Abstract
As an essential vitamin, the role of riboflavin in human diet and health is increasingly being highlighted. Insufficient dietary intake of riboflavin is often reported in nutritional surveys and population studies, even in non-developing countries with abundant sources of riboflavin-rich dietary products. A latent subclinical riboflavin deficiency can result in a significant clinical phenotype when combined with inborn genetic disturbances or environmental and physiological factors like infections, exercise, diet, aging and pregnancy. Riboflavin, and more importantly its derivatives, flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD), play a crucial role in essential cellular processes including mitochondrial energy metabolism, stress responses, vitamin and cofactor biogenesis, where they function as cofactors to ensure the catalytic activity and folding/stability of flavoenzymes. Numerous inborn errors of flavin metabolism and flavoenzyme function have been described, and supplementation with riboflavin has in many cases been shown to be lifesaving or to mitigate symptoms. This review discusses the environmental, physiological and genetic factors that affect cellular riboflavin status. We describe the crucial role of riboflavin for general human health, and the clear benefits of riboflavin treatment in patients with inborn errors of metabolism.
1. Introduction
B-vitamins are an important group of vitamins that are vital for crucial cellular functions, such as energy production, and in the formation of other vitamins. One of the key players in these processes is vitamin B2, or riboflavin, and its derivatives, flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD). The derivatives FMN and FAD play crucial roles as cofactors for enzyme-catalyzed reactions. Riboflavin is a water-soluble essential vitamin present in dietary sources such as meats, milk, fatty fish, nuts, eggs and vegetables, such as spinach and beans, as well as some fruits [1]. Furthermore, riboflavin is frequently added to vitamin enriched products, such as baby foods and cereals, and it has been suggested that some bacteria in the human microbiome are able to produce riboflavin [2].
Dietary sources contain free riboflavin, but also contain flavoproteins with bound FMN and FAD that need to be hydrolyzed to riboflavin before this can be absorbed [3]. The absorption and transportation of riboflavin is performed by a group of transporters from the solute carrier family SLC52. The majority of riboflavin is absorbed in the small intestine, but absorption also occurs in the stomach, duodenum, colon and rectum, through an active carrier-mediated transport by riboflavin transporter 3 (RFVT3), illustrated in Figure 1. The main function of RFVT3 is to absorb riboflavin from the diet. Once riboflavin is absorbed, it can be transformed into FMN and FAD, and used by the epithelial cells of the gastrointestinal tract, or it can be transported by riboflavin transporter 1 (RFVT1) or riboflavin transporter 2 (RFVT2) to the bloodstream and distributed to tissues, where it is needed. RFVT2 is distributed all over the body and can be detected in high amounts in the brain, but also in endocrine tissues, liver and muscle. Just like the other two transporters, RFVT1 is expressed in the gastrointestinal tract, but one of its most important roles is to transport maternal riboflavin to the fetus in the placenta. When riboflavin reaches the target cells, it is transformed into its active forms, FMN by riboflavin kinase (RFK), and in turn to FAD by FAD synthase (FADS) [3,4], as illustrated in Figure 1. Abundant amounts of flavins are used by the mitochondria, and both FMN and FAD can be transported inside the mitochondrion by the mitochondrial folate transporter (MFT) [5]. Several studies indicate that the human mitochondrion itself has the ability to produce FAD. In support of this theory, several FAD synthase isoforms have been reported, including an isoform with a mitochondrial targeting sequence [6,7,8,9].
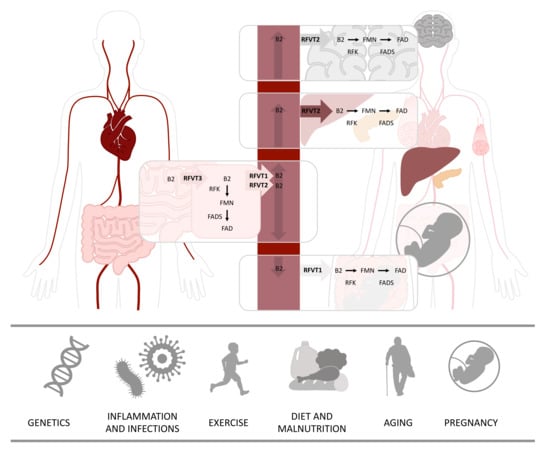
Figure 1.
Riboflavin metabolism and its interaction with environmental factors. Riboflavin is absorbed from the gastrointestinal tract predominantly by riboflavin transporter 3 (RFVT3). Inside the gastrointestinal cells, riboflavin can either be further metabolized to flavin mononucleotide (FMN) by riboflavin kinase (RFK) or to flavin adenine dinucleotide (FAD) by FAD synthase (FADS) or transported to the bloodstream by riboflavin transporter 1 (RFVT1) and riboflavin transporter 2 (RFVT2). Riboflavin is distributed via the bloodstream to its destination cells. In addition to being expressed in the gastrointestinal system, RFVT1 is expressed in the placenta, where it carries riboflavin from maternal bloodstream to fetal bloodstream. RFVT2 is expressed all over the body and highly expressed in the brain, endocrine organs, such as pancreas, but also in the liver and muscle tissue. Inside the destination cells, riboflavin is used directly or transformed into either FMN or FAD, which are used as cofactors for several processes. Several factors can affect human riboflavin status, hereunder, genetics, inflammation and infections, exercise, diet and nutrition, aging and pregnancy.
The daily dietary requirements of riboflavin range from 1.0 to 1.3 mg/d in adults, and even higher amounts are advised to young adults, pregnant and lactating women, athletes and elderly people. The excess riboflavin is excreted in the urine as riboflavin or as riboflavin-derived metabolites, 7-hydroxymethylriboflavin and lumiflavin. To date, there are no reported complications associated with riboflavin supplementation, even when supplied in very high doses [3,10]. Several factors can affect human riboflavin status, of which diet has the largest impact in the general population. However, other factors such as pregnancy, exercise, aging, infections—and in rare cases genetic variations—can also affect riboflavin status (Figure 1).
2. Riboflavin’s Importance for Cellular Functions
In 1975, Hoppel and Tandler illustrated the importance of riboflavin in the mitochondrial energy metabolism by showing that riboflavin depletion in mice led to a wide impairment of mitochondrial function [11]. Despite its central role in the mitochondrial energy metabolism, other metabolic pathways strongly depend on riboflavin. In fact, the human genome contains 90 genes encoding flavin-dependent proteins, which together form the human flavoproteome [12]. Flavoproteins are involved in various cellular functions, as briefly described below.
A wide number of flavoenzymes are involved in lipid and protein metabolism. Seven mitochondrial acyl-CoA dehydrogenases (ACADs) and three peroxisomal acyl-CoA oxidases (ACOX) are involved in the catabolism of lipids. Flavoenzymes are active also in lipid anabolism, in particular, they participate in fatty acid desaturation and cholesterol metabolism in the endoplasmic reticulum and in ether lipid synthesis in peroxisomes [12,13]. Four ACADs are involved in the mitochondrial catabolism of various amino acids, and choline dehydrogenase (CHDH), dimethylglycine dehydrogenase (DMGDH) and sarcosine dehydrogenase (SARDH) process choline in mitochondria. Flavoenzymes are involved also in the oxidative folding of proteins in the endoplasmic reticulum [12,14].
Riboflavin is crucial for mitochondrial electron transport chain (ETC) function. In fact, complex I and complex II harbor FMN and FAD, respectively, and the mitochondrial glycerol-3-phosphate dehydrogenase (GPDM), the sulfide:quinone oxidoreductase (SQOR), the electron transfer flavoprotein (ETF) and the electron transfer flavoprotein-ubiquinone oxidoreductase (ETF-QO), which shuttle electrons to CoQ10, are all flavoenzymes [12].
In addition, flavoenzymes play a pivotal role in cellular stress responses. Glutathione reductase is a flavoenzyme, which catalyzes the reduction of glutathione disulfide to glutathione, a key cellular antioxidant peptide [12,15]. Flavoenzymes are also involved in cellular apoptosis. The apoptosis inducing factor (AIF) is a mitochondrial flavoenzyme, which during apoptosis is translocated to the nucleus, where it is incorporated into a chromatin-degrading complex, ultimately leading to cell death [16]. The apoptosis-inducing factor-homologous mitochondrion-associated inducer of death (AMID) is a newly characterized cytosolic flavoenzyme involved in caspase-independent apoptosis [12,17]. Finally, flavoenzymes participate in drug metabolism, since both mitochondrial and microsomal cytochromes P450 receive electrons from flavoenzymes [12,18], and in epigenetic regulations, with lysine-specific demethylase 1 (LSD1) utilizing FAD as cofactor [12,19].
The Role of Riboflavin in Homocysteine, Folic Acid and the Metabolism of Other Vitamins
Homocysteine is an amino acid derived from methionine, and high levels of homocysteine are associated with a risk of developing cardiovascular diseases [20]. Metabolizing homocysteine can be performed in two ways, either transsulfuration, depending on vitamin B6 (pyridoxine), or remethylation, depending on vitamin B9 (folic acid), vitamin B12 (cyanocobalamin) and riboflavin [21]. In the metabolic regulation of homocysteine and the metabolism of folate, the methylenetetrahydrofolate reductase (MFTHR) is a key enzyme. MFTHR uses FAD as a cofactor to metabolize vitamin B9 in the homocysteine methylation. Studies have shown that 5–30% of the general population is homozygous for a common genetic variant (c.677C>T) that is associated with increased levels of homocysteine [22]. Further investigations of this common variant have shown that a daily dose of riboflavin (1.6mg/d) for a 12-week period significantly decreases the amount of homocysteine in plasma in individuals homozygous for the c.677C>T variant. The decrease of homocysteine level could lower the risk of developing cardiovascular disease in the c.677C>T homozygous individuals [23]. Similar to MFTHR, pyridoxine 5′-phosphate oxidase (PNPO), converting vitamin B6 to the biologically active form, pyridoxal phosphate, depends on a riboflavin derivative, in this case FMN [24], and the methionine synthase reductase (MTRR), involved in the regeneration of vitamin B12, uses both FMN and FAD [12]. Even in the biosynthesis of D vitamin, flavoenzymes have important roles [13].
3. Riboflavin Deficiency and its Significance in the General Population
3.1. Riboflavin and Diet
Riboflavin deficiency is the most common vitamin deficiency in developing countries with diets lacking riboflavin-rich products, such as milk and meat [25]. Clinical symptoms of riboflavin deficiency appear only after several months of insufficient riboflavin intake, and vary from milder symptoms as sore throat, loss of hair, and skin inflammation, to severe symptoms as swollen tongue, anemia and impaired nerve functions. Although these symptoms are rarely seen in non-developing countries and well-nourished societies, dietary insufficiency and subclinical riboflavin deficiency is detected in remarkably large groups in the population [26,27,28,29]. Several population studies of vitamin status report on riboflavin insufficiency in children and young adults, especially in young women [30]. A national survey performed in the United Kingdom, investigating the biochemical riboflavin status in 2127 schoolchildren, revealed a poor riboflavin status that increased with age. In boys, from 59% insufficient riboflavin intake in 4–6-year-olds, to 78% in 7–10-year-olds, but the largest group with riboflavin insufficiency were the 15–18-year-old girls. The survey revealed that 95% of the 15–18-year-old girls had an insufficient intake of riboflavin [30] and an increasing risk of developing riboflavin deficiency. The increase of riboflavin insufficiency in both boys and girls is comparable to a declined consumption of milk, from 25% of the daily riboflavin intake in the 4–6-year-olds to only 10% of the daily riboflavin intake in the 15–18-year-olds. The implications for this riboflavin insufficiency, especially in young girls, are not fully known, but it has been shown that subclinical riboflavin deficiency could influence iron handling and that a daily supplement with riboflavin (2 or 4 mg) for 8 weeks significantly improves the hematologic status, with an increase in circulating red blood cells and hemoglobin concentrations in young women, even without an additional iron supplementation [28].
Most of the reported population studies performed on riboflavin status are older studies and the recent year’s changes in lifestyle, especially in well-nourished societies, with diets based on less dairy and meat products in combination with more exercise (see below), could potentially increase the risk of riboflavin deficiency. In this context, studies on the dietary intake of riboflavin in well-nourished countries amongst people that follow a vegan diet without meat, dairy products and eggs, have shown that up to 48% have lower than the recommended daily intake of riboflavin, and thereby an increasing risk for developing riboflavin deficiency [31,32,33].
3.2. Riboflavin and Aging
In addition, in the elderly population a general riboflavin insufficiency has been described. Studies in the United Kingdom and the United States indicate that 10–41% of the elderly population have an insufficient riboflavin intake [34,35] and are at risk of developing riboflavin deficiency, based on dietary reports [36]. The deficiency observed in the elderly population in these studies can partly be explained by a decreased intake of milk and other dairy products. However, the most plausible explanation is that the elderly population displays a reduced efficiency in the absorption of riboflavin that increases with aging [3,37]. In addition, riboflavin deficiency and deficiency of other B vitamins in the elderly have been linked to depression and changes in cognitive function, and it has been shown that riboflavin supplementation in elderly people could work as a neuroprotective agent and prevent disorders such as dementia, Parkinson’s and Alzheimer’s disease [38,39,40,41,42].
3.3. Riboflavin and Exercise
Athletes and people with high physical activity could be at risk of developing riboflavin deficiency. Studies in healthy men with a moderate activity level and biochemical signs of riboflavin deficiency have shown that even short periods with increased physical activity deteriorate riboflavin levels further. The deterioration in riboflavin is caused by the metabolic stress that occurs during periods of increased physical activity [43,44].
3.4. Riboflavin Pre- and Postpartum
One of the groups at high risk of developing riboflavin deficiency is women during pregnancy, where the needs of riboflavin rise with fetal development and continue postpartum. Riboflavin deficiency may not only affect the mother but also her child.
In 1943, the first connections between women’s diet and their pregnancy were reported by Burke et al. [45]. They observed that maternal nutritional status affected the infant’s condition at birth. Today, the connection between maternal nutritional status and normal fetus development and growth is well described, and it is known that many vitamins are of huge importance during pregnancy, including riboflavin. Riboflavin is essential for normal fetal development, and animal studies have shown that severe riboflavin deficiency in pregnant mice and chicken leads to abnormal fetal development and termination of pregnancy [46,47]. In humans, most studies documenting riboflavin deficiency have been performed in societies with low riboflavin intake. The studies have shown that the risk of riboflavin deficiency in pregnant women is especially high during the third trimester, approaching parturition and during lactation. During pregnancy, the metabolic needs increase, with an average of 230 kcal pr. day, which for most women is more than 10% of their total daily kcal intake. In the first two trimesters the anabolism is dominating, there is an increased insulin sensitivity, and the maternal fat deposits increase [48]. At the beginning of the third trimester, hormones from the placenta cause an increasing insulin resistance and catabolism in the mother, that enable nutrients for fetal growth, and an increasing need for energy rich nutrients, such as fatty acids and vitamins, including riboflavin, to ensure mitochondrial energy metabolism [49,50]. The increased need of nutrients and supporting vitamins, such as riboflavin, continues during parturition and postpartum. During parturition, riboflavin has an important antioxidant function, based on FAD being an essential cofactor for glutathione. Glutathione is crucial for counteracting the peroxidation reactions triggered by the rapid change from a hypoxic to a hyperoxic environment during birth [51]. Riboflavin is important also postpartum. Studies have shown that light therapy, that is frequently used for treating hyperbilirubinemia in infants shortly after birth, can cause riboflavin deficiency and lowers riboflavin levels to 50% within hours [52]. Moreover, maternal nutrient and riboflavin status is of great importance during breastfeeding. For infants that are exclusively breastfed, maternal milk is the only source of riboflavin. Riboflavin is mostly present as FAD in human milk and maternal riboflavin deficiency is rapidly reflected in low flavin concentration in the milk [53]. The postpartum breastfeeding of the infant is of great importance to ensure nutrient needs, but also to develop the infant’s immune response.
3.5. Riboflavin and the Immune Response
Riboflavin, FMN and FAD play a key role in immune functions and responses as described recently by Suwannasom et al. [54]. FMN and FAD are important cofactors for the human energy metabolism that is closely connected to the cellular immune responses. The immune response requires tremendous amounts of ATP since a large number of cells need to either be differentiated, proliferated or activated in order to perform their function. Both FMN and FAD are exceedingly involved in the production of ATP, as cofactors for crucial flavoenzymes in the oxidation of fatty acids and branched-chain amino acids, in the Krebs cycle and in the ETC. Recently, riboflavin was shown to have an important role in macrophage function, and that riboflavin deficiency causes disruption in the activation of macrophages that ultimately leads to a decreasing recognition of pathogens and a failed activation of immune responses [55]. Additionally, the key producers of ROS in the immune response, the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX), require binding of FAD. The production of ROS is crucial for destroying pathogen DNA, RNA and proteins, and ROS have an important role as cellular signaling molecules of immune cell function [56,57].
Several studies have investigated the functions of riboflavin in the immune responses, and riboflavin has been reported to have anti-inflammatory effects by lowering several proinflammatory cytokines, namely TNF-α, IL-1ß and IL-6 [58,59]. Moreover, riboflavin can reduce mortality in mice with septic shock, and it has been suggested that riboflavin treatment in septic shock could be potentially useful also in humans [58]. Using riboflavin as a treatment in disease is not new, and in the field of inborn errors of metabolism (IEM), riboflavin treatment has been used extensively and shown extraordinary results in some groups of patients.
4. Inborn Errors of Metabolism—Riboflavin as a Potent Treatment
Riboflavin supplementation is a well-established therapy in some IEM, such as multiple acyl-CoA dehydrogenation deficiency (MADD) and riboflavin transporter deficiencies, leading to a significant clinical improvement or stabilization in the majority of patients. Riboflavin supplementation has shown promising results in other IEM, but only single case reports or small-scale studies are reported in the literature. Therefore, further investigations and large-scale clinical trials are needed to confirm riboflavin therapy efficacy in those patients. An overview of IEM benefiting from riboflavin supplementation can be found in Table 1.
Riboflavin responsiveness can be explained by three major molecular mechanisms, although overlapping mechanisms may exist in individual IEM.
- It has been shown that riboflavin-derived cofactors are able to bind apoproteins and chaperone their folding (see below). Therefore, increased cellular levels of FMN and FAD could improve folding and stability of mutant flavoenzymes and decrease their proteolytic degradation. This process is likely to happen in inborn genetic errors of flavoenzymes such as ETF, ETF-QO, and certain ACADs, as well as in inborn genetic errors of ETC function and assembly (Figure 2).
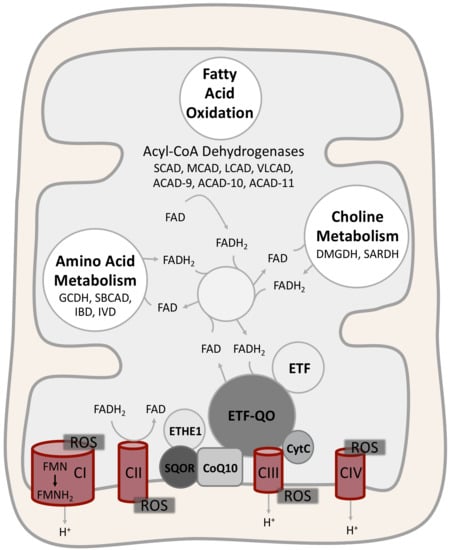 Figure 2. Disease-related flavoenzymes in mitochondrial energy metabolism. Several flavoenzymes are involved in the mitochondrial energy metabolism including fatty acid oxidation, amino acid metabolism and choline metabolism. Acyl-CoA dehydrogenases are involved in fatty acid oxidation and amino acid metabolism, while dimethylglycine dehydrogenase (DMGDH) and sarcosine dehydrogenase (SARDH) participate in choline metabolism. All these dehydrogenases harbor FAD as a redox cofactor, which is reduced to FADH2 during the dehydrogenation reactions. The dehydrogenases donate the electrons obtained from their substrates to the electron transfer flavoprotein (ETF), and finally to the electron transport chain (ETC) through the ETF-ubiquinone oxidoreductase (ETF-QO), which reduces coenzyme Q10 (CoQ10). The ethylmalonic encephalopathy protein 1 (ETHE1) is connected to the electron transport chain via the sulfide:quinone oxidoreductase (SQOR) and donates electrons to CoQ10. In the assembly and function of complex I and complex II, FMN and FAD, respectively, have important functions. Additionally, both FMN and FAD are crucial for folding and stability of all these flavoenzymes, that, in some cases, explains the benefits of riboflavin supplementation in patients with genetic variants causing primary or secondary dysfunction of flavoenzymes. Riboflavin can also compensate secondary flavin homeostasis derangements. Abbreviations: ACAD9, acyl-CoA dehydrogenase family member 9; ACAD10, acyl-CoA dehydrogenase family member 10; ACAD11, acyl-CoA dehydrogenase family member 11; CI, complex I; CII, complex II; CIII, complex III; CIV, complex IV; CoQ10, coenzyme Q10; CytC, cytochrome C; DMGDH, dimethylglycine dehydrogenase; ETF, electron transfer flavoprotein; ETF-QO, ETF-ubiquinone oxidoreductase; ETHE1, ethylmalonic encephalopathy protein 1; GCDH, glutaryl-CoA dehydrogenase; IBD, isobutyryl-CoA dehydrogenase; IVD, isovaleryl-CoA dehydrogenase; LCAD, long-chain acyl-CoA dehydrogenase; MCAD, medium-chain acyl-CoA dehydrogenase; SARDH, sarcosine dehydrogenase; SBCAD, short-branched chain acyl-CoA dehydrogenase; SCAD, short-chain acyl-CoA dehydrogenase; SQOR, sulfide:quinone oxidoreductase; VLCAD, very long-chain acyl-CoA dehydrogenase.
Figure 2. Disease-related flavoenzymes in mitochondrial energy metabolism. Several flavoenzymes are involved in the mitochondrial energy metabolism including fatty acid oxidation, amino acid metabolism and choline metabolism. Acyl-CoA dehydrogenases are involved in fatty acid oxidation and amino acid metabolism, while dimethylglycine dehydrogenase (DMGDH) and sarcosine dehydrogenase (SARDH) participate in choline metabolism. All these dehydrogenases harbor FAD as a redox cofactor, which is reduced to FADH2 during the dehydrogenation reactions. The dehydrogenases donate the electrons obtained from their substrates to the electron transfer flavoprotein (ETF), and finally to the electron transport chain (ETC) through the ETF-ubiquinone oxidoreductase (ETF-QO), which reduces coenzyme Q10 (CoQ10). The ethylmalonic encephalopathy protein 1 (ETHE1) is connected to the electron transport chain via the sulfide:quinone oxidoreductase (SQOR) and donates electrons to CoQ10. In the assembly and function of complex I and complex II, FMN and FAD, respectively, have important functions. Additionally, both FMN and FAD are crucial for folding and stability of all these flavoenzymes, that, in some cases, explains the benefits of riboflavin supplementation in patients with genetic variants causing primary or secondary dysfunction of flavoenzymes. Riboflavin can also compensate secondary flavin homeostasis derangements. Abbreviations: ACAD9, acyl-CoA dehydrogenase family member 9; ACAD10, acyl-CoA dehydrogenase family member 10; ACAD11, acyl-CoA dehydrogenase family member 11; CI, complex I; CII, complex II; CIII, complex III; CIV, complex IV; CoQ10, coenzyme Q10; CytC, cytochrome C; DMGDH, dimethylglycine dehydrogenase; ETF, electron transfer flavoprotein; ETF-QO, ETF-ubiquinone oxidoreductase; ETHE1, ethylmalonic encephalopathy protein 1; GCDH, glutaryl-CoA dehydrogenase; IBD, isobutyryl-CoA dehydrogenase; IVD, isovaleryl-CoA dehydrogenase; LCAD, long-chain acyl-CoA dehydrogenase; MCAD, medium-chain acyl-CoA dehydrogenase; SARDH, sarcosine dehydrogenase; SBCAD, short-branched chain acyl-CoA dehydrogenase; SCAD, short-chain acyl-CoA dehydrogenase; SQOR, sulfide:quinone oxidoreductase; VLCAD, very long-chain acyl-CoA dehydrogenase. - Riboflavin-derived cofactors may counteract secondary impairment of flavoenzymes induced by an IEM. This seems to be the mechanism of riboflavin-responsiveness in genetic disorders of ethylmalonic encephalopathy protein 1 (ETHE1), succinate dehydrogenase assembly factor I (SDHAF1) and mitochondrial tRNALeu(UUR) (MT-TL1).
- Genetic deficiencies in RFVTs, MFT and FADS invariably affect cellular flavin supply and distribution (Figure 1). In this case, riboflavin supplementation could act directly by restoring cellular flavin levels. Secondary derangements of flavin homeostasis, induced by the variant flavoenzymes themselves (FADS and ETF-QO), and/or by external factors as discussed above, could be equally compensated through this mechanism.
Before we summarize evidence of riboflavin therapy in individual IEM, we will discuss the molecular mechanisms by which flavin cofactors maintain the functional stability of flavoproteins and their genetic variants.
4.1. The Role of Flavin Cofactors for Folding and Stability of Flavoenzymes and Their Genetic Variants
The availability of flavin cofactors plays an important role for both folding and cellular stability of flavoenzymes with the two processes being widely intertwined. During folding, the protein forms all the non-covalent interactions between atoms of the polypeptide chain and the flavin cofactor that are present in the native conformation. The sum of these interactions determines the biophysical stability of the protein. High biophysical stability protects proteins from degradation, as intracellular proteases have a strong preference for unfolded or partially unfolded protein molecules [60]. Therefore, the contribution of the interactions between the atoms of the protein and the atoms of cofactors, like FAD and FMN, have a great influence on the intracellular stability and turnover of flavoenzymes.
Atomic interactions of the polypeptide chain with prosthetic groups often also play a crucial role for the folding propensity. According to the ‘nucleation condensation’ theory of protein folding, one important rate-limiting step for the acquisition of the native structure is the formation of the ‘folding nucleus’, a subset of native interactions [61]. Once formed, the folding nucleus speeds up the formation of the remaining interactions. In the 1990s, experimental evidence for the crucial role of FAD for folding, assembly and stability of the ACADs was provided by the Tanaka laboratory. Firstly, import of various ACADs into rat mitochondria resulted in increased degradation of the ACADs when FAD-depleted mitochondria were used, compared to mitochondria with normal FAD levels [62]. Secondly, follow-up investigations with one of the ACADs, medium-chain acyl-CoA dehydrogenase (MCAD), showed that its folding, after import into the mitochondrial matrix, occurs via sequential interactions with the mitochondrial matrix HSP70 chaperone orthologue and HSP60. The authors demonstrated that FAD was required for the formation of the core structure, a kind of folding nucleus, before MCAD monomers were released from the HSP60 complex and then assembled into native tetramers [63]. This clearly showed that the interactions of the folding polypeptide chain with the flavin cofactor is crucial for folding and assembly of these proteins. Using another mitochondrial ACAD, DMGDH, the Barile group showed evidence of a direct interaction between DMGDH and FADS, with cofactor transfer occurring in a sort of “FAD-chaperoning” action catalyzed by FADS. They also showed evidence of an interaction with the HSP60/HSP10 chaperone complex and a possible control exerted by the redox status of FAD, although the exact mechanisms need further investigations [64].
Except for 6 out of the 90 human flavoenzymes, the flavin cofactors FAD or FMN are non-covalently bound to the protein [12]. The FAD dissociation constant for the human ETF, a heterodimer with one non-covalently bound FAD, has been measured to be 0.04 µM [65], i.e., the great majority of ETF molecules have FAD bound under physiological conditions. The melting temperature, a parameter of conformational stability, of some purified ACADs, was shown to be significantly increased in the presence of FAD, suggesting that the availability of another FAD molecule, quickly replacing the dissociated one, protects against denaturation [66]. The presence of the substrates of these enzymes had an additional stabilizing effect. The residual activity of these enzymes at a fever simulating temperature (40 °C) was also stabilized by presence of FAD and additionally by presence of both FAD and the preferred substrate.
The majority of disease-associated point variants in flavoenzymes impair folding and conformational stability [67]. In the light of its role for folding and stability, the levels of the flavin cofactor become even more crucial as this can modulate the residual function of the affected enzymes both by spurring correct folding and by stabilizing the native structure. Two in vitro studies on genetic variants of ETF support this notion. ETF with a disease-causing variant in the beta subunit (ETFβ-p.Asp128Asn) lost the bound FAD more readily at 39 °C, a temperature modeling fever, and the presence of saturating levels of FAD alleviated this [65]. Similar ‘fever’ treatment of two ETF variant proteins differing in a polymorphic site (Ile or Thr at position 171) in the alpha subunit showed that the less frequent variant lost the FAD more readily. Furthermore, the presence of the bacterial HSP60 chaperonin GroEL protected ETF activity under these conditions [68].
In a study from our research lab [69], we used a human HEK-293 cell expression system to investigate the influence of riboflavin and temperature on the steady-state level and the activity of variant ETF-QO proteins, identified in patients with riboflavin-responsive or non-responsive MADD disease. All studied variants were of the missense type and they showed temperature-dependent impairment of ETF-QO protein stability and function to varying degree. However, only those variants (p.Gly429Arg, p.Pro456Leu, p.Pro483Leu) associated with a riboflavin-responsive clinical phenotype could be rescued to wild-type ETF-QO protein and activity levels when expressed at therapeutic concentrations of riboflavin. One of the variants (p.Pro456Leu) showed indications of prolonged association with HSP60, when imported into rat liver mitochondria. The results from these experimental analyses, and presumed structural effects of ETF-QO missense variants [70], are consistent with the idea that the riboflavin response results from the ability of FAD to act as a chemical chaperone that can promote folding and stability of certain variant ETF-QO proteins. This is in accordance with recent biophysical and structural studies of the purified Rhodobacter p.Pro389Leu ETF-QO variant protein, a model for the disease-associated human p.Pro456Leu ETF-QO variant. This variant, as compared to wild-type protein, showed lower stability of FAD binding and increased susceptibility to thermal unfolding, FAD loss and proteolytic degradation [71]. Interestingly, the p.Pro456Leu variant, along with other riboflavin-responsive variants, is located in the ubiquinone-binding domain of ETF-QO. Yet, other riboflavin-responsive variants are located in the FAD binding domain. In the HEK-293 expression study described above, we showed that the effects of amino acid variants in the FAD and ubiquinone binding domain are similar; probably due to cooperative folding of the two domains in the early stages of the folding process [69].
Similar to ETF and ETF-QO variants, FADS variants have been associated with various degree of clinical responsiveness to riboflavin. In particular, patients harboring missense variants in the C-terminal domain of FADS have shown a dramatic response [9]. In vitro overexpression with a molar excess of FAD in E. coli resulted in a fully FAD-bound p.Ser495del FADS protein and rescued its proteolytic degradation, supporting a chaperone-like action of FAD [9].
Taken together, these studies illustrate that high levels of FAD and FMN, spurred by intake of the precursor riboflavin, promote folding and stability of flavoproteins, especially under stress conditions, like fever. Riboflavin supplementation does even allow certain mutant flavoenzymes to reach a folding efficiency and stability that attenuates the enzyme deficiency.
4.2. Inborn Errors of Flavoenzymes
4.2.1. Multiple Acyl-CoA Dehydrogenation Deficiency
The first riboflavin-responsive IEM was described in 1982 as a mild form of MADD (OMIM #231680). MADD is a rare recessive disorder of fatty acid, amino acid, and choline metabolism [72]. Mitochondrial electron transfer from fatty acid, amino acid and choline metabolism depends on a coordinated function of multiple flavoenzymes, many of which are ACADs. The first step of mitochondrial fatty acid oxidation is catalyzed by seven ACADs with different substrate specificity, namely short- (SCAD), medium- (MCAD), long- (LCAD), very long- (VLCAD) chain acyl-CoA dehydrogenase and the more recently characterized acyl-CoA dehydrogenase family members 9, 10 and 11 (ACAD9, ACAD10, ACAD11) [73]. Four ACADs are involved in amino acid metabolism; isovaleryl-CoA dehydrogenase (IVD), isobutyryl-CoA dehydrogenase (IBD), short/branched-chain acyl-CoA dehydrogenase (SBCAD) and glutaryl-CoA dehydrogenase (GCDH) [73]. All the ACADs donate the electrons obtained from their substrates to ETF. ETF transfers the electrons to ETF-QO which in turn reduces CoQ10 in the ETC. Two flavoenzymes involved in choline metabolism (DMGDH and SARDH), also deliver electrons to the ETC through the ETF/ETF-QO enzymes (Figure 2). Inborn errors in ETF subunits (ETFA, ETFB) or in ETF-QO (ETFDH) are the most common causes of MADD. The clinical presentation of MADD is heterogeneous with phenotype severity being related to the effect of variants on protein function. In severe-MADD patients (S-MADD), the disease onset is neonatal, there is a multisystem involvement, and the patients usually die within the first year of life. S-MADD patients are mainly homozygous or compound heterozygous for loss-of-function (LOF) variants. In mild-MADD patients (M-MADD), the disease can have a later onset and it is often triggered by stress conditions. M-MADD patients usually carry missense variants affecting protein folding and stability and have milder clinical symptoms, such as muscle weakness, hypoglycemia, encephalopathy and, more rarely, cardiomyopathies [74]. Interestingly, decreased levels and/or activities of ACADs and ETC complexes, sometimes combined with low levels of flavin cofactors, have been described in patients with ETF or ETF-QO deficiencies, suggesting that a secondary flavin homeostasis derangement occurs [75,76,77,78,79]. However, how ETF/ETF-QO impairment triggers flavin derangement has yet to be established. A possible explanation could be a negative feedback control on riboflavin transport and metabolism, as hypothesized for patients with inborn genetic errors in FADS, and observed in C. elegans experiments (see below). Moreover, CoQ10 depletion and massive ROS production have been measured in MADD patients, suggesting that an even wider metabolic reprogramming occurs [80,81]. Riboflavin supplementation has shown excellent results in M-MADD patients, while it has no or very limited effect in S-MADD patients. Most riboflavin responsive patients harbor ETFDH missense variants, but some responsive patients with ETFA and ETFB variants have been reported [75,82]. Riboflavin supplementation could compensate for disturbances of flavin homeostasis induced by endogenous or external sources, and thereby stabilize mutant ETF/ETF-QO folding, as discussed above [69,77].
4.2.2. Short-Chain Acyl-CoA Dehydrogenase Deficiency
Riboflavin responsiveness in SCAD deficiency (SCADD; OMIM #201470) has been investigated in a few studies. In 1995, Kmoch et al. [83] reported on a 5-years-old patient, who experienced seizures, hypotony, lethargy and cyclic vomiting during stress conditions. Genetic analyses revealed homozygosity for the polymorphic c.625G>A variant, which confers susceptibility to SCADD [84]. When riboflavin supplementation (25 mg/kg/d) was started, the condition of the child improved dramatically and after 2 years of riboflavin treatment, biochemical parameters were normalized. Other single case reports on the beneficial effect of riboflavin supplementation in SCADD patients are available, but genetic characterization of the patients was not reported [85,86]. A wider investigation was performed in 2010 by Van Maldegem et al. [87]. The study included 16 symptomatic SCADD children divided into three groups according to their genotype. The first group included patients homozygous or compound heterozygous for rare missense variants, the second group included compound heterozygous patients with one rare missense variant on one allele and the c.625G>A susceptibility variant on the other allele, the third group included patients homozygous for c.625G>A susceptibility variant. Riboflavin supplementation 10 mg/kg/d (up to 150 mg/d) was administered for 35 days. A mild biochemical improvement was observed in five patients belonging to the second group, and four of them reported a clinical improvement as well. However, the riboflavin effect was questionable because the improved clinical condition persisted after discontinuation of treatment in all four patients. This result is in contrast with the clear clinical amelioration observed in Kmoch et al. patient, suggesting that further studies are needed to clarify the effectiveness of riboflavin supplementation in SCADD patients.
4.2.3. Medium-Chain Acyl-CoA Dehydrogenase Deficiency
Some studies testing riboflavin therapy in patients with MCAD deficiency (MCADD; #201450) have been published. A first study was performed by Duran et al. in 1992 [88]. Five patients were given riboflavin supplementation 50–150 mg/d for 3 weeks and MCAD enzyme activity was measured in lymphocytes before and after riboflavin treatment. Upon treatment with riboflavin, residual MCAD activity was raised from 5.2% to 19% in four patients, and from 11.8% to the heterozygous level in the fifth patient. However, neither the clinical outcome nor genetic characterization of the patients were reported. A second study was published in 2014 by Derks et al. The research group examined lipid and protein oxidative damage and antioxidant defense status in blood samples from 27 MCADD patients harboring homozygous or compound heterozygous missense variants [89]. The patients received either no supplementation (14/27), carnitine supplementation (7/27) or carnitine and riboflavin supplementation (6/27). Riboflavin dosage was 50-150 mg/d. Compared with healthy controls, non-supplemented patients and carnitine-supplemented patients displayed increased protein oxidative damage and altered antioxidant defense, while carnitine and riboflavin-supplemented patients did not display any alteration, suggesting an antioxidant activity of riboflavin. The authors hypothesized that FAD chaperone activity could stabilize mutant MCAD and thereby reduce the amount of acyl-CoA derivatives, which have been shown to cause mitochondrial impairment and ROS production. The clinical outcomes were not evaluated. Therefore, further clinical trials are needed to evaluate whether riboflavin could have a beneficial effect in MCADD patients.
4.2.4. Acyl-CoA Dehydrogenase-9 Deficiency
Riboflavin therapy has been successfully tested in mitochondrial complex I deficiency nuclear type 20 (MC1DN20; OMIM #611126, also known as ACAD9 deficiency). The clinical outcome of ACAD9 deficiency is, in most cases, hypertrophic cardiomyopathy, lactic acidosis and muscle weakness [90,91]. Recently, Repp et al. published an overview of riboflavin therapy efficacy in ACAD9 deficiency [91]. Out of 70 patients analyzed, 31 were following a riboflavin treatment and 20/31 reported a clinical improvement. Details on individual riboflavin dosage are not reported, but the authors advise 20 mg/kg/d, 200 mg/d maximum. Moreover, a significant increase in complex I activity was observed in fibroblasts derived from 9/15 patients, when the cells were supplemented with riboflavin. Interestingly, all patients responding to riboflavin treatment harbored missense variants, suggesting that a FAD chaperoning effect could be the molecular rationale of riboflavin responsiveness, maybe combined with a stabilizing effect of flavins on complex I and complex IV assembly and supercomplexes formation [90,91].
4.3. Inborn Errors Causing Secondary Impairment of Flavoenzymes
4.3.1. Mitochondrial Complex II Deficiency
Riboflavin treatment has been tested also in patients with mitochondrial complex II (or succinate dehydrogenase, SDH) deficiency (OMIM #252011) [92], but only a small number of patients benefited from the treatment, as reviewed in Jain-Ghai et al. [93]. The three patients benefiting from riboflavin treatment [92] carried a homozygous missense variant in SDHAF1, encoding SDHAF1, which contributes to iron-sulfur cluster incorporation into the B subunit of complex II (SDHB) [94]. The mechanism underlying riboflavin responsiveness in these patients was later explained by Maio et al [95]. Experiments in patients’ cells, demonstrated that impaired SDHB biogenesis resulted in lower SDH subunit A (SDHA) flavinylation and protein level. When the cells were supplemented with riboflavin, SDHA protein level and flavinylation increased, together with complex II activity [95]. These experiments demonstrate that riboflavin supplementation is beneficial and should be attempted in patients with complex II deficiency and SDHAF1 variants.
4.3.2. Ethylmalonic Encephalopathy
Pathogenic variants in ETHE1, encoding the mitochondrial persulfide dioxygenase ETHE1 [96], cause ethylmalonic encephalopathy (EE; OMIM #602473), an extremely rare disorder of mitochondrial sulfur metabolism [97]. The disease manifests neonatally, usually with fatal outcome within the first years of life. Patients present with a complex and diverse range of severe symptoms of encephalopathy, vasculopathy and enteropathy, and a characteristic biochemical profile with tissue specific complex IV deficiency, lactic acidemia and elevated excretion of ethylmalonic acid, thiosulfate, C4- and C5-acylglycines and C4- and C5-acylcarnitines [97,98,99]. The etiology of these biochemical features was more than a decade later revealed to be due to secondary inhibition of complex IV and SCAD by the toxic accumulation of the gasotransmitter hydrogen sulfide [97]. A resemblance of some EE biochemical features with ACADs and complex IV deficiency may have encouraged treatment with riboflavin. While some reports find no effect of riboflavin [98,100,101,102,103,104,105,106,107], a number of reports suggest some level of positive effect [99,108,109,110,111,112]. However, the therapeutic effect of riboflavin is difficult to interpret since most studies have used riboflavin in combination with other vitamins or cofactors. A beneficial effect of riboflavin may be well founded. Prior to the reaction catalyzed by ETHE1, sulfide is oxidized to persulfide in a reaction catalyzed by the flavoenzyme SQOR, where it donates electrons directly to CoQ10 in the ETC [113,114]. Proper function of complex IV, and the ETC in general, is therefore instrumental in the immediate first step of sulfide clearance. The accumulation of sulfide both destabilizes and inhibits the activity of complex IV [115], meanwhile, riboflavin supplementation may aid in counteracting this destabilization by providing flavin cofactors needed for complex IV assembly factor stability [116,117]. Moreover, riboflavin supplementation may also stabilize SCAD, counteracting the destabilization induced by sulfide. Today no curative treatment of EE has been described, but multiple complementary life extending treatments for managing EE are available [118,119,120,121,122]. Treatment with riboflavin can be used in combination with these and may be beneficial.
4.3.3. MT-TL1 Deficiency
MT-TL1, encodes the mitochondrial tRNALeu(UUR). Various clinical outcomes of MT-TL1 variants have been described, ranging from pure myopathy to MELAS syndrome (OMIM #540000), and some patients responding to riboflavin have been reported [123,124,125,126]. An interesting study, by Garrido-Maraver et al. [127], demonstrated that riboflavin or CoQ10 supplementation rescued ETC function in fibroblasts obtained from a MELAS patient with a heteroplasmic variant in MT-TL1. Supplementation with either riboflavin or CoQ10 resulted in a significant increase in ATP levels and respiratory chain function, and a considerable decrease in superoxide production and mitochondrial degradation. Beneficial effects of riboflavin and CoQ10 supplementation were observed in MELAS cybrids with the same variant. The beneficial effects of riboflavin or CoQ10 supplementation can be most likely attributed to their antioxidant activity, which prevents ROS production and mitophagy. The results obtained by Garrido-Maraver et al. are very promising, but clinical trials are needed to confirm their in vitro studies.
4.4. Disorders of Flavin Transportation and Biosynthesis
4.4.1. Riboflavin Transporter Deficiencies
Riboflavin transporter deficiency, which includes Brown-Vialetto-Van Laere syndrome (BVVLS1; OMIM #211530 and BVVLS2; OMIM #614707) and Fazio-Londe disease (OMIM #211500), is caused by variants in SLC52A2 and/or SLC52A3, which encode the human riboflavin transporters RFVT2 and RFVT3, respectively. Even if some clinical differences in patients with SLC52A2 or SLC52A3 variants have been reported, the most frequently observed phenotype is a progressive peripheral and cranial neuropathy with vision loss, deafness, sensory ataxia, muscle weakness and respiratory compromise [128]. Some patients show a MADD-like acyl-carnitine profile, reflecting that ETF/ETF-QO and the associated ACADs all depend on riboflavin for proper function [129]. Riboflavin supplementation (10–80 mg/kg/d) is widely used to treat riboflavin transporter deficiencies and significantly improves or stabilizes the phenotype of patients, especially if started as soon as symptoms arise [128], but the molecular basis of riboflavin responsiveness is still unknown. A possible explanation could be that the expression of RFVTs is overlapping in most tissues and if one transporter is not functioning properly, the others can take over.
Variants in SLC52A1, which encodes RFVT1, highly expressed in the placenta, have been reported in two pregnant women, causing transient MADD (OMIM #615026) in their infants [130,131,132]. Both women had a complicated pregnancy with hyperemesis gravidarum. The first woman carried a heterozygous deletion in SLC52A1, which results in a complete disruption of SLC52A1 expression in the affected allele [130,131]. Biochemical characterization of the woman revealed a mild MADD profile, but no clinical symptoms were detected. At birth, her infant similarly showed a MADD-like biochemical profile and complex neurological symptoms, which were fully normalized shortly after riboflavin treatment (100 mg/d). The clinical and biochemical abnormalities did not reappear after riboflavin treatment discontinuation and genetic analyses did not reveal the SLC52A1 deletion found in the mother. During the second pregnancy, the woman was supplemented with riboflavin and the second infant did not experience transient MADD.
In 2017, Mosegaard et al., reported a second case [132]. A heterozygous SLC52A1 intronic variant (c.1134 + 11G>A) was detected both in the mother and in the infant. In vitro studies demonstrated that the variant creates a binding site for the splice inhibitory hnRNP A1 protein, causing exon 4 skipping and most likely nonsense-mediated decay (NMD) of the resulting transcript. At birth, the infant harbored a MADD-like clinical and biochemical phenotype which was corrected with vitamin and carnitine supplementation, including riboflavin (100 mg two times a day), and a low fat and protein diet. The riboflavin treatment was discontinued at 2.5 years of age with no reappearance of MADD metabolites or symptoms, confirming the transient phenotype. The mother showed neither MADD biochemistry nor clinical symptoms, but borderline low FAD/riboflavin levels were detected in her blood shortly after parturition. These two case reports show that low placental transport of riboflavin due to RFVT1 haploinsufficiency, can cause transient MADD in newborns that can be easily corrected, and prevented, by riboflavin supplementation. Catabolic stress, induced by hyperemesis gravidarum, seems to be an important risk factor.
4.4.2. Mitochondrial Folate Transporter Deficiency
Another disorder of flavin transport that can be easily corrected by riboflavin supplementation is riboflavin-responsive exercise intolerance (RREI; OMIM #616839), caused by inborn genetic errors in SLC25A32, encoding MFT. Two patients harboring SLC25A32 variants have been reported up to now. The first patient was reported in 2016 by Schiff et al. [133]. The 14-year-old girl experienced recurrent exercise intolerance and biochemical analyses showed a MADD-like profile, reflecting ETF/ETF-QO and/or ACADs dysfunction. Skeletal-muscle biopsy revealed ragged-red fibers and lipid storage together with a faint succinate dehydrogenase staining. Genetic analyses revealed two heterozygous variants in SLC25A32, one missense variant and one nonsense variant. A second patient was described in 2017 by Hellebrekers et al. The 52-year-old patient experienced ataxia, myoclonia, dysarthria, muscle weakness and exercise intolerance [134]. Similar to Schiff et al., the patient showed a MADD-like biochemical profile and impaired ETC function. Genetic analyses revealed a homozygous deletion-insertion in SLC25A32, which comprehended the start codon. Both patients were treated with riboflavin, which improved their phenotype. Riboflavin supplementation most probably restores mitochondrial FAD levels, but the mechanisms are unclear, especially for the patient harboring a homozygous LOF variant, which is not easily explained unless disrupted mitochondrial FAD import is compensated by mitochondrial FAD synthesis or other mechanisms ensuring mitochondrial FAD supply.
4.4.3. FAD Synthase Deficiency
Riboflavin supplementation is currently used as a treatment also in lipid storage myopathy due to flavin adenine dinucleotide synthetase deficiency (LSMFLAD; OMIM #255100), caused by pathogenic variants in FLAD1, the gene encoding FADS [9]. FADS is a bi-functional enzyme with a N-terminal molybdopterin-binding domain (MPTb) and a C-terminal 3′phosphoadenosine 5′phosphosulfate reductase domain (PAPS). The PAPS domain is able, per se, to catalyze FAD synthesis [135]. Two isoforms, a mitochondrial (FADS1) and a cytosolic (FADS2) one, have been described in detail, and a shorter isoform (FADS6), which contains the sole PAPS domain, has been recently characterized at the protein level and shown to have FAD synthase activity [6,136]. In 2016, FLAD1 variants were outlined in nine individuals with a MADD-like biochemical profile [9,137]. Six novel patients have been reported in the last four years and new patients are under investigation [138,139,140,141,142]. The phenotype of patients is extremely heterogeneous, ranging from early-onset and severe cardiac and respiratory insufficiency, often with a poor prognosis, to late-onset myopathies. Interestingly, patients with biallelic LOF variants in the MPTb domain (exon 2) harbored residual FADS activity and a considerably high cellular FAD level [9,139]. The expression of FADS6, encoded by a downstream ATG in exon 2, could be a possible explanation [9,136]. ETF, ETF-QO, ACADs and ETC decompensation have been observed in the patients at various extent, as a direct consequence of FAD depletion [9,138,139,142]. Interestingly, in LSMFLAD patients both FMN (FADS substrate) and FAD (FADS product) are decreased [9,139]. This suggests that FADS dysfunction could exert a negative feedback control on either RFK or RFVT2, but the molecular mechanisms are unknown [9,139]. FAD, FMN and riboflavin depletion has been observed in C. elegans with silenced flad1 gene (FLAD1 orthologue), further supporting this hypothesis [143].
Riboflavin supplementation has shown to be beneficial in patients harboring mild missense variants in the PAPS domain and a mild myopathic phenotype [9,137,144], while it has no or limited effect in patients with biallelic LOF variants and a severe phenotype [139,140,141,144]. The molecular rationale of riboflavin responsiveness is unclear. In patients with mild missense variants, riboflavin supplementation could rescue folding stability of mutant FADS and/or counteract the negative feedback control on RFK/RFVT2 [9,139]. Boosting FADS6 activity in patients with LOF variants in the MPTb domain could be a possible therapeutic approach in the future [136].

Table 1.
Overview of riboflavin-responsive disorders.
Table 1.
Overview of riboflavin-responsive disorders.
| Disorder | OMIM# | Gene(s) (Gene ID) | Clinical Response | Biochemical Response | References |
|---|---|---|---|---|---|
| Multiple Acyl-CoA Dehydrogenase Deficiency (MADD) | #231680 | ETFA (2108) ETFB (2109) ETFDH (2110) | Several studies documenting complete or partial recovery in patients with mild missense variants. Not effective in patients with more severe missense variants or biallelic LOF variants | Significant reduction or normalization of multiple urinary organic acids and blood acylcarnitine excretion. Significant increases in ETF-QO protein or fatty acid oxidation flux in patients’ cultured fibroblasts upon supplementation with riboflavin | [75,82] |
| Short-Chain Acyl-CoA Dehydrogenase Deficiency (SCADD) | #201470 | ACADS (35) | Two single studies; indications of clinical improvement in 4/16 patients. The four patients were compound heterozygous for c.625G>A susceptibility variant and a rare missense variant. A clear clinical improvement reported in one patient with homozygous c.625G>A susceptibility variant | All patients clinically responding to riboflavin treatment had decreased or normalized urinary organic acids excretion and/or decreased butyrylcarnitine in blood. One patient responded biochemically but not clinically | [83,87] |
| Medium-Chain Acyl-CoA Dehydrogenase Deficiency (MCADD) | #201450 | ACADM (34) | NR | Two studies; significant increase of MCAD enzyme activity in cultured lymphocytes from 5/5 patients. The genetic characterization of the patients was not reported. In a study of 17 patients with ACADM missense variants, patients supplemented with riboflavin and carnitine (6) did not display blood markers of oxidative damage when compared with carnitine supplemented (7) and non-supplemented (14) patients | [88,89] |
| ACAD9 Deficiency or Mitochondrial Complex I Deficiency Nuclear type 20 (MC1DN20) | #611126 | ACAD9 (28976) | Several studies. In one comprehensive study, riboflavin treatment resulted in clinical improvement in 20/31 patients. Responsive patients carried missense variants | Significant increase of complex I activity was seen upon supplementation with riboflavin in fibroblasts derived from 9/15 patients | [91] |
| Mitochondrial Complex II Deficiency | #252011 | SDHAF1 (612848) | Riboflavin treatment resulted in a clinical improvement in 3 patients | Plasma lactate, pyruvate and alanine remained within the control range in two patients. In a third patient, plasma lactate was elevated and normalized after riboflavin treatment | [92,94,95] |
| Ethylmalonic Encephalopathy (EE) | #602473 | ETHE1 (23474) | Riboflavin treatment, in combination with other vitamins and CoQ10, has in a few cases been seen to slightly reduce symptoms. A single study has reported a partial effect of riboflavin alone | Clear biochemical improvement of blood acylcarnitines has been reported using riboflavin in combination with other vitamins, CoQ10, and NAC. A single study has showed some improvements upon treatment with riboflavin alone | [109,111,112,121] |
| Brown-Vialetto-Van Laere Syndrome 1 and 2 (BVVLS1; BVVLS2), Fazio-Londe Disease | #211530 #614707 #615026 | SLC52A3 (113278) SLC52A2 (79581) | Several studies; clinical improvement or stabilization of symptoms observed in almost all patients treated with riboflavin | Some patients have shown abnormalities in blood acylcarnitines, urine organic acids, and plasma flavin content that were improved by riboflavin treatment | [128,129,145] |
| Transient MADD or Riboflavin Deficiency | #615026 | SLC52A1 (607883) | Two cases; clear clinical improvement of both neonates | Biochemical normalization of blood acyl-carnitines and/or urine organic acids in both mothers and neonates | [130,131,132] |
| Mitochondrial Folate Transporter Deficiency or Riboflavin-Responsive Exercise Intolerance (RREI) | #616839 | SLC25A32 (81034) | Clinical improvement in 2/2 patients | NR | [133,134] |
| Lipid Storage Myopathy due to Flavin Adenine Dinucleotide Synthetase Deficiency (LSMFLAD) | #255100 | FLAD1 (80308) | Several cases; clinical improvement in patients harboring missense variants. Patients with biallelic LOF variants have shown only a transient clinical improvement or an alleviation of symptoms | Some decreases in acylcarnitine species and normalization of urine organic acids in patients harboring missense variants | [9,88,137,139,141,144] |
Abbreviations: CoQ10, Coenzyme Q10; LOF, loss of function; NAC, N-acetylcysteine; NR, not reported.
5. Conclusions and Perspectives for Research and Clinical Managements of IEM
Riboflavin deficiency has implications for human health due to the key role of its derivatives, FMN and FAD, in various essential cellular functions and physiological conditions, as described above. Whilst clinical riboflavin deficiency is rare in developed countries, riboflavin insufficiency and subclinical riboflavin deficiency is widespread and could give rise to clinical manifestations when combined with an increased need of riboflavin due to physiological conditions like pregnancy, exercise, infections, or due to genetic disorders of flavin biosynthesis and function.
Since the first riboflavin-responsive metabolic defect was described in 1982 as a mild variant form of MADD [146], an increasing number of riboflavin-responsive IEM have been identified and characterized. The response to riboflavin has been explained by the ability of riboflavin to compensate for genetic deficiencies in riboflavin transport or cofactor synthesis [9,131,133,147,148], or by the ability of riboflavin-derived cofactors to improve folding and stability of individually mutated flavoproteins [9,65,69,149]. In addition, ETFDH and FLAD1 variants could induce secondary disturbances of cellular flavin homeostasis, potentially creating a vicious cycle of impaired mitochondrial electron transfer [9,69,77,143], whose exact mechanism is still under investigation.
Riboflavin deficiency induced by pregnancy and/or low vitamin supply can reveal milder genotypes or carriers, as described for transient MADD [130,132], where both women displayed hyperemesis gravidarum and an increased risk of low nutrient and vitamin supply. Moreover, one of the reported SLC52A1 variants (c.1134 + 11G>A) is a mild splice variant with a rather high allele frequency in the general population (MAF 0.2%). This variant could represent a risk factor for transient MADD in newborns when combined with insufficient riboflavin supply during pregnancy. Similar, heterozygous ETFDH variants were recently reported in two elderly patients with stress-induced myopathy and lack of a previous history of myopathy. The authors speculate that non-genetic factors, such as statin therapy and hypothyroidism, could have acted as precipitating factors and thereby triggered the clinical symptoms [150]. Nevertheless, the clinical symptoms and biochemical defects fully reversed after treatment with riboflavin and carnitine. This, combined with a decreased efficiency of riboflavin absorbance or intake in the elderly population [3,34,35,36,37], could further support the role of disturbed riboflavin supply in the pathogenesis of the disease. Additional cases of clinically manifesting ETFDH carriers, highly responsive to riboflavin therapy, are known to the authors of this paper and may represent an under-recognized phenomenon.
The survival rate of patients with IEM has remarkably improved in the last 40 years due to better diagnosis and treatment options, including the implementation of expanded newborn screening programs. However, elderly people and the majority of women that are currently in their childbearing age, have been born prior to the expanded newborn screening programs. It is also not known to what extent diagnostic metabolites in newborns with riboflavin-responsive genetic variants are initially blurred or masked by transfer of maternally derived riboflavin. The clear benefits, and in some cases lifesaving effects, of high dose riboflavin therapy makes these conditions important to be aware of and to recognize and treat accordingly. A wider use of metabolic and extended genetic screening in suspected cases, together with improved knowledge on the complex gene-environmental interactions that control cellular riboflavin homeostasis, as discussed in the present review, will hopefully add to this.
Author Contributions
Writing—Original draft preparation, visualization and editing, S.M. and G.D.; writing—Review and editing, P.B. and J.C.; supervision and editing, N.G.; writing—0riginal draft preparation, editing and supervision, R.K.J.O. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by The Lundbeck Foundation PhD Scholarship R263-2017-4384 (SM), and Aarhus County Research Initiative.
Acknowledgments
We would like to acknowledge patients and colleagues, who over the years have provided samples and important data and scientific discussions to improve our understanding of the complex interactions between riboflavin and IEM, and to Michele Galluccio and Maria Barile for the invitation to write this review.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ACAD | Acyl-CoA Dehydrogenase |
| AIF | Apoptosis Inducing Factor |
| AMID | Mitochondrion-Associated Inducer of Death |
| CHDH | Choline Dehydrogenase |
| CoQ10 | Coenzyme Q10 |
| CytC | Cytochrome C |
| DMGDH | Dimethylglycine Dehydrogenase |
| ETC | Electron Transport Chain |
| ETHE1 | Ethylmalonic Encephalopathy Protein 1 |
| ETF | Electron Transfer Flavoprotein |
| ETF-QO | Electron Transfer Flavoprotein-ubiquinone Oxidoreductase |
| FAD | Flavin Adenine Dinucleotide |
| FMN | Flavin Mononucleotide |
| GCDH | Glutaryl-CoA Dehydrogenase |
| GPDM | Mitochondrial Glycerol-3-phosphate Dehydrogenase |
| HSP | Heat Shock Protein |
| IBD | Isobutyryl-CoA Dehydrogenase |
| IEM | Inborn Errors of Metabolism |
| IL | Interleukin |
| IVD | Isovaleryl-CoA Dehydrogenase |
| LCAD | Long-Chain Acyl-CoA Dehydrogenase |
| LOF | Loss of Function |
| LSD1 | Lysine-specific Demethylase 1 |
| MADD | Multiple Acyl-CoA Dehydrogenation Deficiency |
| MCAD | Medium-Chain Acyl-CoA Dehydrogenase |
| MFT | Mitochondrial Folate Transporter |
| MFTHR | Methylenetetrahydrofolate Reductase |
| MPTb | Molybdopterin-binding Domain |
| MTRR | Methionine Synthase Reductase |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NMD | Nonsense-mediated Decay |
| NOX | NADPH oxidases |
| PNPO | Pyridoxine 5´-phosphate Oxidase |
| RFK | Riboflavin Kinase |
| RFVT | Riboflavin Transporter |
| ROS | Reactive Oxygen Species |
| SARDH | Sarcosine Dehydrogenase |
| SBCAD | Short/Branched-Chain Acyl-CoA Dehydrogenase |
| SCAD | Short-Chain Acyl-CoA Dehydrogenase |
| SLC | Solute Carrier |
| SQOR | Sulfide:Quinone Oxidoreductase |
| TNF-α | Tumor Necrosis Factor Alpha |
| VLCAD | Very Long-Chain Acyl-CoA Dehydrogenase |
References
- Auclair, O.; Han, Y.; Burgos, S.A. Consumption of Milk and Alternatives and Their Contribution to Nutrient Intakes among Canadian Adults: Evidence from the 2015 Canadian Community Health Survey-Nutrition. Nutrients 2019, 11, 1948. [Google Scholar] [CrossRef] [PubMed]
- Thakur, K.; Tomar, S.K.; De, S. Lactic acid bacteria as a cell factory for riboflavin production. Microb. Biotechnol. 2016, 9, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Powers, H.J. Riboflavin (vitamin B-2) and health. Am. J. Clin. Nutr. 2003, 77, 1352–1360. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, A.V. Riboflavin metabolism—Relevance to human nutrition. Indian J. Med. Res. 1998, 108, 182–190. [Google Scholar] [PubMed]
- Spaan, A.N.; Ijlst, L.; Van Roermund, C.W.T.; Wijburg, F.A.; Wanders, R.J.A.; Waterham, H.R. Identification of the human mitochondrial FAD transporter and its potential role in multiple acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 2005, 86, 441–447. [Google Scholar] [CrossRef]
- Brizio, C.; Galluccio, M.; Wait, R.; Torchetti, E.M.; Bafunno, V.; Accardi, R.; Gianazza, E.; Indiveri, C.; Barile, M. Over-expression in Escherichia coli and characterization of two recombinant isoforms of human FAD synthetase. Biochem. Biophys. Res. Commun. 2006, 344, 1008–1016. [Google Scholar] [CrossRef]
- Torchetti, E.M.; Brizio, C.; Colella, M.; Galluccio, M.; Giancaspero, T.A.; Indiveri, C.; Roberti, M.; Barile, M. Mitochondrial localization of human FAD synthetase isoform 1. Mitochondrion 2010, 10, 263–273. [Google Scholar] [CrossRef]
- Barile, M.; Giancaspero, T.A.; Leone, P.; Galluccio, M.; Indiveri, C. Riboflavin transport and metabolism in humans. J. Inherit. Metab. Dis. 2016, 39, 545–557. [Google Scholar] [CrossRef]
- Olsen, R.K.J.; Koňaříková, E.; Giancaspero, T.A.; Mosegaard, S.; Boczonadi, V.; Mataković, L.; Veauville-Merllié, A.; Terrile, C.; Schwarzmayr, T.; Haack, T.B.; et al. Riboflavin-Responsive and -Non-Responsive Mutations in FAD Synthase Cause Multiple Acyl-CoA Dehydrogenase and Combined Respiratory-Chain Deficiency. Am. J. Hum. Genet. 2016, 98, 1130–1145. [Google Scholar] [CrossRef]
- Schoenen, J.; Lenaerts, M.; Bastings, E. High-dose riboflavin as a prophylactic treatment of migraine: Results of an open pilot study. Cephalalgia 1994, 14, 328–329. [Google Scholar] [CrossRef]
- Hoppel, C.L.; Tandler, B. Riboflavin and Mouse Hepatic Cell Structure and Function. Mitochondrial Oxidative Metabolism in Severe Deficiency States. J. Nutr. 1975, 105, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Lienhart, W.D.; Gudipati, V.; MacHeroux, P. The human flavoproteome. Arch. Biochem. Biophys. 2013, 535, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.T.; Cooper, A.J.L. From Cholesterogenesis to Steroidogenesis: Role of Riboflavin and Flavoenzymes in the Biosynthesis of Vitamin D. Adv. Nutr. 2014, 5, 144–163. [Google Scholar] [CrossRef] [PubMed]
- Kodali, V.K.; Thorpe, C. Oxidative protein folding and the quiescin-sulfhydryl oxidase family of flavoproteins. Antioxidants Redox Signal. 2010, 13, 1217–1230. [Google Scholar] [CrossRef] [PubMed]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Bano, D.; Prehn, J.H.M. Apoptosis-Inducing Factor (AIF) in Physiology and Disease: The Tale of a Repented Natural Born Killer. EBioMedicine 2018, 30, 29–37. [Google Scholar] [CrossRef]
- Marshall, K.R.; Gong, M.; Wodke, L.; Lamb, J.H.; Jones, D.J.L.; Farmer, P.B.; Scrutton, N.S.; Munro, A.W. The human apoptosis-inducing protein AMID is an oxidoreductase with a modified flavin cofactor and DNA binding activity. J. Biol. Chem. 2005, 280, 30735–30740. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochromes P450, drugs, and diseases. Mol. Interv. 2003, 3, 194–204. [Google Scholar] [CrossRef]
- Gu, F.; Lin, Y.; Wang, Z.; Wu, X.; Ye, Z.; Wang, Y.; Lan, H. Biological roles of LSD1 beyond its demethylase activity. Cell. Mol. Life Sci. 2020, 1–10. [Google Scholar] [CrossRef]
- Refsum, H.; Ueland, P.M.; Nygard, O.; Vollset, S.E. Homocysteine and cardiovascular disease. Annu. Rev. Med. 1998, 49, 31–62. [Google Scholar] [CrossRef]
- Škovierová, H.; Vidomanová, E.; Mahmood, S.; Sopková, J.; Drgová, A.; Červeňová, T.; Halašová, E.; Lehotský, J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int. J. Mol. Sci. 2016, 17, 1733. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Wong, P.W.; Susmano, A.; Sora, J.; Norusis, M.; Ruggie, N. Thermolabile methylenetetrahydrofolate reductase: an inherited risk factor for coronary artery disease. Am. J. Hum. Genet. 1991, 48, 536–545. [Google Scholar] [PubMed]
- McNulty, H.; Dowey, L.R.C.; Strain, J.J.; Dunne, A.; Ward, M.; Molloy, A.M.; McAnena, L.B.; Hughes, J.P.; Hannon-Fletcher, M.; Scott, J.M. Riboflavin lowers homocysteine in individuals homozygous for the MTHFR 677C->T polymorphism. Circulation 2006, 113, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Shane, B. Folate and vitamin B12 metabolism: overview and interaction with riboflavin, vitamin B6, and polymorphisms. Food Nutr. Bull. 2008, 29, S5–S16. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, J.G.; Laino, J.E.; del Valle, M.J.; Vannini, V.; van Sinderen, D.; Taranto, M.P.; de Valdez, G.F.; de Giori, G.S.; Sesma, F. B-group vitamin production by lactic acid bacteria—Current knowledge and potential applications. J. Appl. Microbiol. 2011, 111, 1297–1309. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, M.M.; Kiely, M.; Harrington, K.E.; Robson, P.J.; Strain, J.J.; Flynn, A. The North/South Ireland Food Consumption Survey: vitamin intakes in 18-64-year-old adults. Public Health Nutr. 2001, 4, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.J.B.; Suchindran, C.M.; Roggenkamp, K.J. Micronutrient intakes in two US populations of older adults: lipid research clinics program prevalence study findings. J. Nutr. Health Aging 2009, 13, 595–600. [Google Scholar] [CrossRef]
- Powers, H.J.; Hill, M.H.; Mushtaq, S.; Dainty, J.R.; Majsak-Newman, G.; Williams, E.A. Correcting a marginal riboflavin deficiency improves hematologic status in young women in the United Kingdom (RIBOFEM). Am. J. Clin. Nutr. 2011, 93, 1274–1284. [Google Scholar] [CrossRef]
- Preziosi, P.; Galan, P.; Deheeger, M.; Yacoub, N.; Drewnowski, A.; Hercberg, S. Breakfast type, daily nutrient intakes and vitamin and mineral status of French children, adolescents, and adults. J. Am. Coll. Nutr. 1999, 18, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Smithers, G.; Gregory, J.R.; Bates, C.J.; Prentice, A.; Jackson, L.V.; Wenlock, R. The National Diet and Nutrition Survey: young people aged 4–18 years. Nutr. Bull. 2000, 25, 105–111. [Google Scholar] [CrossRef]
- Waldmann, A.; Koschizke, J.W.; Leitzmann, C.; Hahn, A. Dietary intakes and lifestyle factors of a vegan population in Germany: results from the German Vegan Study. Eur. J. Clin. Nutr. 2003, 57, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Larsson, C.L.; Johansson, G.K. Dietary intake and nutritional status of young vegans and omnivores in Sweden. Am. J. Clin. Nutr. 2002, 76, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Majchrzak, D.; Singer, I.; Manner, M.; Rust, P.; Genser, D.; Wagner, K.-H.; Elmadfa, I. B-vitamin status and concentrations of homocysteine in Austrian omnivores, vegetarians and vegans. Ann. Nutr. Metab. 2006, 50, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Fanelli, M.T.; Woteki, C.E. Nutrient intakes and health status of older Americans. Data from the NHANES II. Ann. N. Y. Acad. Sci. 1989, 561, 94–103. [Google Scholar] [CrossRef]
- Bates, C.J.; Prentice, A.; Cole, T.J.; van der Pols, J.C.; Doyle, W.; Finch, S.; Smithers, G.; Clarke, P.C. Micronutrients: highlights and research challenges from the 1994-5 National Diet and Nutrition Survey of people aged 65 years and over. Br. J. Nutr. 1999, 82, 7–15. [Google Scholar] [CrossRef]
- Flynn, A.; Moreiras, O.; Stehle, P.; Fletcher, R.J.; Muller, D.J.G.; Rolland, V. Vitamins and minerals: A model for safe addition to foods. Eur. J. Nutr. 2003, 42, 118–130. [Google Scholar] [CrossRef]
- Boisvert, W.A.; Mendoza, I.; Castaneda, C.; De Portocarrero, L.; Solomons, N.W.; Gershoff, S.N.; Russell, R.M. Riboflavin requirement of healthy elderly humans and its relationship to macronutrient composition of the diet. J. Nutr. 1993, 123, 915–925. [Google Scholar] [CrossRef]
- Mikkelsen, K.; Stojanovska, L.; Tangalakis, K.; Bosevski, M.; Apostolopoulos, V. Cognitive decline: A vitamin B perspective. Maturitas 2016, 93, 108–113. [Google Scholar] [CrossRef]
- Mikkelsen, K.; Stojanovska, L.; Apostolopoulos, V. The Effects of Vitamin B in Depression. Curr. Med. Chem. 2016, 23, 4317–4337. [Google Scholar] [CrossRef]
- Seidl, S.E.; Santiago, J.A.; Bilyk, H.; Potashkin, J.A. The emerging role of nutrition in Parkinson’s disease. Front. Aging Neurosci. 2014, 6, 36. [Google Scholar] [CrossRef]
- Synofzik, M.; Schule, R. Overcoming the divide between ataxias and spastic paraplegias: Shared phenotypes, genes, and pathways. Mov. Disord. 2017, 32, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Udhayabanu, T.; Manole, A.; Rajeshwari, M.; Varalakshmi, P.; Houlden, H.; Ashokkumar, B. Riboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Clin. Med. 2017, 6, 52. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.J.; Satyanarayana, K.; Bamji, M.S.; Jacob, C.M.; Ramana, Y.V.; Rao, S.S. The effect of exercise on the riboflavin status of adult men. Br. J. Nutr. 1993, 69, 541–551. [Google Scholar] [CrossRef]
- Rodriguez, N.R.; Di Marco, N.M.; Langley, S. American College of Sports Medicine position stand. Nutrition and athletic performance. Med. Sci. Sports Exerc. 2009, 41, 709–731. [Google Scholar] [CrossRef] [PubMed]
- Burke, B.S.; Beal, V.A.; Kirkwood, S.B.; Stuart, H.C. The Influence of Nutrition During Pregnancy Upon the Condition of the Infant at Birth. J. Nutr. 1943, 26, 569–583. [Google Scholar] [CrossRef]
- Natraj, U.; Kumar R, A.; Kadam, P. Termination of pregnancy in mice with antiserum to chicken riboflavin-carrier protein. Biol. Reprod. 1987, 36, 677–685. [Google Scholar] [CrossRef]
- White, H.B.; Merrill, A.H.J. Riboflavin-binding proteins. Annu. Rev. Nutr. 1988, 8, 279–299. [Google Scholar] [CrossRef]
- King, J.C. Physiology of pregnancy and nutrient metabolism. Am. J. Clin. Nutr. 2000, 71, 1218S–1225S. [Google Scholar] [CrossRef]
- Herrera, E.; Desoye, G. Maternal and fetal lipid metabolism under normal and gestational diabetic conditions. Horm. Mol. Biol. Clin. Investig. 2016, 26, 109–127. [Google Scholar] [CrossRef]
- Bates, C.J.; Prentice, A.M.; Paul, A.A.; Sutcliffe, B.A.; Watkinson, M.; Whitehead, R.G. Riboflavin status in Gambian pregnant and lactating women and its implications for Recommended Dietary Allowances. Am. J. Clin. Nutr. 1981, 34, 928–935. [Google Scholar] [CrossRef]
- Bohles, H. Antioxidative vitamins in prematurely and maturely born infants. Int. J. Vitam. Nutr. Res. 1997, 67, 321–328. [Google Scholar] [PubMed]
- Gromisch, D.S.; Lopez, R.; Cole, H.S.; Cooperman, J.M. Light (phototherapy)-induced riboflavin deficiency in the neonate. J. Pediatr. 1977, 90, 118–122. [Google Scholar] [CrossRef]
- Allen, L.H. B vitamins in breast milk: Relative importance of maternal status and intake, and effects on infant status and function. Adv. Nutr. 2012, 3, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Suwannasom, N.; Kao, I.; Pruss, A.; Georgieva, R.; Baumler, H. Riboflavin: The Health Benefits of a Forgotten Natural Vitamin. Int. J. Mol. Sci. 2020, 21, 950. [Google Scholar] [CrossRef] [PubMed]
- Mazur-Bialy, A.I.; Pochec, E.; Plytycz, B. Immunomodulatory effect of riboflavin deficiency and enrichment-reversible pathological response versus silencing of inflammatory activation. J. Physiol. Pharmacol. 2015, 66, 793–802. [Google Scholar]
- Angajala, A.; Lim, S.; Phillips, J.B.; Kim, J.-H.; Yates, C.; You, Z.; Tan, M. Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Front. Immunol. 2018, 9, 1605. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef]
- Toyosawa, T.; Suzuki, M.; Kodama, K.; Araki, S. Effects of intravenous infusion of highly purified vitamin B2 on lipopolysaccharide-induced shock and bacterial infection in mice. Eur. J. Pharmacol. 2004, 492, 273–280. [Google Scholar] [CrossRef]
- Toyosawa, T.; Suzuki, M.; Kodama, K.; Araki, S. Potentiation by amino acid of the therapeutic effect of highly purified vitamin B2 in mice with lipopolysaccharide-induced shock. Eur. J. Pharmacol. 2004, 493, 177–182. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef]
- Gomes Faísca, P.F.N.C.M. Protein Folding—An Introduction; Springer International Publishing: Cham, Switzerland, 2019; ISBN 978-3-319-00882-0. [Google Scholar]
- Nagao, M.; Tanaka, K. FAD-dependent regulation of transcription, translation, post-translational processing, and post-processing stability of various mitochondrial acyl-CoA dehydrogenases and of electron transfer flavoprotein and the site of holoenzyme formation. J. Biol. Chem. 1992, 267, 17925–17932. [Google Scholar] [PubMed]
- Saijo, T.; Tanaka, K. Isoalloxazine ring of FAD is required for the formation of the core in the Hsp60-assisted folding of medium chain Acyl-CoA dehydrogenase subunit into the assembly competent conformation in mitochondria. J.Biol.Chem. 1995, 270, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Giancaspero, T.A.; Colella, M.; Brizio, C.; Difonzo, G.; Fiorino, G.M.; Leone, P.; Brandsch, R.; Bonomi, F.; Iametti, S.; Barile, M. Remaining challenges in cellular flavin cofactor homeostasis and flavoprotein biogenesis. Front. Chem. 2015, 3, 30. [Google Scholar] [CrossRef] [PubMed]
- Henriques, B.J.; Rodrigues, J.V.; Olsen, R.K.; Bross, P.; Gomes, C.M. Role of flavinylation in a mild variant of multiple acyl-CoA dehydrogenation deficiency: A molecular rationale for the effects of riboflavin supplementation. J.Biol.Chem. 2009, 284, 4222–4229. [Google Scholar] [CrossRef] [PubMed]
- Lucas, T.G.; Henriques, B.J.; Rodrigues, J.V.; Bross, P.; Gregersen, N.; Gomes, C.M. Cofactors and metabolites as potential stabilizers of mitochondrial acyl-CoA dehydrogenases. Biochim. Biophys. Acta 2011, 1812, 1658–1663. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.; Corydon, T.J.; Andresen, B.S.; Jørgensen, M.M.; Bolund, L.; Gregersen, N. Protein misfolding and degradation in genetic diseases. Hum. Mutat. 1999, 14, 186–198. [Google Scholar] [CrossRef]
- Henriques, B.J.; Fisher, M.T.; Bross, P.; Gomes, C.M. A polymorphic position in electron transfer flavoprotein modulates kinetic stability as evidenced by thermal stress. FEBS Lett. 2011, 585, 505–510. [Google Scholar] [CrossRef][Green Version]
- Cornelius, N.; Frerman, F.E.; Corydon, T.J.; Palmfeldt, J.; Bross, P.; Gregersen, N.; Olsen, R.K.J. Molecular mechanisms of riboflavin responsiveness in patients with ETF-QO variations and multiple acyl-CoA dehydrogenation deficiency. Hum. Mol. Genet. 2012, 21, 3435–3448. [Google Scholar] [CrossRef]
- Er, T.-K.; Chen, C.-C.; Liu, Y.-Y.; Chang, H.-C.; Chien, Y.-H.; Chang, J.-G.; Hwang, J.-K.; Jong, Y.-J. Computational analysis of a novel mutation in ETFDH gene highlights its long-range effects on the FAD-binding motif. BMC Struct. Biol. 2011, 11, 43. [Google Scholar] [CrossRef]
- Lucas, T.G.; Henriques, B.J.; Gomes, C.M. Conformational analysis of the riboflavin-responsive ETF:QO-p.Pro456Leu variant associated with mild multiple acyl-CoA dehydrogenase deficiency. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140393. [Google Scholar] [CrossRef]
- Gregersen, N.; Wintzensen, H.; Kølvraa, S.; Christensen, E.; Christensen, M.F.; Brandt, N.J.; Rasmussen, K. C6-c10-dicarboxylic aciduria: Investigations of a patient with riboflavin responsive multiple acyl- CoA dehydrogenation defects. Pediatr. Res. 1982, 16, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Lund, M.; Olsen, R.K.J.; Gregersen, N. A short introduction to acyl-CoA dehydrogenases; deficiencies and novel treatment strategies. Expert Opin. Orphan Drugs 2015, 3, 1375–1386. [Google Scholar] [CrossRef]
- Frerman, F.E.; Goodman, S.I. Defects of Electron Transfer Flavoprotein and Electron Transfer Flavoprotein-Ubiquinone Oxidoreductase: Glutaric Acidemia Type II. In The Metabolic and Molecular Basis of Inherited Diseases; Valle, D., Beaudet, A.L., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A., Gibson, K.M.G., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 2357–2365. [Google Scholar]
- Olsen, R.K.J.; Olpin, S.E.; Andresen, B.S.; Miedzybrodzka, Z.H.; Pourfarzam, M.; Merinero, B.; Frerman, F.E.; Beresford, M.W.; Dean, J.C.S.; Cornelius, N.; et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007, 130, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, L.A.; He, M.; Vockley, J.; Payne, N.; Rhead, W.; Hoppel, C.; Spector, E.; Gernert, K.; Gibson, K.M. Novel ETF dehydrogenase mutations in a patient with mild glutaric aciduria type II and complex II-III deficiency in liver and muscle. J. Inherit. Metab. Dis. 2010, 33. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Li, D.; Lv, J.; Xu, X.; Wen, B.; Lin, P.; Liu, F.; Ji, K.; Shan, J.; Li, H.; et al. ETFDH Mutations and Flavin Adenine Dinucleotide Homeostasis Disturbance Are Essential for Developing Riboflavin-Responsive Multiple Acyl-Coenzyme A Dehydrogenation Deficiency. Ann. Neurol. 2018, 84, 659–673. [Google Scholar] [CrossRef]
- Rhead, W.; Roettger, V.; Marshall, T.; Amendt, B. Multiple acyl-coenzyme A dehydrogenation disorder responsive to riboflavin: Substrate oxidation, flavin metabolism, and flavoenzyme activities in fibroblasts. Pediatr. Res. 1993, 33, 129–135. [Google Scholar] [CrossRef]
- Amendt, B.A.; Rhead, W.J. The multiple acyl-coenzyme A dehydrogenation disorders, glutaric aciduria type II and ethylmalonic-adipic aciduria. Mitochondrial fatty acid oxidation, acyl-coenzyme A dehydrogenase, and electron transfer flavoprotein activities in fibroblasts. J. Clin. Invest. 1986, 78, 205–213. [Google Scholar] [CrossRef]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.H.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef]
- Cornelius, N.; Byron, C.; Hargreaves, I.; Guerra, P.F.; Furdek, A.K.; Land, J.; Radford, W.W.; Frerman, F.; Corydon, T.J.; Gregersen, N.; et al. Secondary coenzyme Q10 deficiency and oxidative stress in cultured fibroblasts from patients with riboflavin responsive multiple Acyl-CoA dehydrogenation deficiency. Hum. Mol. Genet. 2013, 22, 3819–3827. [Google Scholar] [CrossRef]
- Grünert, S.C. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency. Orphanet J. Rare Dis. 2014, 9, 117. [Google Scholar] [CrossRef]
- Kmoch, S.; Zeman, J.; Hrebíček, M.; Ryba, L.; Kristensen, M.J.; Gregersen, N. Riboflavin-responsive epilepsy in a patient with SER209 variant form of short-chain acyl-CoA dehydrogenase. J. Inherit. Metab. Dis. 1995, 18, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Nochi, Z.; Olsen, R.K.J.; Gregersen, N. Short-chain acyl-CoA dehydrogenase deficiency: From gene to cell pathology and possible disease mechanisms. J. Inherit. Metab. Dis. 2017, 40, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Sewell, A.C.; Herwig, J.; Böhles, H.; Rinaldo, P.; Bhala, A.; Hale, D.E. A new case of short-chain acyl-CoA dehydrogenase deficiency with isolated ethylmalonic aciduria. Eur. J. Pediatr. 1993, 152, 922–924. [Google Scholar] [CrossRef] [PubMed]
- Dawson, D.B.; Waber, L.; Hale, D.E.; Bennett, M.J. Transient organic aciduria and persistent lacticacidemia in a patient with short-chain acyl-coenzyme A dehydrogenase deficiency. J. Pediatr. 1995, 126, 69–71. [Google Scholar] [CrossRef]
- Van Maldegem, B.T.; Duran, M.; Wanders, R.J.A.; Waterham, H.R.; Wijburg, F.A. Flavin adenine dinucleotide status and the effects of high-dose riboflavin treatment in short-chain Acyl-CoA dehydrogenase deficiency. Pediatr. Res. 2010, 67, 304–308. [Google Scholar] [CrossRef]
- Duran, M.; Cleutjens, C.B.J.M.; Ketting, D.; Dorland, L.; de Klerk, J.B.C.; van Sprang, F.J.; Berger, R. Diagnosis of medium-chain acyl-CoA dehydrogenase deficiency in lymphocytes and liver by a gas chromatographic method: The effect of oral riboflavin supplementation. Pediatr. Res. 1992, 31, 39–42. [Google Scholar] [CrossRef][Green Version]
- Derks, T.G.J.; Touw, C.M.L.; Ribas, G.S.; Biancini, G.B.; Vanzin, C.S.; Negretto, G.; Mescka, C.P.; Reijngoud, D.J.; Smit, G.P.A.; Wajner, M.; et al. Experimental evidence for protein oxidative damage and altered antioxidant defense in patients with medium-chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2014, 37, 783–789. [Google Scholar] [CrossRef]
- Gerards, M.; Van Den Bosch, B.J.C.; Danhauser, K.; Serre, V.; Van Weeghel, M.; Wanders, R.J.A.; Nicolaes, G.A.F.; Sluiter, W.; Schoonderwoerd, K.; Scholte, H.R.; et al. Riboflavin-responsive oxidative phosphorylation complex i deficiency caused by defective ACAD9: New function for an old gene. Brain 2011, 134, 210–219. [Google Scholar] [CrossRef]
- Repp, B.M.; Mastantuono, E.; Alston, C.L.; Schiff, M.; Haack, T.B.; Rötig, A.; Ardissone, A.; Lombès, A.; Catarino, C.B.; Diodato, D.; et al. Clinical, biochemical and genetic spectrum of 70 patients with ACAD9 deficiency: Is riboflavin supplementation effective? Orphanet J. Rare Dis. 2018, 13, 1–10. [Google Scholar] [CrossRef]
- Bugiani, M.; Lamantea, E.; Invernizzi, F.; Moroni, I.; Bizzi, A.; Zeviani, M.; Uziel, G. Effects of riboflavin in children with complex II deficiency. Brain Dev. 2006, 28, 576–581. [Google Scholar] [CrossRef]
- Jain-Ghai, S.; Cameron, J.M.; Al Maawali, A.; Blaser, S.; Mackay, N.; Robinson, B.; Raiman, J. Complex II deficiency-A case report and review of the literature. Am. J. Med. Genet. Part A 2013, 161, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, D.; Goffrini, P.; Uziel, G.; Horvath, R.; Klopstock, T.; Lochmüller, H.; D’Adamo, P.; Gasparini, P.; Strom, T.M.; Prokisch, H.; et al. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat. Genet. 2009, 41, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Maio, N.; Ghezzi, D.; Verrigni, D.; Rizza, T.; Bertini, E.; Martinelli, D.; Zeviani, M.; Singh, A.; Carrozzo, R.; Rouault, T.A. Disease-causing SDHAF1 mutations impair transfer of Fe-S clusters to SDHB. Cell Metab. 2016, 23, 292–302. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; D’Adamo, P.; Briem, E.; Ferrari, G.; Mineri, R.; Lamantea, E.; Mandel, H.; Balestri, P.; Garcia-Silva, M.-T.; Vollmer, B.; et al. Ethylmalonic Encephalopathy Is Caused by Mutations in ETHE1, a Gene Encoding a Mitochondrial Matrix Protein. Am. J. Hum. Genet. 2004, 74, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Viscomi, C.; Hildebrandt, T.; Di Meo, I.; Mineri, R.; Tiveron, C.; Levitt, M.D.; Prelle, A.; Fagiolari, G.; Rimoldi, M.; et al. Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 2009, 15, 200–205. [Google Scholar] [CrossRef]
- Burlina, A.B.; Dionisi-Vici, C.; Bennett, M.J.; Gibson, K.M.; Servidei, S.; Bertini, E.; Hale, D.E.; Schmidt-Sommerfeld, E.; Sabetta, G.; Zacchello, F. A new syndrome with ethylmalonic aciduria and normal fatty acid oxidation in fibroblasts. J. Pediatr. 1994, 124, 79–86. [Google Scholar] [CrossRef]
- Garcia-Silva, M.T.; Ribes, A.; Campos, Y.; Garavaglia, B.; Arenas, J. Syndrome of encephalopathy, petechiae, and ethylmalonic aciduria. Pediatr. Neurol. 1997, 17, 165–170. [Google Scholar] [CrossRef]
- Zhang, K.; Huang, Y.; Gai, Z.; Liu, Y. Clinical and genetic analysis of a case with atypical ethyl malonate encephalopathy. Chinese J. Med. Genet. 2018, 35, 694–698. [Google Scholar] [CrossRef]
- Papetti, L.; Garone, G.; Schettini, L.; Giordano, C.; Nicita, F.; Papoff, P.; Zeviani, M.; Leuzzi, V.; Spalice, A. Severe early onset ethylmalonic encephalopathy with West syndrome. Metab. Brain Dis. 2015, 30, 1537–1545. [Google Scholar] [CrossRef]
- Pigeon, N.; Campeau, P.M.; Cyr, D.; Lemieux, B.; Clarke, J.T.R. Clinical heterogeneity in ethylmalonic encephalopathy. J. Child Neurol. 2009, 24, 991–996. [Google Scholar] [CrossRef]
- Merinero, B.; Perez-Cerda, C.; Ruiz Sala, P.; Ferrer, I.; Garcia, M.J.; Martinez Pardo, M.; Belanger-Quintana, A.; de la Mota, J.L.; Martin-Hernandez, E.; Vianey-Saban, C.; et al. Persistent increase of plasma butyryl/isobutyrylcarnitine concentrations as marker of SCAD defect and ethylmalonic encephalopathy. J. Inherit. Metab. Dis. 2006, 29, 685. [Google Scholar] [CrossRef] [PubMed]
- Heberle, L.C.; Al Tawari, A.A.; Ramadan, D.G.; Ibrahim, J.K. Ethylmalonic encephalopathy-report of two cases. Brain Dev. 2006, 28, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Ozand, P.T.; Rashed, M.; Millington, D.S.; Sakati, N.; Hazzaa, S.; Rahbeeni, Z.; al Odaib, A.; Youssef, N.; Mazrou, A.; Gascon, G.G. Ethylmalonic aciduria: An organic acidemia with CNS involvement and vasculopathy. Brain Dev. 1994, 16 (Suppl. 12–22). [Google Scholar] [CrossRef]
- Pavlou, E.; Augoustides-Savvopoulou, P.; Gregersen, N.; Haas, D.; Gkampeta, A.; Athanassiadou-Piperopoulou, F. An infant with ethylmalonic encephalopathy masquerading as a hematologic disorder. J. Child Neurol. 2013, 28, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Hack, A.; Klein, K.; Wößner, R.; Kress, W.; Straßburg, H.M. A severe case of encephalopathy with ethylmalonic aciduria and generalized vasculopathy. Neuropediatrics 2008, 39, P47. [Google Scholar] [CrossRef]
- Govindaraj, P.; Parayil Sankaran, B.; Nagappa, M.; Arvinda, H.R.; Deepha, S.; Jessiena Ponmalar, J.N.; Sinha, S.; Gayathri, N.; Taly, A.B. Child Neurology: Ethylmalonic encephalopathy. Neurology 2020, 94, e1336–e1339. [Google Scholar] [CrossRef]
- Kilic, M.; Dedeoglu, O.; Gocmen, R.; Kesici, S.; Yuksel, D. Successful treatment of a patient with ethylmalonic encephalopathy by intravenous N-acetylcysteine. Metab. Brain Dis. 2017, 32, 293–296. [Google Scholar] [CrossRef]
- Yis, U.; Polat, I.; Karakaya, P.; Ayanoglu, M.; Hiz, A.S. Importance of acrocyanosis in delayed walking. J. Pediatr. Neurosci. 2015, 10, 80–81. [Google Scholar]
- Yoon, H.R.; Hahn, S.H.; Ahn, Y.M.; Jang, S.H.; Shin, Y.J.; Lee, E.H.; Ryu, K.H.; Eun, B.L.; Rinaldo, P.; Yamaguchi, S. Therapeutic trial in the first three Asian cases of ethylmalonic encephalopathy: Response to riboflavin. J. Inherit. Metab. Dis. 2001, 24, 870–873. [Google Scholar] [CrossRef]
- Zafeiriou, D.I.; Augoustides-Savvopoulou, P.; Haas, D.; Smet, J.; Triantafyllou, P.; Vargiami, E.; Tamiolaki, M.; Gombakis, N.; van Coster, R.; Sewell, A.C.; et al. Ethylmalonic encephalopathy: Clinical and biochemical observations. Neuropediatrics 2007, 38, 78–82. [Google Scholar] [CrossRef]
- Libiad, M.; Yadav, P.K.; Vitvitsky, V.; Martinov, M.; Banerjee, R. Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J. Biol. Chem. 2014, 289, 30901–30910. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, T.M.; Grieshaber, M.K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008, 275, 3352–3361. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, I.; Fagiolari, G.; Prelle, A.; Viscomi, C.; Zeviani, M.; Tiranti, V. Chronic exposure to sulfide causes accelerated degradation of cytochrome c oxidase in ethylmalonic encephalopathy. Antioxid. Redox Signal. 2011, 15, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Chaban, Y.; Boekema, E.J.; Dudkina, N. V Structures of mitochondrial oxidative phosphorylation supercomplexes and mechanisms for their stabilisation. Biochim. Biophys. Acta 2014, 1837, 418–426. [Google Scholar] [CrossRef]
- Dionisi-Vici, C.; Diodato, D.; Torre, G.; Picca, S.; Pariante, R.; Giuseppe Picardo, S.; Di Meo, I.; Rizzo, C.; Tiranti, V.; Zeviani, M.; et al. Liver transplant in ethylmalonic encephalopathy: A new treatment for an otherwise fatal disease. Brain 2016, 139, 1045–1051. [Google Scholar] [CrossRef]
- Boyer, M.; Sowa, M.; Di Meo, I.; Eftekharian, S.; Steenari, M.R.; Tiranti, V.; Abdenur, J.E. Response to medical and a novel dietary treatment in newborn screen identified patients with ethylmalonic encephalopathy. Mol. Genet. Metab. 2018, 124, 57–63. [Google Scholar] [CrossRef]
- Viscomi, C.; Burlina, A.B.; Dweikat, I.; Savoiardo, M.; Lamperti, C.; Hildebrandt, T.; Tiranti, V.; Zeviani, M. Combined treatment with oral metronidazole and N-acetylcysteine is effective in ethylmalonic encephalopathy. Nat. Med. 2010, 16, 869–871. [Google Scholar] [CrossRef]
- Kitzler, T.M.; Gupta, I.R.; Osterman, B.; Poulin, C.; Trakadis, Y.; Waters, P.J.; Buhas, D.C. Acute and Chronic Management in an Atypical Case of Ethylmalonic Encephalopathy. JIMD Rep. 2019, 45, 57–63. [Google Scholar] [CrossRef]
- Tam, A.; AlDhaheri, N.S.; Mysore, K.; Tessier, M.E.; Goss, J.; Fernandez, L.A.; D’Alessandro, A.M.; Schwoerer, J.S.; Rice, G.M.; Elsea, S.H.; et al. Improved clinical outcome following liver transplant in patients with ethylmalonic encephalopathy. Am. J. Med. Genet. A 2019, 179, 1015–1019. [Google Scholar] [CrossRef]
- Penn, A.M.W.; Lee, J.W.K.; Thuillier, P.; Wagner, M.; Maclure, K.M.; Menard, M.R.; Hall, L.D.; Kennaway, N.G. MELAS syndrome with mitochondrial tRNA Leu(UUR) mutation: Correlation of clinical state, nerve conduction, and muscle 31P magnetic resonance spectroscopy during treatment with nicotinamide and riboflavin. Neurology 1992, 42, 2147–2152. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.; Salvetti, S.; Vista, M.; Lombardi, V.; Siciliano, G.; Giraldi, C. Long-term treatment with idebenone and riboflavin in a patient with MELAS. Neurol. Sci. 2000, 21, 981–982. [Google Scholar] [CrossRef] [PubMed]
- Darin, N.; Hedberg-Oldfors, C.; Kroksmark, A.K.; Moslemi, A.R.; Kollberg, G.; Oldfors, A. Benign mitochondrial myopathy with exercise intolerance in a large multigeneration family due to a homoplasmic m.3250T>C mutation in MTTL1. Eur. J. Neurol. 2017, 24, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Bernsen, P.L.J.A.; Gabreëls, F.J.M.; Ruitenbeek, W.; Hamburger, H.L. Treatment of complex I deficiency with riboflavin. J. Neurol. Sci. 1993, 118, 181–187. [Google Scholar] [CrossRef]
- Garrido-Maraver, J.; Cordero, M.D.; Moñino, I.D.; Pereira-Arenas, S.; Lechuga-Vieco, A.V.; Cotán, D.; De La Mata, M.; Oropesa-Ávila, M.; De Miguel, M.; Bautista Lorite, J.; et al. Screening of effective pharmacological treatments for MELAS syndrome using yeasts, fibroblasts and cybrid models of the disease. Br. J. Pharmacol. 2012, 167, 1311–1328. [Google Scholar] [CrossRef]
- O’Callaghan, B.; Bosch, A.M.; Houlden, H. An update on the genetics, clinical presentation, and pathomechanisms of human riboflavin transporter deficiency. J. Inherit. Metab. Dis. 2019, 42, 598–607. [Google Scholar] [CrossRef]
- Bosch, A.M.; Abeling, N.G.G.M.; Ijlst, L.; Knoester, H.; Van Der Pol, W.L.; Stroomer, A.E.M.; Wanders, R.J.; Visser, G.; Wijburg, F.A.; Duran, M.; et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: A new inborn error of metabolism with potential treatment. J. Inherit. Metab. Dis. 2011, 34, 159–164. [Google Scholar] [CrossRef]
- Chiong, M.A.; Sim, K.G.; Carpenter, K.; Rhead, W.; Ho, G.; Olsen, R.K.J.; Christodoulou, J. Transient multiple acyl-CoA dehydrogenation deficiency in a newborn female caused by maternal riboflavin deficiency. Mol. Genet. Metab. 2007, 92, 109–114. [Google Scholar] [CrossRef]
- Ho, G.; Yonezawa, A.; Masuda, S.; Inui, K.; Sim, K.G.; Carpenter, K.; Olsen, R.K.J.; Mitchell, J.J.; Rhead, W.J.; Peters, G.; et al. Maternal riboflavin deficiency, resulting in transient neonatal-onset glutaric aciduria Type 2, is caused by a microdeletion in the riboflavin transporter gene GPR172B. Hum. Mutat. 2011, 32, E1976–E1984. [Google Scholar] [CrossRef]
- Mosegaard, S.; Bruun, G.H.; Flyvbjerg, K.F.; Bliksrud, Y.T.; Gregersen, N.; Dembic, M.; Annexstad, E.; Tangeraas, T.; Olsen, R.K.J.; Andresen, B.S. An intronic variation in SLC52A1 causes exon skipping and transient riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Mol. Genet. Metab. 2017, 122, 182–188. [Google Scholar] [CrossRef]
- Schiff, M.; Veauville-Merllié, A.; Su, C.H.; Tzagoloff, A.; Rak, M.; De Baulny, H.O.; Boutron, A.; Smedts-Walters, H.; Romero, N.B.; Rigal, O.; et al. SLC25A32 mutations and riboflavin-responsive exercise intolerance. N. Engl. J. Med. 2016, 374, 795–797. [Google Scholar] [CrossRef] [PubMed]
- Hellebrekers, D.M.E.I.; Sallevelt, S.C.E.H.; Theunissen, T.E.J.; Hendrickx, A.T.M.; Gottschalk, R.W.; Hoeijmakers, J.G.J.; Habets, D.D.; Bierau, J.; Schoonderwoerd, K.G.; Smeets, H.J.M. Novel SLC25A32 mutation in a patient with a severe neuromuscular phenotype. Eur. J. Hum. Genet. 2017, 25, 886–888. [Google Scholar] [CrossRef] [PubMed]
- Miccolis, A.; Galluccio, M.; Giancaspero, T.A.; Indiveri, C.; Barile, M. Bacterial over-expression and purification of the 3’phosphoadenosine 5’phosphosulfate (PAPS) reductase domain of human FAD synthase: Functional characterization and homology modeling. Int. J. Mol. Sci. 2012, 13, 16880–16898. [Google Scholar] [CrossRef]
- Leone, P.; Galluccio, M.; Barbiroli, A.; Eberini, I.; Tolomeo, M.; Vrenna, F.; Gianazza, E.; Iametti, S.; Bonomi, F.; Indiveri, C.; et al. Bacterial production, characterization and protein modeling of a novel monofuctional isoform of FAD synthase in humans: An emergency protein? Molecules 2018, 23, 116. [Google Scholar] [CrossRef] [PubMed]
- Auranen, M.; Paetau, A.; Piirilä, P.; Pohju, A.; Salmi, T.; Lamminen, A.; Löfberg, M.; Mosegaard, S.; Olsen, R.K.; Tyni, T. Patient with multiple acyl-CoA dehydrogenation deficiency disease and FLAD1 mutations benefits from riboflavin therapy. Neuromuscul. Disord. 2017, 27, 581–584. [Google Scholar] [CrossRef]
- García-Villoria, J.; De Azua, B.; Tort, F.; Mosegaard, S.; Ugarteburu, O.; Texidó, L.; Morales-Romero, B.; Olsen, R.K.J.; Ribes, A. FLAD1, encoding FAD synthase, is mutated in a patient with myopathy, scoliosis and cataracts. Clin. Genet. 2018, 94, 592–593. [Google Scholar] [CrossRef]
- Ryder, B.; Tolomeo, M.; Nochi, Z.; Colella, M.; Barile, M.; Olsen, R.K.; Inbar-Feigenberg, M. A Novel Truncating FLAD1 Variant, Causing Multiple Acyl-CoA Dehydrogenase Deficiency (MADD) in an 8-Year-Old Boy. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2018; Volume 45, pp. 37–44. [Google Scholar]
- Yıldız, Y.; Olsen, R.K.J.; Sivri, H.S.; Akçören, Z.; Nygaard, H.H.; Tokatlı, A. Post-mortem detection of FLAD1 mutations in 2 Turkish siblings with hypotonia in early infancy. Neuromuscul. Disord. 2018, 28, 787–790. [Google Scholar] [CrossRef]
- Yamada, K.; Ito, M.; Kobayashi, H.; Hasegawa, Y.; Fukuda, S.; Yamaguchi, S.; Taketani, T. Flavin adenine dinucleotide synthase deficiency due to FLAD1 mutation presenting as multiple acyl-CoA dehydrogenation deficiency-like disease: A case report. Brain Dev. 2019, 41, 638–642. [Google Scholar] [CrossRef]
- Muru, K.; Reinson, K.; Künnapas, K.; Lilleväli, H.; Nochi, Z.; Mosegaard, S.; Pajusalu, S.; Olsen, R.K.J.; Õunap, K. FLAD1-associated multiple acyl-CoA dehydrogenase deficiency identified by newborn screening. Mol. Genet. Genomic Med. 2019, 7. [Google Scholar] [CrossRef]
- Liuzzi, V.C.; Giancaspero, T.A.; Gianazza, E.; Banfi, C.; Barile, M.; De Giorgi, C. Silencing of FAD synthase gene in Caenorhabditis elegans upsets protein homeostasis and impacts on complex behavioral patterns. Biochim. Biophys. Acta - Gen. Subj. 2012, 1820, 521–531. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Christodoulou, J.; Rahman, S. Disorders of riboflavin metabolism. J. Inherit. Metab. Dis. 2019, 42, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Makowski, C.; Yao, Y.; Graf, E.; Hempel, M.; Wieland, T.; Tauer, U.; Ahting, U.; Mayr, J.A.; Freisinger, P.; et al. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J. Inherit. Metab. Dis. 2012, 35, 943–948. [Google Scholar] [CrossRef]
- Gregersen, N.; Koelvraa, S.; Mortensen, P.B.; Rasmussen, K. C6-C10-Dicarboxylic aciduria: biochemical considerations in relation to diagnosis of β-oxidation defects. Scand. J. Clin. Lab. Investig. Suppl. 1982, 42, 15–27. [Google Scholar] [CrossRef]
- Johnson, J.O.; Gibbs, J.R.; Megarbane, A.; Urtizberea, J.A.; Hernandez, D.G.; Foley, A.R.; Arepalli, S.; Pandraud, A.; Simón-Sánchez, J.; Clayton, P.; et al. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain 2012, 135, 2875–2882. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Haberberger, B.; Frisch, E.-M.; Wieland, T.; Iuso, A.; Gorza, M.; Strecker, V.; Graf, E.; Mayr, J.A.; Herberg, U.; et al. Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing. J. Med. Genet. 2012, 49, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.; Pedersen, P.; Winter, V.; Nyholm, M.; Johansen, B.N.; Olsen, R.K.; Corydon, M.J.; Andresen, B.S.; Eiberg, H.; Kolvraa, S.; et al. A polymorphic variant in the human electron transfer flavoprotein alpha-chain (alpha-T171) displays decreased thermal stability and is overrepresented in very-long-chain acyl-CoA dehydrogenase-deficient patients with mild childhood presentation. Mol. Genet. Metab. 1999, 67, 138–147. [Google Scholar] [CrossRef]
- Macchione, F.; Salviati, L.; Bordugo, A.; Vincenzi, M.; Camilot, M.; Teofoli, F.; Pancheri, E.; Zordan, R.; Bertolin, C.; Rossi, S.; et al. Multiple acyl-COA dehydrogenase deficiency in elderly carriers. J. Neurol. 2020. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).