis of the human kinome. Mol. Cell Proteomics 2009, 8, 1751–1764. [Google Scholar] [CrossRef] [PubMed]
- Rane, C.K.; Minden, A. P21 activated kinases: Structure, regulation, and functions. Small GTPases 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.M.; Jones, G.E. The emerging importance of group II PAKs. Biochem. J. 2010, 425, 465–473. [Google Scholar] [CrossRef]
- Arias-Romero, L.E.; Chernoff, J. A tale of two Paks. Biol. Cell 2008, 100, 97–108. [Google Scholar] [CrossRef]
- Callow, M.G.; Zozulya, S.; Gishizky, M.L.; Jallal, B.; Smeal, T. PAK4 mediates morphological changes through the regulation of GEF-H1. J. Cell Sci. 2005, 118, 1861–1872. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Z.; Viklund, E.K.; Stromblad, S. P21-activated kinase 4 interacts with integrin alpha v beta 5 and regulates alpha v beta 5-mediated cell migration. J. Cell Biol. 2002, 158, 1287–1297. [Google Scholar] [CrossRef]
- Kumar, R.; Sanawar, R.; Li, X.; Li, F. Structure, biochemistry, and biology of PAK kinases. Gene 2017, 605, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Abo, A.; Qu, J.; Cammarano, M.S.; Dan, C.; Fritsch, A.; Baud, V.; Belisle, B.; Minden, A. PAK4, a novel effector for Cdc42Hs, is implicated in the reorganization of the actin cytoskeleton and in the formation of filopodia. EMBO J. 1998, 17, 6527–6540. [Google Scholar] [CrossRef]
- Dan, C.; Nath, N.; Liberto, M.; Minden, A. PAK5, a new brain-specific kinase, promotes neurite outgrowth in N1E-115 cells. Mol. Cell Biol. 2002, 22, 567–577. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Shea, K.; Masters, J.R.; Jones, G.E.; Wells, C.M. A PAK4-LIMK1 pathway drives prostate cancer cell migration downstream of HGF. Cell Signal 2008, 20, 1320–1328. [Google Scholar] [CrossRef] [PubMed]
- Wells, C.M.; Abo, A.; Ridley, A.J. PAK4 is activated via PI3K in HGF-stimulated epithelial cells. J. Cell Sci. 2002, 115, 3947–3956. [Google Scholar] [CrossRef] [PubMed]
- King, H.; Thillai, K.; Whale, A.; Arumugam, P.; Eldaly, H.; Kocher, H.M.; Wells, C.M. PAK4 interacts with p85 alpha: Implications for pancreatic cancer cell migration. Sci. Rep. 2017, 7, 42575. [Google Scholar] [CrossRef] [PubMed]
- Paliouras, G.N.; Naujokas, M.A.; Park, M. Pak4, a novel Gab1 binding partner, modulates cell migration and invasion by the Met receptor. Mol. Cell. Biol. 2009, 29, 3018–3032. [Google Scholar] [CrossRef]
- Pandey, A.; Dan, I.; Kristiansen, T.Z.; Watanabe, N.M.; Voldby, J.; Kajikawa, E.; Khosravi-Far, R.; Blagoev, B.; Mann, M. Cloning and characterization of PAK5, a novel member of mammalian p21-activated kinase-II subfamily that is predominantly expressed in brain. Oncogene 2002, 21, 3939–3948. [Google Scholar] [CrossRef][Green Version]
- Thillai, K.; Lam, H.; Sarker, D.; Wells, C.M. Deciphering the link between PI3K and PAK: An opportunity to target key pathways in pancreatic cancer? Oncotarget 2017, 8, 14173–14191. [Google Scholar] [CrossRef]
- Hannigan, M.; Zhan, L.; Li, Z.; Ai, Y.; Wu, D.; Huang, C.K. Neutrophils lacking phosphoinositide 3-kinase gamma show loss of directionality during N-formyl-Met-Leu-Phe-induced chemotaxis. Proc. Natl. Acad. Sci. USA 2002, 99, 3603–3608. [Google Scholar] [CrossRef]
- Merlot, S.; Firtel, R.A. Leading the way: Directional sensing through phosphatidylinositol 3-kinase and other signaling pathways. J. Cell Sci. 2003, 116, 3471–3478. [Google Scholar] [CrossRef] [PubMed]
- Babbin, B.A.; Jesaitis, A.J.; Ivanov, A.I.; Kelly, D.; Laukoetter, M.; Nava, P.; Parkos, C.A.; Nusrat, A. Formyl peptide receptor-1 activation enhances intestinal epithelial cell restitution through phosphatidylinositol 3-kinase-dependent activation of Rac1 and Cdc42. J. Immunol. 2007, 179, 8112–8121. [Google Scholar] [CrossRef] [PubMed]
- Pavone, L.M.; Cattaneo, F.; Rea, S.; De Pasquale, V.; Spina, A.; Sauchelli, E.; Mastellone, V.; Ammendola, R. Intracellular signaling cascades triggered by the NK1 fragment of hepatocyte growth factor in human prostate epithelial cell line PNT1A. Cell Signal 2011, 23, 1961–1971. [Google Scholar] [CrossRef] [PubMed]
- Tinti, M.; Madeira, F.; Murugesan, G.; Hoxhaj, G.; Toth, R.; Mackintosh, C. ANIA: ANnotation and Integrated Analysis of the 14-3-3 interactome. Database (Oxford) 2014, 2014, bat085. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.G.; Ning, K.; Li, F. Group II p21-activated kinases as therapeutic targets in gastrointestinal cancer. World J. Gastroenterol 2016, 22, 1224–1235. [Google Scholar] [CrossRef]
- Dan, C.; Kelly, A.; Bernard, O.; Minden, A. Cytoskeletal changes regulated by the PAK4 serine/threonine kinase are mediated by LIM kinase 1 and cofilin. J. Biol. Chem. 2001, 276, 32115–32121. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H.; Lundin, L.; Thullberg, M.; Liu, Y.; Wang, Y.; Claesson-Welsh, L.; Stromblad, S. p21-activated kinase 4 phosphorylation of integrin beta5 Ser-759 and Ser-762 regulates cell migration. J. Biol. Chem. 2010, 285, 23699–23710. [Google Scholar] [CrossRef]
- Wong, L.E.; Reynolds, A.B.; Dissanayaka, N.T.; Minden, A. p120-catenin is a binding partner and substrate for Group B Pak kinases. J. Cell. Biochem. 2010, 110, 1244–1254. [Google Scholar] [CrossRef]
- Guo, Q.; Su, N.; Zhang, J.; Li, X.; Miao, Z.; Wang, G.; Cheng, M.; Xu, H.; Cao, L.; Li, F. PAK4 kinase-mediated SCG10 phosphorylation involved in gastric cancer metastasis. Oncogene 2014, 33, 3277–3287. [Google Scholar] [CrossRef]
- Li, Y.; Shao, Y.; Tong, Y.; Shen, T.; Zhang, J.; Li, Y.; Gu, H.; Li, F. Nucleo-cytoplasmic shuttling of PAK4 modulates beta-catenin intracellular translocation and signaling. Biochim. Biophys. Acta 2012, 1823, 465–475. [Google Scholar] [CrossRef]
- Wang, C.; Li, Y.; Zhang, H.; Liu, F.; Cheng, Z.; Wang, D.; Wang, G.; Xu, H.; Zhao, Y.; Cao, L.; et al. Oncogenic PAK4 regulates Smad2/3 axis involving gastric tumorigenesis. Oncogene 2014, 33, 3473–3484. [Google Scholar] [CrossRef] [PubMed]
- Mayya, V.; Lundgren, D.H.; Hwang, S.I.; Rezaul, K.; Wu, L.; Eng, J.K.; Rodionov, V.; Han, D.K. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci. Signal 2009, 2, ra46. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Song, C.; Cheng, K.; Dong, M.; Wang, F.; Huang, J.; Sun, D.; Wang, L.; Ye, M.; Zou, H. An enzyme assisted RP-RPLC approach for in-depth analysis of human liver phosphoproteome. J. Proteomics 2014, 96, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Kuramochi, S.; Moriguchi, T.; Kuida, K.; Endo, J.; Semba, K.; Nishida, E.; Karasuyama, H. LOK is a novel mouse STE20-like protein kinase that is expressed predominantly in lymphocytes. J. Biol. Chem. 1997, 272, 22679–22684. [Google Scholar] [CrossRef] [PubMed]
- Leberer, E.; Dignard, D.; Harcus, D.; Thomas, D.Y.; Whiteway, M. The protein kinase homologue Ste20p is required to link the yeast pheromone response G-protein beta gamma subunits to downstream signalling components. EMBO J. 1992, 11, 4815–4824. [Google Scholar] [CrossRef] [PubMed]
- Herskowitz, I. MAP kinase pathways in yeast: For mating and more. Cell 1995, 80, 187–197. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays 1996, 18, 567–577. [Google Scholar] [CrossRef]
- Sells, M.A.; Chernoff, J. Emerging from the Pak: The p21-activated protein kinase family. Trends Cell Biol. 1997, 7, 162–167. [Google Scholar] [CrossRef]
- Kuramochi, S.; Matsuda, Y.; Okamoto, M.; Kitamura, F.; Yonekawa, H.; Karasuyama, H. Molecular cloning of the human gene STK10 encoding lymphocyte-oriented kinase, and comparative chromosomal mapping of the human, mouse, and rat homologues. Immunogenetics 1999, 49, 369–375. [Google Scholar] [CrossRef]
- Walter, S.A.; Cutler, R.E., Jr.; Martinez, R.; Gishizky, M.; Hill, R.J. Stk10, a new member of the polo-like kinase kinase family highly expressed in hematopoietic tissue. J. Biol. Chem. 2003, 278, 18221–18228. [Google Scholar] [CrossRef]
- Viswanatha, R.; Ohouo, P.Y.; Smolka, M.B.; Bretscher, A. Local phosphocycling mediated by LOK/SLK restricts ezrin function to the apical aspect of epithelial cells. J. Cell. Biol. 2012, 199, 969–984. [Google Scholar] [CrossRef]
- Arora, S.; Gonzales, I.M.; Hagelstrom, R.T.; Beaudry, C.; Choudhary, A.; Sima, C.; Tibes, R.; Mousses, S.; Azorsa, D.O. RNAi phenotype profiling of kinases identifies potential therapeutic targets in Ewing’s sarcoma. Mol. Cancer 2010, 9, 218. [Google Scholar] [CrossRef] [PubMed]
- Bignell, G.; Smith, R.; Hunter, C.; Stephens, P.; Davies, H.; Greenman, C.; Teague, J.; Butler, A.; Edkins, S.; Stevens, C.; et al. Sequence analysis of the protein kinase gene family in human testicular germ-cell tumors of adolescents and adults. Genes Chromosomes Cancer 2006, 45, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, K.; Yamashita, Y.; Kawazu, M.; Sai, E.; Fujiwara, S.; Nakamura, N.; Takeuchi, K.; Ando, M.; Miyazono, K.; Ueno, T.; et al. STK10 missense mutations associated with anti-apoptotic function. Oncol. Rep. 2013, 30, 1542–1548. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, Y.; Wang, L.; Yao, B.; Chen, T.; Li, Q.; Liu, Z.; Liu, R.; Niu, Y.; Song, T.; et al. Resolvin D1 prevents epithelial-mesenchymal transition and reduces the stemness features of hepatocellular carcinoma by inhibiting paracrine of cancer-associated fibroblast-derived COMP. J. Exp. Clin. Cancer Res. 2019, 38, 170. [Google Scholar] [CrossRef]
- Lu, J.; Zhao, J.; Jia, C.; Zhou, L.; Cai, Y.; Ni, J.; Ma, J.; Zheng, M.; Lu, A. FPR2 enhances colorectal cancer progression by promoting EMT process. Neoplasma 2019, 66, 785–791. [Google Scholar] [CrossRef]
- Bozinovski, S.; Vlahos, R.; Anthony, D.; McQualter, J.; Anderson, G.; Irving, L.; Steinfort, D. COPD and squamous cell lung cancer: Aberrant inflammation and immunity is the common link. Br. J. Pharmacol. 2016, 173, 635–648. [Google Scholar] [CrossRef]
- Cussell, P.J.G.; Howe, M.S.; Illingworth, T.A.; Gomez Escalada, M.; Milton, N.G.N.; Paterson, A.W.J. The formyl peptide receptor agonist FPRa14 induces differentiation of Neuro2a mouse neuroblastoma cells into multiple distinct morphologies which can be specifically inhibited with FPR antagonists and FPR knockdown using siRNA. PLoS ONE 2019, 14, e0217815. [Google Scholar] [CrossRef]
- Xiang, Y.; Yao, X.; Chen, K.; Wang, X.; Zhou, J.; Gong, W.; Yoshimura, T.; Huang, J.; Wang, R.; Wu, Y.; et al. The G-protein coupled chemoattractant receptor FPR2 promotes malignant phenotype of human colon cancer cells. Am. J. Cancer Res. 2016, 6, 2599–2610. [Google Scholar]
- Leroy, C.; Belkina, N.V.; Long, T.; Deruy, E.; Dissous, C.; Shaw, S.; Tulasne, D. Caspase Cleavages of the Lymphocyte-oriented Kinase Prevent Ezrin, Radixin, and Moesin Phosphorylation during Apoptosis. J. Biol. Chem. 2016, 291, 10148–10161. [Google Scholar] [CrossRef]
- Pelaseyed, T.; Viswanatha, R.; Sauvanet, C.; Filter, J.J.; Goldberg, M.L.; Bretscher, A. Ezrin activation by LOK phosphorylation involves a PIP2-dependent wedge mechanism. Elife 2017, 6. [Google Scholar] [CrossRef]
- Niggli, V.; Andreoli, C.; Roy, C.; Mangeat, P. Identification of a phosphatidylinositol-4,5-bisphosphate-binding domain in the N-terminal region of ezrin. FEBS Lett. 1995, 376, 172–176. [Google Scholar] [CrossRef]
- Pelaseyed, T.; Bretscher, A. Regulation of actin-based apical structures on epithelial cells. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [PubMed]
- Pike, A.C.; Rellos, P.; Niesen, F.H.; Turnbull, A.; Oliver, A.W.; Parker, S.A.; Turk, B.E.; Pearl, L.H.; Knapp, S. Activation segment dimerization: A mechanism for kinase autophosphorylation of non-consensus sites. EMBO J. 2008, 27, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Carrascal, M.; Ovelleiro, D.; Casas, V.; Gay, M.; Abian, J. Phosphorylation analysis of primary human T lymphocytes using sequential IMAC and titanium oxide enrichment. J. Proteome Res. 2008, 7, 5167–5176. [Google Scholar] [CrossRef]
- Zahedi, R.P.; Lewandrowski, U.; Wiesner, J.; Wortelkamp, S.; Moebius, J.; Schutz, C.; Walter, U.; Gambaryan, S.; Sickmann, A. Phosphoproteome of resting human platelets. J. Proteome Res. 2008, 7, 526–534. [Google Scholar] [CrossRef]
- Beck, F.; Geiger, J.; Gambaryan, S.; Solari, F.A.; Dell’Aica, M.; Loroch, S.; Mattheij, N.J.; Mindukshev, I.; Potz, O.; Jurk, K.; et al. Temporal quantitative phosphoproteomics of ADP stimulation reveals novel central nodes in platelet activation and inhibition. Blood 2017, 129, e1–e12. [Google Scholar] [CrossRef]
- Brott, B.K.; Alessandrini, A.; Largaespada, D.A.; Copeland, N.G.; Jenkins, N.A.; Crews, C.M.; Erikson, R.L. MEK2 is a kinase related to MEK1 and is differentially expressed in murine tissues. Cell Growth Differ. 1993, 4, 921–929. [Google Scholar]
- Srinivas, N.R. Pharmacology of Pimasertib, A Selective MEK1/2 Inhibitor. Eur. J. Drug Metab. Pharmacokinet. 2018, 43, 373–382. [Google Scholar] [CrossRef]
- Zhao, Y.; Adjei, A.A. The clinical development of MEK inhibitors. Nat. Rev. Clin. Oncol. 2014, 11, 385–400. [Google Scholar] [CrossRef]
- Robinson, M.J.; Cobb, M.H. Mitogen-activated protein kinase pathways. Curr. Opin Cell. Biol. 1997, 9, 180–186. [Google Scholar] [CrossRef]
- Coffelt, S.B.; Tomchuck, S.L.; Zwezdaryk, K.J.; Danka, E.S.; Scandurro, A.B. Leucine leucine-37 uses formyl peptide receptor-like 1 to activate signal transduction pathways, stimulate oncogenic gene expression, and enhance the invasiveness of ovarian cancer cells. Mol. Cancer. Res. 2009, 7, 907–915. [Google Scholar] [CrossRef]
- Kam, A.Y.; Liu, A.M.; Wong, Y.H. Formyl peptide-receptor like-1 requires lipid raft and extracellular signal-regulated protein kinase to activate inhibitor-kappa B kinase in human U87 astrocytoma cells. J. Neurochem. 2007, 103, 1553–1566. [Google Scholar] [CrossRef] [PubMed]
- Kam, A.Y.; Tse, T.T.; Kwan, D.H.; Wong, Y.H. Formyl peptide receptor like 1 differentially requires mitogen-activated protein kinases for the induction of glial fibrillary acidic protein and interleukin-1alpha in human U87 astrocytoma cells. Cell Signal 2007, 19, 2106–2117. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.L.; Ji, C.D.; Tang, J.; Wang, Y.X.; Xiang, D.F.; Li, H.Q.; Liu, W.W.; Wang, J.X.; Yan, H.Z.; Wang, Y.; et al. FPR2 promotes invasion and metastasis of gastric cancer cells and predicts the prognosis of patients. Sci. Rep. 2017, 7, 3153. [Google Scholar] [CrossRef] [PubMed]
- Fischmann, T.O.; Smith, C.K.; Mayhood, T.W.; Myers, J.E.; Reichert, P.; Mannarino, A.; Carr, D.; Zhu, H.; Wong, J.; Yang, R.S.; et al. Crystal structures of MEK1 binary and ternary complexes with nucleotides and inhibitors. Biochem. 2009, 48, 2661–2674. [Google Scholar] [CrossRef]
- Liang, H.; Liu, T.; Chen, F.; Liu, Z.; Liu, S. A full-length 3D structure for MAPK/ERK kinase 2 (MEK2). Sci Chin. Life. Sci. 2011, 54, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Ohren, J.F.; Chen, H.; Pavlovsky, A.; Whitehead, C.; Zhang, E.; Kuffa, P.; Yan, C.; McConnell, P.; Spessard, C.; Banotai, C.; et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat. Struct. Mol. Biol. 2004, 11, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Liang, R.Y.; Liu, B.H.; Huang, C.J.; Lin, K.T.; Ko, C.C.; Huang, L.L.; Hsu, B.; Wu, C.Y.; Chuang, S.M. MEK2 is a critical modulating mechanism to down-regulate GCIP stability and function in cancer cells. FASEB J. 2020, 34, 1958–1969. [Google Scholar] [CrossRef]
- Downey, G.P.; Butler, J.R.; Brumell, J.; Borregaard, N.; Kjeldsen, L.; Sue, A.Q.A.K.; Grinstein, S. Chemotactic peptide-induced activation of MEK-2, the predominant isoform in human neutrophils. Inhibition by wortmannin. J. Biol. Chem. 1996, 271, 21005–21011. [Google Scholar] [CrossRef] [PubMed]
- Krump, E.; Sanghera, J.S.; Pelech, S.L.; Furuya, W.; Grinstein, S. Chemotactic peptide N-formyl-met-leu-phe activation of p38 mitogen-activated protein kinase (MAPK) and MAPK-activated protein kinase-2 in human neutrophils. J. Biol Chem 1997, 272, 937–944. [Google Scholar] [CrossRef]
- Clark, R.A.; Volpp, B.D.; Leidal, K.G.; Nauseef, W.M. Two cytosolic components of the human neutrophil respiratory burst oxidase translocate to the plasma membrane during cell activation. J. Clin. Investig. 1990, 85, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Dang, P.M.; Cross, A.R.; Quinn, M.T.; Babior, B.M. Assembly of the neutrophil respiratory burst oxidase: A direct interaction between p67PHOX and cytochrome b558 II. Proc. Natl. Acad. Sci. USA 2002, 99, 4262–4265. [Google Scholar] [CrossRef]
- Iaccio, A.; Collinet, C.; Gesualdi, N.M.; Ammendola, R. Protein kinase C-alpha and -delta are required for NADPH oxidase activation in WKYMVm-stimulated IMR90 human fibroblasts. Arch. Biochem. Biophys. 2007, 459, 288–294. [Google Scholar] [CrossRef]
- Benna, J.E.; Dang, P.M.; Gaudry, M.; Fay, M.; Morel, F.; Hakim, J.; Gougerot-Pocidalo, M.A. Phosphorylation of the respiratory burst oxidase subunit p67(phox) during human neutrophil activation. Regulation by protein kinase C-dependent and independent pathways. J. Biol. Chem. 1997, 272, 17204–17208. [Google Scholar] [CrossRef]
- Dang, P.M.; Morel, F.; Gougerot-Pocidalo, M.A.; El Benna, J. Phosphorylation of the NADPH oxidase component p67(PHOX) by ERK2 and P38MAPK: Selectivity of phosphorylated sites and existence of an intramolecular regulatory domain in the tetratricopeptide-rich region. Biochem. 2003, 42, 4520–4526. [Google Scholar] [CrossRef] [PubMed]
- Dang, P.M.; Raad, H.; Derkawi, R.A.; Boussetta, T.; Paclet, M.H.; Belambri, S.A.; Makni-Maalej, K.; Kroviarski, Y.; Morel, F.; Gougerot-Pocidalo, M.A.; et al. The NADPH oxidase cytosolic component p67phox is constitutively phosphorylated in human neutrophils: Regulation by a protein tyrosine kinase, MEK1/2 and phosphatases 1/2A. Biochem. Pharmacol. 2011, 82, 1145–1152. [Google Scholar] [CrossRef]
- Tsai, Y.R.; Wang, Y.J.; Lee, M.R.; Hsu, M.F.; Wang, J.P. p38 Mitogen-activated protein kinase and extracellular signal-regulated kinase signaling pathways are not essential regulators of formyl peptide-stimulated p47(phox) activation in neutrophils. Eur. J. Pharmacol. 2013, 701, 96–105. [Google Scholar] [CrossRef]
- Hazan-Halevy, I.; Levy, T.; Wolak, T.; Lubarsky, I.; Levy, R.; Paran, E. Stimulation of NADPH oxidase by angiotensin II in human neutrophils is mediated by ERK, p38 MAP-kinase and cytosolic phospholipase A2. J. Hypertens. 2005, 23, 1183–1190. [Google Scholar] [CrossRef] [PubMed]
- Leevers, S.J.; Paterson, H.F.; Marshall, C.J. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature 1994, 369, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Marais, R.; Light, Y.; Paterson, H.F.; Marshall, C.J. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 1995, 14, 3136–3145. [Google Scholar] [CrossRef] [PubMed]
- Wetzker, R.; Bohmer, F.D. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat. Rev. Mol. Cell. Biol. 2003, 4, 651–657. [Google Scholar] [CrossRef]
- Faure, M.; Voyno-Yasenetskaya, T.A.; Bourne, H.R. cAMP and beta gamma subunits of heterotrimeric G proteins stimulate the mitogen-activated protein kinase pathway in COS-7 cells. J. Biol. Chem. 1994, 269, 7851–7854. [Google Scholar]
- Zhong, D.; Xiong, L.; Liu, T.; Liu, X.; Liu, X.; Chen, J.; Sun, S.Y.; Khuri, F.R.; Zong, Y.; Zhou, Q.; et al. The glycolytic inhibitor 2-deoxyglucose activates multiple prosurvival pathways through IGF1R. J. Biol. Chem. 2009, 284, 23225–23233. [Google Scholar] [CrossRef]
- Gryshkova, V.; Cotter, M.; McGhan, P.; Obajdin, J.; Fleurance, R.; da Costa, A.N. Phosphoprotein expression profiles in rat kidney injury: Source for potential mechanistic biomarkers. J. Cell. Mol. Med. 2019, 23, 2251–2255. [Google Scholar] [CrossRef]
- Ohki, S.; Eto, M.; Kariya, E.; Hayano, T.; Hayashi, Y.; Yazawa, M.; Brautigan, D.; Kainosho, M. Solution NMR structure of the myosin phosphatase inhibitor protein CPI-17 shows phosphorylation-induced conformational changes responsible for activation. J. Mol. Biol. 2001, 314, 839–849. [Google Scholar] [CrossRef]
- Eto, M.; Kitazawa, T.; Matsuzawa, F.; Aikawa, S.; Kirkbride, J.A.; Isozumi, N.; Nishimura, Y.; Brautigan, D.L.; Ohki, S.Y. Phosphorylation-induced conformational switching of CPI-17 produces a potent myosin phosphatase inhibitor. Structure 2007, 15, 1591–1602. [Google Scholar] [CrossRef]
- Eto, M. Regulation of cellular protein phosphatase-1 (PP1) by phosphorylation of the CPI-17 family, C-kinase-activated PP1 inhibitors. J. Biol. Chem. 2009, 284, 35273–35277. [Google Scholar] [CrossRef]
- Dippold, R.P.; Fisher, S.A. A bioinformatic and computational study of myosin phosphatase subunit diversity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R256–R270. [Google Scholar] [CrossRef] [PubMed]
- Eto, M.; Brautigan, D.L. Endogenous inhibitor proteins that connect Ser/Thr kinases and phosphatases in cell signaling. IUBMB Life 2012, 64, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Eto, M.; Senba, S.; Morita, F.; Yazawa, M. Molecular cloning of a novel phosphorylation-dependent inhibitory protein of protein phosphatase-1 (CPI17) in smooth muscle: Its specific localization in smooth muscle. FEBS Lett. 1997, 410, 356–360. [Google Scholar] [CrossRef]
- Eto, M.; Bock, R.; Brautigan, D.L.; Linden, D.J. Cerebellar long-term synaptic depression requires PKC-mediated activation of CPI-17, a myosin/moesin phosphatase inhibitor. Neuron 2002, 36, 1145–1158. [Google Scholar] [CrossRef]
- Kim, J.I.; Urban, M.; Young, G.D.; Eto, M. Reciprocal regulation controlling the expression of CPI-17, a specific inhibitor protein for the myosin light chain phosphatase in vascular smooth muscle cells. Am. J. Physiol. Cell. Physiol. 2012, 303, C58–C68. [Google Scholar] [CrossRef] [PubMed]
- Eto, M.; Kitazawa, T. Diversity and plasticity in signaling pathways that regulate smooth muscle responsiveness: Paradigms and paradoxes for the myosin phosphatase, the master regulator of smooth muscle contraction. J. Smooth Muscle Res. 2017, 53, 1–19. [Google Scholar] [CrossRef]
- Eto, M.; Kitazawa, T.; Yazawa, M.; Mukai, H.; Ono, Y.; Brautigan, D.L. Histamine-induced vasoconstriction involves phosphorylation of a specific inhibitor protein for myosin phosphatase by protein kinase C alpha and delta isoforms. J. Biol. Chem. 2001, 276, 29072–29078. [Google Scholar] [CrossRef]
- Kitazawa, T.; Eto, M.; Woodsome, T.P.; Brautigan, D.L. Agonists trigger G protein-mediated activation of the CPI-17 inhibitor phosphoprotein of myosin light chain phosphatase to enhance vascular smooth muscle contractility. J. Biol. Chem. 2000, 275, 9897–9900. [Google Scholar] [CrossRef]
- Jin, H.; Sperka, T.; Herrlich, P.; Morrison, H. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature 2006, 442, 576–579. [Google Scholar] [CrossRef]
- Huang, J.; Mahavadi, S.; Sriwai, W.; Hu, W.; Murthy, K.S. Gi-coupled receptors mediate phosphorylation of CPI-17 and MLC20 via preferential activation of the PI3K/ILK pathway. Biochem. J. 2006, 396, 193–200. [Google Scholar] [CrossRef]
- Zemlickova, E.; Johannes, F.J.; Aitken, A.; Dubois, T. Association of CPI-17 with protein kinase C and casein kinase I. Biochem. Biophys. Res. Commun. 2004, 316, 39–47. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, J.A.; Eto, M.; Borman, M.A.; Brautigan, D.L.; Haystead, T.A. Dual Ser and Thr phosphorylation of CPI-17, an inhibitor of myosin phosphatase, by MYPT-associated kinase. FEBS Lett. 2001, 493, 91–94. [Google Scholar] [CrossRef]
- Takizawa, N.; Koga, Y.; Ikebe, M. Phosphorylation of CPI17 and myosin binding subunit of type 1 protein phosphatase by p21-activated kinase. Biochem. Biophys. Res. Commun. 2002, 297, 773–778. [Google Scholar] [CrossRef]
- Koyama, M.; Ito, M.; Feng, J.; Seko, T.; Shiraki, K.; Takase, K.; Hartshorne, D.J.; Nakano, T. Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett. 2000, 475, 197–200. [Google Scholar] [CrossRef]
- Hamaguchi, T.; Ito, M.; Feng, J.; Seko, T.; Koyama, M.; Machida, H.; Takase, K.; Amano, M.; Kaibuchi, K.; Hartshorne, D.J.; et al. Phosphorylation of CPI-17, an inhibitor of myosin phosphatase, by protein kinase N. Biochem Biophys Res. Commun 2000, 274, 825–830. [Google Scholar] [CrossRef]
- Dimopoulos, G.J.; Semba, S.; Kitazawa, K.; Eto, M.; Kitazawa, T. Ca2+-dependent rapid Ca2+ sensitization of contraction in arterial smooth muscle. Circ. Res. 2007, 100, 121–129. [Google Scholar] [CrossRef]
- Eto, M.; Kirkbride, J.A.; Chugh, R.; Karikari, N.K.; Kim, J.I. Nuclear localization of CPI-17, a protein phosphatase-1 inhibitor protein, affects histone H3 phosphorylation and corresponds to proliferation of cancer and smooth muscle cells. Biochem. Biophys. Res. Commun. 2013, 434, 137–142. [Google Scholar] [CrossRef]
- Dubois, T.; Howell, S.; Zemlickova, E.; Learmonth, M.; Cronshaw, A.; Aitken, A. Novel in vitro and in vivo phosphorylation sites on protein phosphatase 1 inhibitor CPI-17. Biochem. Biophys. Res. Commun. 2003, 302, 186–192. [Google Scholar] [CrossRef]
- Eto, M.; Ohmori, T.; Suzuki, M.; Furuya, K.; Morita, F. A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. Isolation from porcine aorta media and characterization. J. Biochem. 1995, 118, 1104–1107. [Google Scholar] [CrossRef]
- Bhetwal, B.P.; Sanders, K.M.; An, C.; Trappanese, D.M.; Moreland, R.S.; Perrino, B.A. Ca2+ sensitization pathways accessed by cholinergic neurotransmission in the murine gastric fundus. J. Physiol. 2013, 591, 2971–2986. [Google Scholar] [CrossRef]
- Simoes, R.L.; Fierro, I.M. Involvement of the Rho-kinase/myosin light chain kinase pathway on human monocyte chemotaxis induced by ATL-1, an aspirin-triggered lipoxin A4 synthetic analog. J. Immunol. 2005, 175, 1843–1850. [Google Scholar] [CrossRef]
- Bokoch, G.M. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 2005, 15, 163–171. [Google Scholar] [CrossRef]
- Eichel, K.; Jullie, D.; von Zastrow, M. beta-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol. 2016, 18, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteomics 2014, 13, 1690–1704. [Google Scholar] [CrossRef] [PubMed]
- Dakshinamurti, S.; Mellow, L.; Stephens, N.L. Regulation of pulmonary arterial myosin phosphatase activity in neonatal circulatory transition and in hypoxic pulmonary hypertension: A role for CPI-17. Pediatr. Pulmonol. 2005, 40, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Sakai, H.; Chiba, Y.; Hirano, T.; Misawa, M. Possible involvement of CPI-17 in augmented bronchial smooth muscle contraction in antigen-induced airway hyper-responsive rats. Mol. Pharmacol. 2005, 68, 145–151. [Google Scholar] [CrossRef]
- Morin, C.; Sirois, M.; Echave, V.; Rousseau, E. CPI-17 silencing-reduced responsiveness in control and TNF-alpha-treated human bronchi. Am. J. Respir. Cell. Mol. Biol. 2008, 39, 638–643. [Google Scholar] [CrossRef]
- Ohama, T.; Hori, M.; Sato, K.; Ozaki, H.; Karaki, H. Chronic treatment with interleukin-1beta attenuates contractions by decreasing the activities of CPI-17 and MYPT-1 in intestinal smooth muscle. J. Biol. Chem. 2003, 278, 48794–48804. [Google Scholar] [CrossRef]
- Xie, Z.; Su, W.; Guo, Z.; Pang, H.; Post, S.R.; Gong, M.C. Up-regulation of CPI-17 phosphorylation in diabetic vasculature and high glucose cultured vascular smooth muscle cells. Cardiovasc. Res. 2006, 69, 491–501. [Google Scholar] [CrossRef]
- Chang, S.; Hypolite, J.A.; DiSanto, M.E.; Changolkar, A.; Wein, A.J.; Chacko, S. Increased basal phosphorylation of detrusor smooth muscle myosin in alloxan-induced diabetic rabbit is mediated by upregulation of Rho-kinase beta and CPI-17. Am. J. Physiol. Renal Physiol. 2006, 290, F650–F656. [Google Scholar] [CrossRef] [PubMed][Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Annunziata, M.C.; Parisi, M.; Esposito, G.; Fabbrocini, G.; Ammendola, R.; Cattaneo, F. Phosphorylation Sites in Protein Kinases and Phosphatases Regulated by Formyl Peptide Receptor 2 Signaling. Int. J. Mol. Sci. 2020, 21, 3818. https://doi.org/10.3390/ijms21113818
Annunziata MC, Parisi M, Esposito G, Fabbrocini G, Ammendola R, Cattaneo F. Phosphorylation Sites in Protein Kinases and Phosphatases Regulated by Formyl Peptide Receptor 2 Signaling. International Journal of Molecular Sciences. 2020; 21(11):3818. https://doi.org/10.3390/ijms21113818
Chicago/Turabian StyleAnnunziata, Maria Carmela, Melania Parisi, Gabriella Esposito, Gabriella Fabbrocini, Rosario Ammendola, and Fabio Cattaneo. 2020. "Phosphorylation Sites in Protein Kinases and Phosphatases Regulated by Formyl Peptide Receptor 2 Signaling" International Journal of Molecular Sciences 21, no. 11: 3818. https://doi.org/10.3390/ijms21113818
APA StyleAnnunziata, M. C., Parisi, M., Esposito, G., Fabbrocini, G., Ammendola, R., & Cattaneo, F. (2020). Phosphorylation Sites in Protein Kinases and Phosphatases Regulated by Formyl Peptide Receptor 2 Signaling. International Journal of Molecular Sciences, 21(11), 3818. https://doi.org/10.3390/ijms21113818