Targeting Wnt Signaling for the Treatment of Gastric Cancer



Abstract
:1. Gastric Cancer
2. Wnt Signaling
3. Deregulated Wnt Components in GC
4. Fzd7 in Gastric Cancer
5. Targeting Wnt Signaling at the Receptor Level in Cells with Downstream Mutations
6. Wnt Signaling in Metastatic GC
7. Clinical Applications of Wnt Inhibitors for GC
8. Conclusions
Funding
Conflicts of Interest
Abbreviations
| APC | Adenomatous polyposis coli |
| β-TrCP | Beta-Transducin Repeat Containing E3 Ubiquitin Protein Ligase |
| CAMKII | calmodulin dependent protein kinase II |
| CBP | CREB-binding protein |
| CDC | Cell Division Cycle 42 |
| CDK | Cyclin Dependent Kinase |
| CK1α | Casein kinase 1α |
| CREB | CAMP Responsive Element Binding Protein |
| DAAM | Dishevelled Associated Activator Of Morphogenesis |
| DAG | 1,2-diacylglycerol |
| DGO | Diego |
| DKK | Dickkopf |
| DVL | Dishevelled |
| FZD | Frizzled |
| FMI | Flamingo |
| GC | gastric cancer |
| GSK3α/β | Glycogen synthase 3 |
| IP3 | Inositol 1,4,5-triphosphate |
| JUN | Jun Proto-Oncogene, AP-1 Transcription Factor Subunit |
| LGR | Leucine-rich repeat-containing G-protein-coupled receptor |
| LRP | Low-density lipoprotein receptor-related protein |
| NFκB | Nuclear Factor Kappa B Subunit 1 |
| OC | Oesophageal cancer |
| PK | LIM domain protein Prickle |
| PLC | Phospholipase C |
| pTNM stage | Pathological Tumor-Node-Metastasis stage |
| RAC | Rac Family Small GTPase |
| RHO | Rhodopsin |
| ROCK | Rho Associated Coiled-Coil Containing Protein Kinase |
| ROR | Receptor Tyrosine Kinase Like Orphan Receptor |
| RNF | Ring finger |
| Ryk | Receptor Like Tyrosine Kinase |
| R | SPO—Rspondin |
| SFRP | Secreted Frizzled-related protein |
| SDC | Syndican4 |
| TAZ | Taffazin |
| TCF/LEF | T-cell factor/lymphoid enhancer factor |
| TLE | Groucho/ transducin-like Enhancer of Split |
| VANG | Van Gogh transmembrane protein |
| YAP | Yes associated protein |
| VANG | Van Gogh transmembrane protein |
| YAP | Yes associated protein |
References
- B Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asplund, J.; Kauppila, J.H.; Mattsson, F.; Lagergren, J. Survival Trends in Gastric Adenocarcinoma: A Population-Based Study in Sweden. Ann. Surg. Oncol. 2018, 25, 2693–2702. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Edge, S.; Greene, F.; Byrd, D.; Brookland, R.; Washington, M.; Gershenwald, J.; Compton, C.; Hess, K.R.; Sullivan, D.; et al. AJCC Cancer Staging Manual, 8th ed.; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar]
- Powell, A.; Wheat, J.; Patel, N.; Chan, D.; Foliaki, A.; Roberts, S.A.; Lewis, W.G. Value of individual surgeon performance metrics as quality assurance measures in oesophagogastric cancer surgery. Bjs Open 2020, 4, 91–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Carcas, L.P. Gastric cancer review. J. Carcinog. 2014, 13, 14. [Google Scholar] [CrossRef]
- Piazuelo, M.B.; Correa, P. Gastric cáncer: Overview. Colomb Med. 2013, 44, 192–201. [Google Scholar]
- Russo, A.E.; Strong, V.E. Gastric Cancer Etiology and Management in Asia and the West. Annu. Rev. Med. 2019, 70, 353–367. [Google Scholar] [CrossRef]
- Macdonald, J.S.; Smalley, S.R.; Benedetti, J.; Hundahl, S.A.; Estes, N.C.; Stemmermann, G.N.; Haller, D.G.; Ajani, J.A.; Gunderson, L.L.; Jessup, J.M.; et al. Chemoradiotherapy after surgery compared with surgery alone for adenocarcinoma of the stomach or gastroesophageal junction. N. Engl. J. Med. 2001, 345, 725–730. [Google Scholar] [CrossRef]
- Cunningham, D.; Allum, W.H.; Stenning, S.P.; Thompson, J.N.; Van de Velde, C.J.; Nicolson, M.; Scarffe, J.H.; Lofts, F.J.; Falk, S.J.; Iveson, T.J.; et al. Perioperative chemotherapy versus surgery alone for resectable gastroesophageal cancer. N. Engl. J. Med. 2006, 355, 11–20. [Google Scholar] [CrossRef]
- Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric Cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signalling in cancer. Oncogene 2016, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Et. Biophys. Acta 2003, 1653, 1–24. [Google Scholar] [CrossRef]
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R.; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003, 423, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [Green Version]
- Bryja, V.; Červenka, I.; Čajánek, L. The connections of Wnt pathway components with cell cycle and centrosome: Side effects or a hidden logic? Crit Rev. Biochem. Mol. Biol. 2017, 52, 614–637. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, D.J.; Austin, C.R.; Vincan, E.; Phesse, T.J. Wnt Signalling in Gastrointestinal Epithelial Stem Cells. Genes 2018, 9, 178. [Google Scholar] [CrossRef] [Green Version]
- Slusarski, D.C.; Yang-Snyder, J.; Busa, W.B.; Moon, R.T. Modulation of embryonic intracellular Ca2+ signaling by Wnt-5A. Dev. Biol. 1997, 182, 114–120. [Google Scholar] [CrossRef] [Green Version]
- Kreusser, M.M.; Backs, J. Integrated mechanisms of CaMKII-dependent ventricular remodeling. Front. Pharmacol. 2014, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, K.A.; Fuerer, C.; Ching, W.; Fish, M.; Willert, K.; Nusse, R. Secreted Wingless-interacting molecule (Swim) promotes long-range signaling by maintaining Wingless solubility. Proc. Natl. Acad. Sci. USA 2012, 109, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Mii, Y.; Taira, M. Secreted Frizzled-related proteins enhance the diffusion of Wnt ligands and expand their signalling range. Development 2009, 136, 4083–4088. [Google Scholar] [CrossRef] [Green Version]
- Gross, J.C.; Chaudhary, V.; Bartscherer, K.; Boutros, M. Active Wnt proteins are secreted on exosomes. Nat. Cell Biol. 2012, 14, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Stanganello, E.; Scholpp, S. Role of cytonemes in Wnt transport. J. Cell Sci. 2016, 129, 665–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattes, B.; Dang, Y.; Greicius, G.; Kaufmann, L.T.; Prunsche, B.; Rosenbauer, J.; Stegmaier, J.; Mikut, R.; Ozbek, S.; Nienhaus, G.U.; et al. Wnt/PCP controls spreading of Wnt/beta-catenin signals by cytonemes in vertebrates. eLife 2018, 7, e36953. [Google Scholar] [CrossRef] [PubMed]
- Ooi, C.H.; Ivanova, T.; Wu, J.; Lee, M.; Tan, I.B.; Tao, J.; Ward, L.; Koo, J.H.; Gopalakrishnan, V.; Zhu, Y.; et al. Oncogenic pathway combinations predict clinical prognosis in gastric cancer. Plos Genet. 2009, 5, e1000676. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Fan, S.; Ma, W.; Fan, P.; Wang, B.; Zhang, J.; Wang, H.; Tang, B.; Zhang, Q.; Yu, X.; et al. Roles of Wnt/beta-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis 2014, 5, e1039. [Google Scholar] [CrossRef] [Green Version]
- Katoh, M.; Kirikoshi, H.; Terasaki, H.; Shiokawa, K. WNT2B2 mRNA, up-regulated in primary gastric cancer, is a positive regulator of the WNT- beta-catenin-TCF signaling pathway. Biochem Biophys Res. Commun 2001, 289, 1093–1098. [Google Scholar] [CrossRef]
- Saitoh, T.; Mine, T.; Katoh, M. Frequent up-regulation of WNT5A mRNA in primary gastric cancer. Int. J. Mol. Med. 2002, 9, 515–519. [Google Scholar] [CrossRef]
- Yuan, G.; Regel, I.; Lian, F.; Friedrich, T.; Hitkova, I.; Hofheinz, R.D.; Strobel, P.; Langer, R.; Keller, G.; Rocken, C.; et al. WNT6 is a novel target gene of caveolin-1 promoting chemoresistance to epirubicin in human gastric cancer cells. Oncogene 2013, 32, 375–387. [Google Scholar] [CrossRef] [Green Version]
- Kirikoshi, H.; Sekihara, H.; Katoh, M. Up-regulation of WNT10A by tumor necrosis factor alpha and Helicobacter pylori in gastric cancer. Int. J. Oncol. 2001, 19, 533–536. [Google Scholar]
- Flanagan, D.J.; Barker, N.; Costanzo, N.S.D.; Mason, E.A.; Gurney, A.; Meniel, V.S.; Koushyar, S.; Austin, C.R.; Ernst, M.; Pearson, H.B.; et al. Frizzled-7 Is Required for Wnt Signaling in Gastric Tumors with and Without Apc Mutations. Cancer Res. 2019, 79, 970–981. [Google Scholar] [CrossRef] [Green Version]
- Nojima, M.; Suzuki, H.; Toyota, M.; Watanabe, Y.; Maruyama, R.; Sasaki, S.; Sasaki, Y.; Mita, H.; Nishikawa, N.; Yamaguchi, K.; et al. Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene 2007, 26, 4699–4713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.K.; Liu, J.; Liu, C.; Wang, F.Y.; Chen, C.Y.; Zhang, X.H. Hypermethylation of adenomatous polyposis coli gene promoter is associated with novel Wnt signaling pathway in gastric adenomas. J. Gastroenterol. Hepatol. 2012, 27, 1629–1634. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Huebner, A.J.; Sulahian, R.; Anselmo, A.; Xu, X.; Flattery, K.; Desai, N.; Sebastian, C.; Yram, M.A.; Arnold, K.; et al. Sox2 Suppresses Gastric Tumorigenesis in Mice. Cell Rep. 2016, 16, 1929–1941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phesse, T.J.; Sansom, O.J. Lgr5 joins the club of gastric stem cell markers in the corpus. Nat. Cell Biol. 2017, 19, 752–754. [Google Scholar] [CrossRef]
- Flanagan, D.J.; Vincan, E.; Phesse, T.J. Winding back Wnt signalling: Potential therapeutic targets for treating gastric cancers. Br. J. Pharmacol. 2017, 174, 4666–4683. [Google Scholar] [CrossRef] [Green Version]
- Radulescu, S.; Ridgway, R.A.; Cordero, J.; Athineos, D.; Salgueiro, P.; Poulsom, R.; Neumann, J.; Jung, A.; Patel, S.; Woodgett, J.; et al. Acute WNT signalling activation perturbs differentiation within the adult stomach and rapidly leads to tumour formation. Oncogene 2013, 32, 2048–2057. [Google Scholar] [CrossRef] [Green Version]
- Kurayoshi, M.; Oue, N.; Yamamoto, H.; Kishida, M.; Inoue, A.; Asahara, T.; Yasui, W.; Kikuchi, A. Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res. 2006, 66, 10439–10448. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, H.; Kitadai, Y.; Yamamoto, H.; Oue, N.; Ohdan, H.; Yasui, W.; Kikuchi, A. Lamininγ2 Mediates Wnt5a-Induced Invasion of Gastric Cancer Cells. Gastroenterology 2009, 137, 242–252.e6. [Google Scholar] [CrossRef]
- Hanaki, H.; Yamamoto, H.; Sakane, H.; Matsumoto, S.; Ohdan, H.; Sato, A.; Kikuchi, A. An Anti-Wnt5a Antibody Suppresses Metastasis of Gastric Cancer Cells by Inhibiting Receptor-Mediated Endocytosis. Mol. Cancer Ther. 2012, 11, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Astudillo, P. Wnt5a Signaling in Gastric Cancer. Front. Cell Dev. Biol. 2020, 8, 110. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, D.J.; Vincan, E.; Phesse, T.J. Wnt signaling in cancer: Not a binary ON:OFF switch. Cancer Res. 2019, 79, 5901–5906. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Duan, X.L.; Qi, X.L.; Meng, L.; Xu, Y.S.; Wu, T.; Dai, P.G. Concurrent Hypermethylation of SFRP2 and DKK2 Activates the Wnt/beta-Catenin Pathway and Is Associated with Poor Prognosis in Patients with Gastric Cancer. Mol. Cells 2017, 40, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.Y.; Yu, J.; Wong, Y.P.; Man, E.P.; To, K.F.; Jin, V.X.; Li, J.; Tao, Q.; Sung, J.J.; Chan, F.K.; et al. Frequent epigenetic inactivation of secreted frizzled-related protein 2 (SFRP2) by promoter methylation in human gastric cancer. Br. J. Cancer 2007, 97, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.; Yang, H.; Woo Ho Kim, H.; Kim, Y.; Kook, M.; Park, Y.; Kim, H.; Lee, H.; Lee, K.; Gu, M.; et al. Predictive test for chemotherapy response in resectable gastric cancer: A multi-cohort, retrospective analysis. Lancet Oncol. 2018, 19, 629–638. [Google Scholar] [CrossRef]
- Wang, B.; Liu, J.; Ma, L.N.; Xiao, H.L.; Wang, Y.Z.; Li, Y.; Wang, Z.; Fan, L.; Lan, C.; Yang, M.; et al. Chimeric 5/35 adenovirus-mediated Dickkopf-1 overexpression suppressed tumorigenicity of CD44(+) gastric cancer cells via attenuating Wnt signaling. J. Gastroenterol. 2013, 48, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.A.; Yoo, S.H.; Lee, H.H.; Sun, S.; Won, H.S.; Kim, O.; Ko, Y.H. Prognostic value of Dickkopf-1 and ss-catenin expression in advanced gastric cancer. Bmc Cancer 2018, 18, 506. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Chen, Y.; Huang, J.; Cai, Z.; Wang, Y. RYK, a receptor of noncanonical Wnt ligand Wnt5a, is positively correlated with gastric cancer tumorigenesis and potential of liver metastasis. Am. J. Physiol Gastrointest. Liver Physiol. 2020, 318, G352–G360. [Google Scholar] [CrossRef]
- Niu, L.; Qin, H.Z.; Xi, H.Q.; Wei, B.; Xia, S.Y.; Chen, L. RNF43 Inhibits Cancer Cell Proliferation and Could be a Potential Prognostic Factor for Human Gastric Carcinoma. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2015, 36, 1835–1846. [Google Scholar] [CrossRef]
- Koo, B.-K.; van Es, J.H.; van den Born, M.; Clevers, H. Porcupine inhibitor suppresses paracrine Wnt-driven growth of Rnf43;Znrf3-mutant neoplasia. Proc. Natl. Acad. Sci. USA 2015, 112, 7548–7550. [Google Scholar] [CrossRef] [Green Version]
- Kirikoshi, H.; Sekihara, H.; Katoh, M. Expression profiles of 10 members of Frizzled gene family in human gastric cancer. Int. J. Oncol. 2001, 19, 767–771. [Google Scholar] [CrossRef]
- Li, G.; Su, Q.; Liu, H.; Wang, D.; Zhang, W.; Lu, Z.; Chen, Y.; Huang, X.; Li, W.; Zhang, C.; et al. Frizzled7 Promotes Epithelial-to-mesenchymal Transition and Stemness Via Activating Canonical Wnt/β-catenin Pathway in Gastric Cancer. Int. J. Biol. Sci. 2018, 14, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, E.; Buchert, M.; Putoczki, T.; Thiem, S.; Farid, R.; Elzer, J.; Huszar, D.; Waring, P.M.; Phesse, T.J.; Ernst, M. Therapeutic inhibition of Jak activity inhibits progression of gastrointestinal tumors in mice. Mol. Cancer Ther. 2014, 13, 468–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito-Diaz, K.; Benchabane, H.; Tiwari, A.; Tian, A.; Li, B.; Thompson, J.J.; Hyde, A.S.; Sawyer, L.M.; Jodoin, J.N.; Santos, E.; et al. APC Inhibits Ligand-Independent Wnt Signaling by the Clathrin Endocytic Pathway. Dev. Cell 2018, 44, 566–581.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flanagan, D.J.; Barker, N.; Nowell, C.; Clevers, H.; Ernst, M.; Phesse, T.J.; Vincan, E. Loss of the Wnt receptor frizzled 7 in the mouse gastric epithelium is deleterious and triggers rapid repopulation in vivo. Dis. Models Mech. 2017, 10, 971–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Lu, X.; Wu, X.; Xue, L.; Wang, X.; Xu, J. MicroRNA-27b suppresses Helicobacter pylori-induced gastric tumorigenesis through negatively regulating Frizzled7. Oncol. Rep. 2016, 35, 2441–2450. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Liu, G.; Liu, S.; Chen, R.; Wang, N.; Liu, Z.; Zhang, X.; Xiao, Z.; Liu, L. Helicobacter pylori upregulates TRPC6 via Wnt/β-catenin signaling to promote gastric cancer migration and invasion. Onco. Targets 2019, 12, 5269–5279. [Google Scholar] [CrossRef] [Green Version]
- Muncan, V.; Sansom, O.J.; Tertoolen, L.; Phesse, T.J.; Begthel, H.; Sancho, E.; Cole, A.M.; Gregorieff, A.; de Alboran, I.M.; Clevers, H.; et al. Rapid loss of intestinal crypts upon conditional deletion of the Wnt/Tcf-4 target gene c-Myc. Mol. Cell. Biol. 2006, 26, 8418–8426. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, D.; Barker, N.; Ernst, M.; Vincan, E.; Phesse, T. The Function of Lgr5+ Cells in the Gastric Antrum Does Not Require Fzd7 or Myc In Vivo. Biomedines 2019, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Liu, T.; Zhou, X.; Dang, Y.; Yin, C.; Zhang, G. FZD6, targeted by miR-21, represses gastric cancer cell proliferation and migration via activating non-canonical wnt pathway. Am. J. Transl. Res. 2016, 8, 2354–2364. [Google Scholar]
- Brabletz, T.; Jung, A.; Hermann, K.; Günther, K.; Hohenberger, W.; Kirchner, T. Nuclear overexpression of the oncoprotein beta-catenin in colorectal cancer is localized predominantly at the invasion front. Pathol Res. Pr. 1998, 194, 701–704. [Google Scholar] [CrossRef]
- Johansson, J.; Naszai, M.; Hodder, M.C.; Pickering, K.A.; Miller, B.W.; Ridgway, R.A.; Yu, Y.; Peschard, P.; Brachmann, S.; Campbell, A.D.; et al. RAL GTPases Drive Intestinal Stem Cell Function and Regeneration through Internalization of WNT Signalosomes. Cell Stem Cell 2019, 24, 592–607.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatoff, E.M.; Goswami, S.; Zafra, M.P.; Foronda, M.; Shusterman, M.; Leach, B.I.; Katti, A.; Diaz, B.J.; Dow, L.E. Distinct Colorectal Cancer–Associated APC Mutations Dictate Response to Tankyrase Inhibition. Cancer Discov. 2019, 9, 1358–1371. [Google Scholar] [CrossRef] [PubMed]
- Guner, A.; Yildirim, R. Surgical management of metastatic gastric cancer: Moving beyond the guidelines. Transl. Gastroenterol. Hepatol. 2019, 4, 58. [Google Scholar] [CrossRef] [PubMed]
- Riihimaki, M.; Hemminki, A.; Sundquist, K.; Sundquist, J.; Hemminki, K. Metastatic spread in patients with gastric cancer. Oncotarget 2016, 7, 52307–52316. [Google Scholar] [CrossRef] [Green Version]
- Nwabo Kamdje, A.H.; Takam Kamga, P.; Tagne Simo, R.; Vecchio, L.; Seke Etet, P.F.; Muller, J.M.; Bassi, G.; Lukong, E.; Kumar Goel, R.; Mbo Amvene, J.; et al. Developmental pathways associated with cancer metastasis: Notch, Wnt, and Hedgehog. Cancer Biol Med. 2017, 14, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wang, D.; Sun, X.; Zhang, Y.; Wang, L.; Suo, J. ADAM17 promotes lymph node metastasis in gastric cancer via activation of the Notch and Wnt signaling pathways. Int. J. Mol. Med. 2018, 43, 914–926. [Google Scholar] [CrossRef]
- Yanaka, Y.; Muramatsu, T.; Uetake, H.; Kozaki, K.-i.; Inazawa, J. miR-544a induces epithelial–mesenchymal transition through the activation of WNT signaling pathway in gastric cancer. Carcinogenesis 2015, 36, 1363–1371. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, X.; Liu, Y. LGR5 regulates gastric adenocarcinoma cell proliferation and invasion via activating Wnt signaling pathway. Oncogenesis 2018, 7, 57. [Google Scholar] [CrossRef] [Green Version]
- Cromwell, D.; Wahedally, H.; Park, M. National Oesophago-Gastric Cancer Audit. Available online: https://www.nogca.org.uk/content/uploads/2019/12/REF150_NOGCA_2019-Annual-Report-FINAL_19Dec.pdf (accessed on 21 May 2020).
- Powell, A.; Parkinson, D.; Patel, N.; Chan, D.; Christian, A.; Lewis, W.G. Prognostic Significance of Serum Inflammatory Markers in Gastric Cancer. J. Gastrointest. Surg. Off. J. Soc. Surg. Aliment. Tract 2018, 22, 595–605. [Google Scholar] [CrossRef]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef]
- Dotan, E.; Cardin, D.B.; Lenz, H.-J.; Messersmith, W.A.; O’Neil, B.; Cohen, S.J.; Denlinger, C.S.; Shahda, S.; Kapoun, A.M.; Brachmann, R.K.; et al. Phase Ib study of WNT inhibitor ipafricept (IPA) with nab-paclitaxel (Nab-P) and gemcitabine (G) in patients (pts) with previously untreated stage IV pancreatic cancer (mPC). J. Clin. Oncol. 2019, 37, 369. [Google Scholar] [CrossRef]
- Pfeiffer, P.; Yilmaz, M.; Möller, S.; Maltha, L.; Krogh, M.; Zitnjak, D.; Szameitat, C.L.; Hejlesen, F.; Thomsen, K.G.; Qvortrup, C. Randomized study evaluating trifluridine/tipiracil (TAS-102) versus + trifluridine/tipiracil + bevacizumab as last-line therapy in patients with chemorefractory unresectable metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2019, 37, 637. [Google Scholar] [CrossRef]
- Smith, D.C.; Rosen, L.S.; Chugh, R.; Goldman, J.W.; Xu, L.; Kapoun, A.; Brachmann, R.K.; Dupont, J.; Stagg, R.J.; Tolcher, A.W.; et al. First-in-human evaluation of the human monoclonal antibody vantictumab (OMP-18R5; anti-Frizzled) targeting the WNT pathway in a phase I study for patients with advanced solid tumors. J. Clin. Oncol. 2013, 31, 2540. [Google Scholar] [CrossRef]
- Akrami, H.; Moradi, B.; Borzabadi Farahani, D.; Mehdizadeh, K. Ibuprofen reduces cell proliferation through inhibiting Wnt/β catenin signaling pathway in gastric cancer stem cells. Cell Biol. Int. 2018, 42, 949–958. [Google Scholar] [CrossRef]
- US National Library of Medicine. ClinicalTrials.gov NCT01351103. Available online: https://clinicaltrials.gov/ct2/show/NCT01351103(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT02278133. Available online: https://clinicaltrials.gov/ct2/show/NCT02278133(2017) (accessed on 28 May 2020).
- US National Library of Medicine. Clinical Trials.gov. NCT02521844. Available online: https://clinicaltrials.gov/ct2/show/NCT02521844(2015) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT02482441. Available online: https://clinicaltrials.gov/ct2/show/NCT02482441(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT02020291. Available online: https://clinicaltrials.gov/ct2/show/NCT02020291(2016) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT02655952. Available online: https://clinicaltrials.gov/ct2/show/NCT02655952(2016) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT01608867. Available online: https://clinicaltrials.gov/ct2/show/NCT01608867(2016) (accessed on 28 May 2020).
- Jimeno, A.; Gordon, M.; Chugh, R.; Messersmith, W.; Mendelson, D.; Dupont, J.; Stagg, R.; Kapoun, A.M.; Xu, L.; Uttamsingh, S.; et al. A First-in-Human Phase I Study of the Anticancer Stem Cell Agent Ipafricept (OMP-54F28), a Decoy Receptor for Wnt Ligands, in Patients with Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 7490–7497. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.N.; Gunderson, C.C.; Sabbatini, P.; McMeekin, D.S.; Mantia-Smaldone, G.; Burger, R.A.; Morgan, M.A.; Kapoun, A.M.; Brachmann, R.K.; Stagg, R.; et al. A phase 1b dose escalation study of ipafricept (OMP54F28) in combination with paclitaxel and carboplatin in patients with recurrent platinum-sensitive ovarian cancer. Gynecol. Oncol. 2019, 154, 294–301. [Google Scholar] [CrossRef]
- US National Library of Medicine. ClinicalTrials.gov NCT02092363. Available online: https://clinicaltrials.gov/ct2/show/NCT02092363(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT02069145. Available online: https://clinicaltrials.gov/ct2/show/NCT02069145(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT02050178. Available online: https://clinicaltrials.gov/ct2/show/NCT02050178(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT01345201. Available online: https://clinicaltrials.gov/ct2/show/NCT01345201(2016) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT01957007. Available online: https://clinicaltrials.gov/ct2/show/NCT01957007(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT01973309. Available online: https://clinicaltrials.gov/ct2/show/NCT01973309(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT02005315. Available online: https://clinicaltrials.gov/ct2/show/NCT02005315(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT01469975. Available online: https://clinicaltrials.gov/ct2/show/NCT01469975(2017) (accessed on 28 May 2020).
- Giraudet, A.; Badel, J.; Cassier, P.; Desuzinges, C.; Kriza, D.; Perol, D.; Blay, J.Y. SYNFRIZZ-A phase Ia/Ib of a radiolabelled monoclonal AB for the treatment of relapsing synovial sarcoma. J. Nucl. Med. 2014, 55, 223. [Google Scholar]
- US National Library of Medicine. ClinicalTrials.gov NCT02222688. Available online: https://clinicaltrials.gov/ct2/show/NCT02222688(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT02860676. Available online: https://clinicaltrials.gov/ct2/show/NCT02860676(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT03088878. Available online: https://clinicaltrials.gov/ct2/show/NCT03088878(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT02776917. Available online: https://clinicaltrials.gov/ct2/show/NCT02776917(2017) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT01302405. Available online: https://clinicaltrials.gov/ct2/show/NCT01302405(2015) (accessed on 28 May 2020).
- El-Khoueiry, A.B.; Ning, Y.; Yang, D.; Cole, S.; Kahn, M.; Zoghbi, M.; Berg, J.; Fujimori, M.; Inada, T.; Kouji, H.; et al. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2013, 31, 2501. [Google Scholar] [CrossRef]
- US National Library of Medicine. ClinicalTrials.gov NCT01764477. Available online: https://clinicaltrials.gov/ct2/show/NCT01764477(2015) (accessed on 28 May 2020).
- US National Library of Medicine. ClinicalTrials.gov NCT01606579. Available online: https://clinicaltrials.gov/ct2/show/NCT01606579(2017) (accessed on 28 May 2020).
- U.S. National Library of Medicine. Clinical Trials.gov. NCT03604445. Available online: https://clinicaltrials.gov/ct2/show/NCT03604445 (accessed on 28 May 2020).
- Anderson, R.L.; Balasas, T.; Callaghan, J.; Coombes, R.C.; Evans, J.; Hall, J.A.; Kinrade, S.; Jones, D.; Jones, P.S.; Jones, R.; et al. A framework for the development of effective anti-metastatic agents. Nat. Reviews. Clin. Oncol. 2019, 16, 185–204. [Google Scholar] [CrossRef] [Green Version]
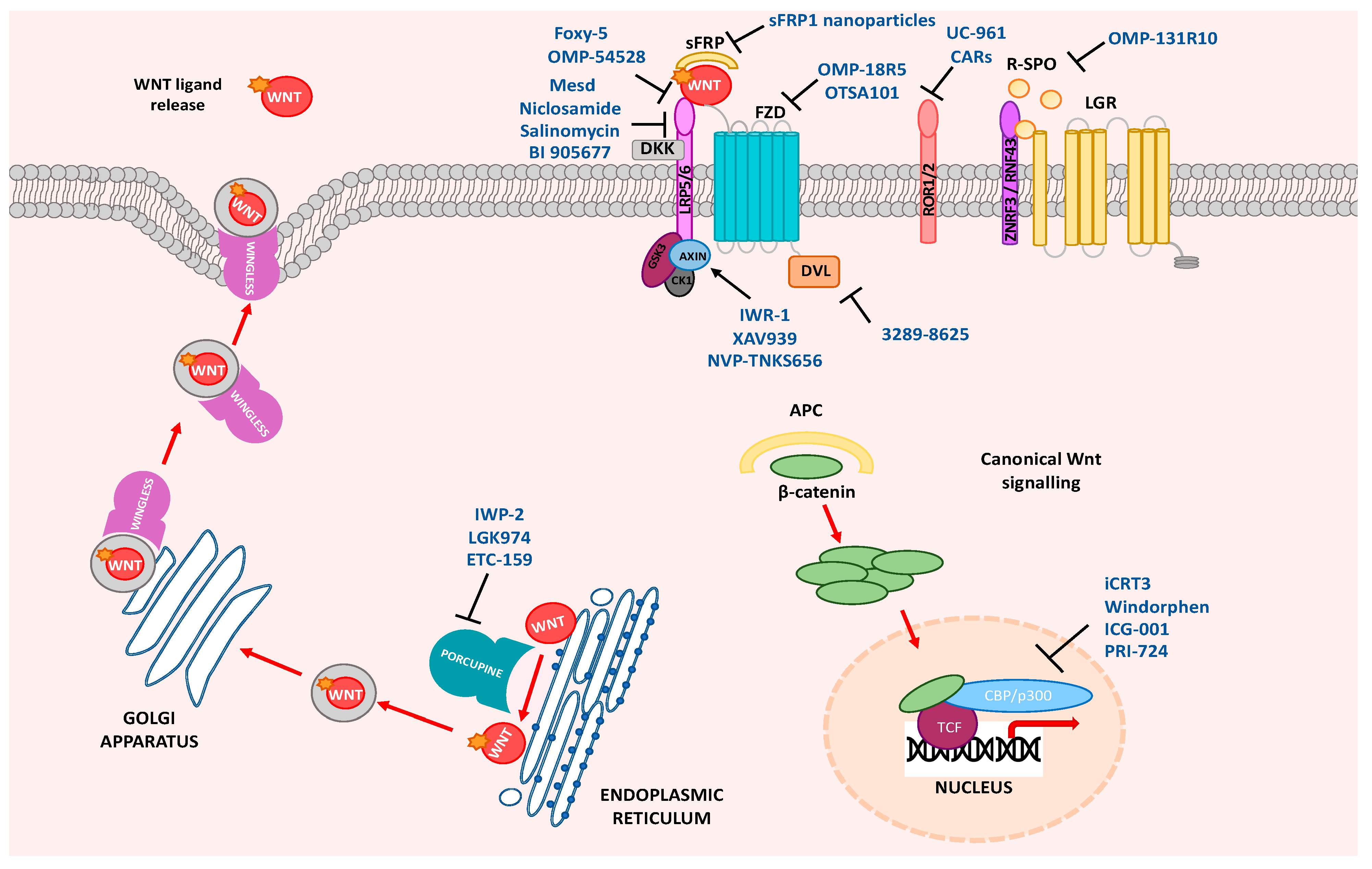

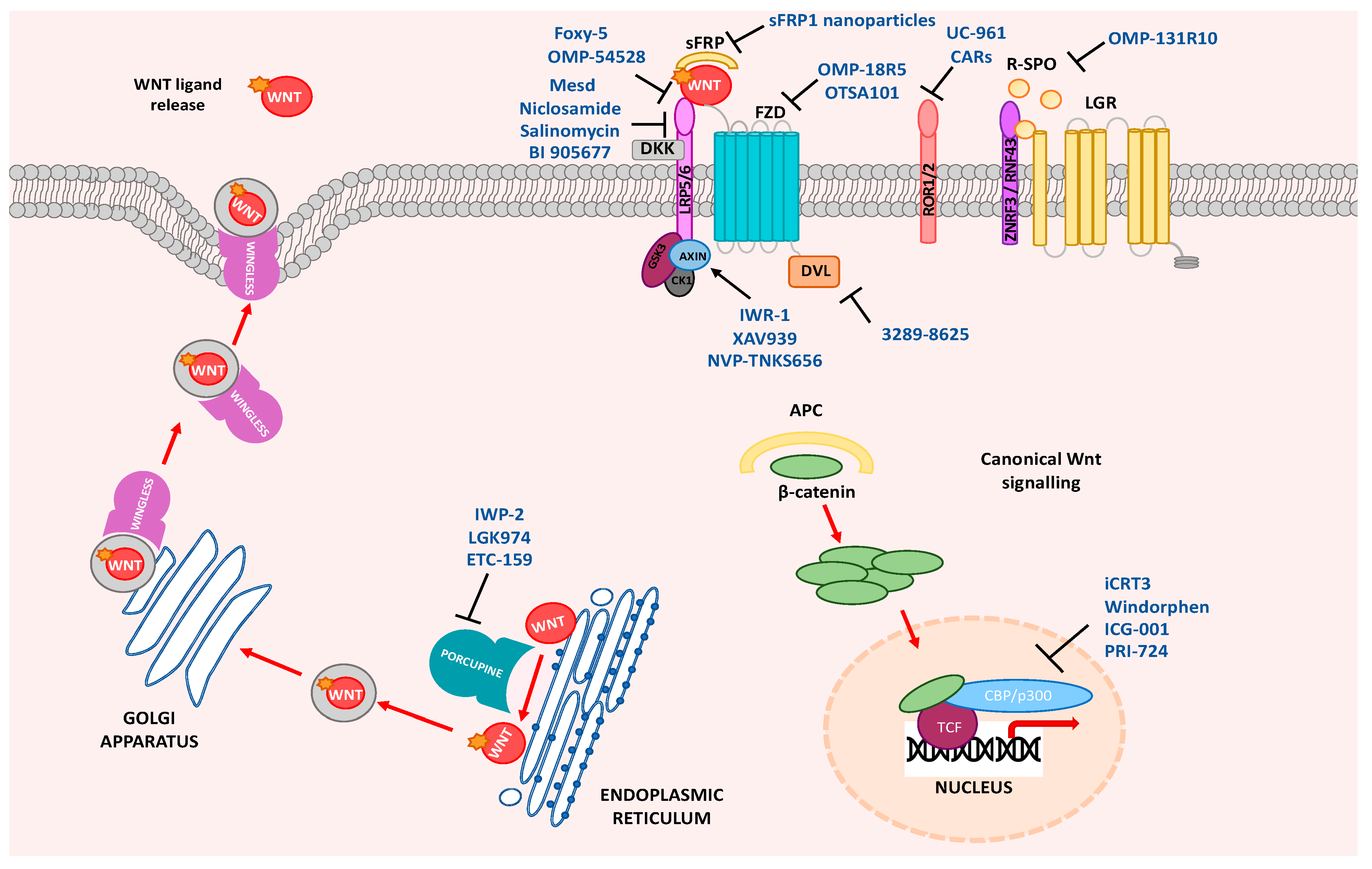{kind=link}
| Wnt Component | Role in GC |
|---|---|
| Cytoplasmic | |
| APC | Mutated/deep deletion in GC patient datasets [31]. Promoter hypermethylation in high grade gastric adenomas [33]. |
| β-catenin | Endogenous nuclear expression seen in 13/15 GC cell lines with a subsequent increase in TCF/LEF transcriptional activity [32]. Abnormal nuclear expression seen in high grade gastric adenomas [33]. |
| AXIN2 | miR-544a targeted protein downregulation in GC cells [68]. 30% of MSI high GCs have a frameshift mutation [36]. |
| Gsk3β | Genetic deletion causes rapid gastric tumor formation in mice [37]. |
| Wnt target genes | |
| MYC | Gene amplification in GC patient samples [36]. GC cells and mouse adenoma show gene upregulation in an Fzd7-dependent manner [31]. |
| LGR5 | Overexpression regulates GC cell proliferation, migration, and invasion [69]. |
| Wnt ligands | |
| WNT1 | Upregulated in human GC tissue. Overexpression accelerates gastric cancer stem cells [26]. |
| WNT2b | Upregulated in GC tissue [27]. |
| Wnt3a | Upregulated in gp130F/F gastric tumors [31]. |
| WNT5a | High protein expression in GC patient samples, positively associated with the depth of tumor invasion and degree of lymph node metastasis [28]. |
| WNT6 | Upregulated in GC patient samples and GC cell lines. Expression positively correlated with tumor stage and node status [29]. |
| WNT10a | Upregulated in GC cells and primary GC tissue [30]. |
| Wnt antagonists | |
| DKK1 | Hypermethylated in GC patient samples [46]. |
| DKK2 | Hypermethylated in GC patient samples + gene transcripts lower in GC patient samples [43]. |
| sFRP2 | Concurrently hypermethylated with DKK2 + gene transcripts lower in GC patient samples [43]. |
| Wnt receptors | |
| RYK co-receptor | High expression correlated with poor differentiation, high TNM stage and liver metastasis in GC patients [48]. |
| RNF43 | Truncating mutation in MSI GC tumors [5]. Protein expression is significantly lower in GC cells than normal gastric epithelial cells [49]. |
| FZD2 | Upregulated in GC cells (TMK1, MKN7, MKN28, MKN45, MKN74, and KATO-III) and in 4/10 primary GC tissue [51]. |
| FZD5 | Upregulated in GC cells (MKN45) [51]. |
| FZD7 | Overexpression is seen in late-stage clinical GC, correlating with a decrease in survival time [52]. Knockdown significantly reduces GC cell proliferation, migration, EMT, and expression of stem cell markers [52]. |
| FZD8 | Upregulated in 4/10 primary GC tissue [51]. |
| FZD9 | Upregulated in 2/10 primary GC tissue [51]. |
| WNT PATHWAY TARGET | DRUG | PHASE AND CLINICAL TRIAL | CANCER TYPE | |
|---|---|---|---|---|
| PORCUPINE | LGK974 (WNT974) | Phase I NCT01351103 Phase I/II NCT02278133 | [77] | Pancreatic Cancer, BRAF mutant CRC, Melanoma, Triple negative Breast Cancer, Head and Neck Squamous Cell Cancer, Cervical Squamous Cell Cancer, Esophageal Squamous Cell Cancer, Lung Squamous Cell Cancer Metastatic Colorectal cancer |
| ETC-1922159 | Phase IA/B NCT02521844 | [78,79] | Advanced or metastatic solid tumors | |
| RSPO3 | OMP131R10 | Phase I NCT02482441 | [80] | Metastatic Colorectal Cancer, advanced relapsed or refractory solid tumors |
| WNT5A RECEPTOR | Foxy-5 | Phase I NCT02020291 | [81] | Metastatic Breast, Colon or Prostate cancer (loss or reduced Wnt5a on IHC) |
| Phase I NCT02655952 | [82] | Metastatic Breast, Colon or Prostate cancer (loss or reduced Wnt5a on IHC) | ||
| WNT FAMILY | OMP-54F28 (Ipafricept) | Phase I NCT01608867 | [83] | Metastatic or unresectable solid tumors |
| Phase I NCT02092363 | [84] | Ovarian, primary peritoneal or fallopian tube cancer | ||
| Phase I NCT02069145 | [85,86] | Locally advanced or metastatic Hepatocellular Carcinoma | ||
| Phase I NCT02050178 | [87,88] | TNM stage IV Ductal adenocarcinoma of the pancreas | ||
| FZD1,2,5,7,8 | OMP-18R5 (Vantictumab) | Phase I NCT01345201 | [89] | Metastatic solid tumors with no other standard treatment options |
| Phase I NCT01957007 | [90] | Recurrent of TNM stage IV Non-small cell lung cancer | ||
| Phase I NCT01973309 | [91] | Recurrent or metastatic breast cancer (HER2 overexpression not eligible) | ||
| Phase I NCT02005315 | [92] | TNM stage IV Ductal adenocarcinoma of the pancreas | ||
| FZD10 | OTSA101 | Phase I NCT01469975 | [93,94] | Progressive synovial sarcoma |
| ROR1 | UC-961 (Cirmtuzumab) | Phase I NCT02222688 | [95] | Relapsed or refractory B cell Chronic Lymphocytic Leukemia (CLL) |
| Phase I NCT02860676 | [96] | Relapsed or refractory B cell CLL | ||
| Phase I/II NCT03088878 | [97] | B Cell CLL, Small Cell Lymphocytic Lymphoma, Mantle Cell Lymphoma | ||
| Phase I NCT02776917 | [98] | Metastatic or locally advanced HER2 negative breast cancer | ||
| CREB BINDING PROTEIN | PRI-724 | Phase I NCT01302405 | [99] | Metastatic or unresectable solid tumors |
| Phase I NCT01764477 | [100] | Relapsed, locally advanced or metastatic pancreatic adenocarcinoma | ||
| Phase I/II NCT01606579 | [101,102] | Relapse or refractory Acute Myeloid Leukemia, advanced Chronic Myeloid Leukemia | ||
| LRP5/6 | BI 905677 | Phase 1 NCT03604445 | [103] | Metastatic or unresectable solid tumors |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koushyar, S.; Powell, A.G.; Vincan, E.; Phesse, T.J. Targeting Wnt Signaling for the Treatment of Gastric Cancer. Int. J. Mol. Sci. 2020, 21, 3927. https://doi.org/10.3390/ijms21113927
Koushyar S, Powell AG, Vincan E, Phesse TJ. Targeting Wnt Signaling for the Treatment of Gastric Cancer. International Journal of Molecular Sciences. 2020; 21(11):3927. https://doi.org/10.3390/ijms21113927
Chicago/Turabian StyleKoushyar, Sarah, Arfon G. Powell, Elizabeth Vincan, and Toby J. Phesse. 2020. "Targeting Wnt Signaling for the Treatment of Gastric Cancer" International Journal of Molecular Sciences 21, no. 11: 3927. https://doi.org/10.3390/ijms21113927
APA StyleKoushyar, S., Powell, A. G., Vincan, E., & Phesse, T. J. (2020). Targeting Wnt Signaling for the Treatment of Gastric Cancer. International Journal of Molecular Sciences, 21(11), 3927. https://doi.org/10.3390/ijms21113927