Tau Gene Deletion Does Not Influence Axonal Regeneration and Retinal Neuron Survival in the Injured Mouse Visual System

Abstract
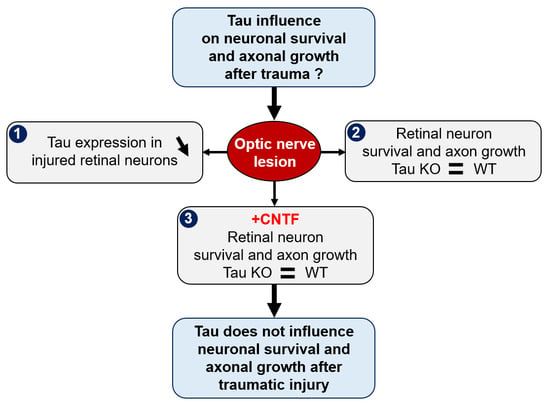
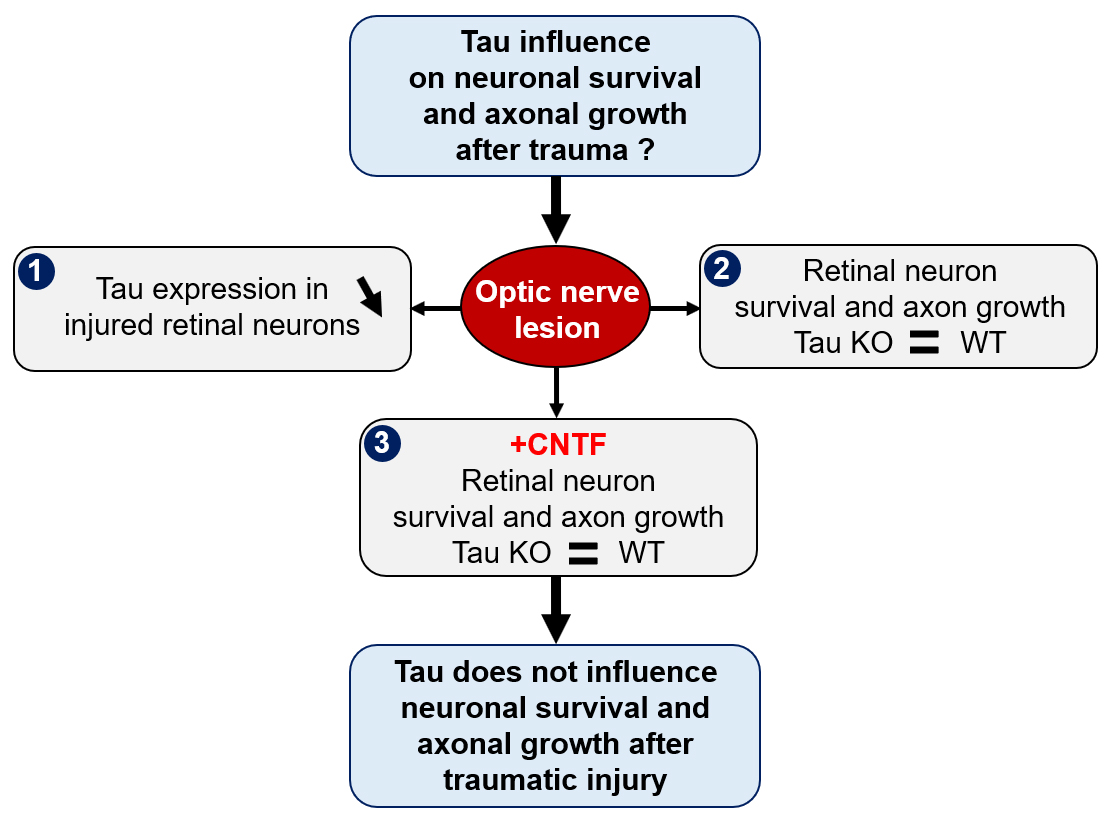{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
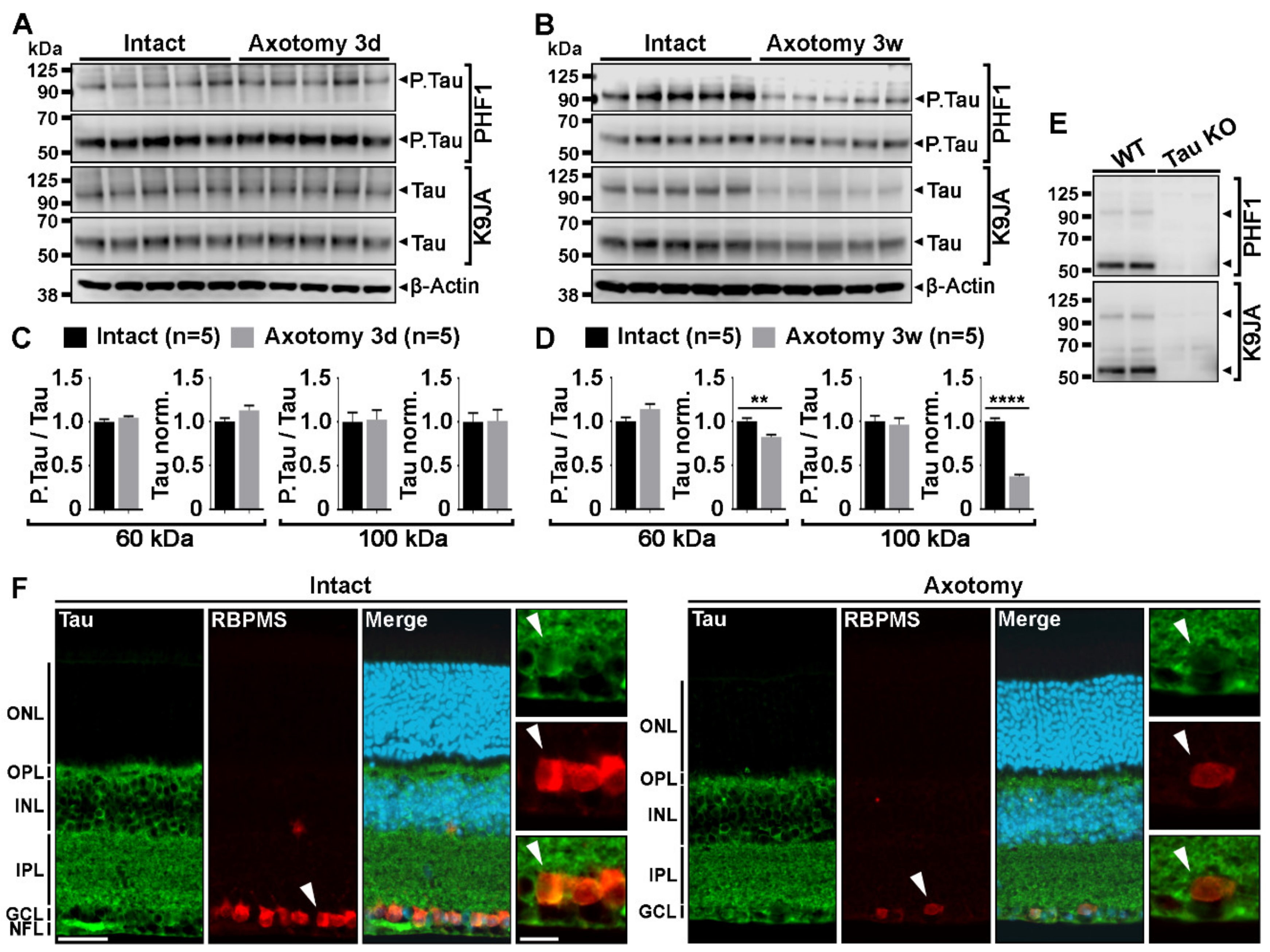
2.1. Optic Nerve Injury Decreases the Level of Tau Protein in the Retina
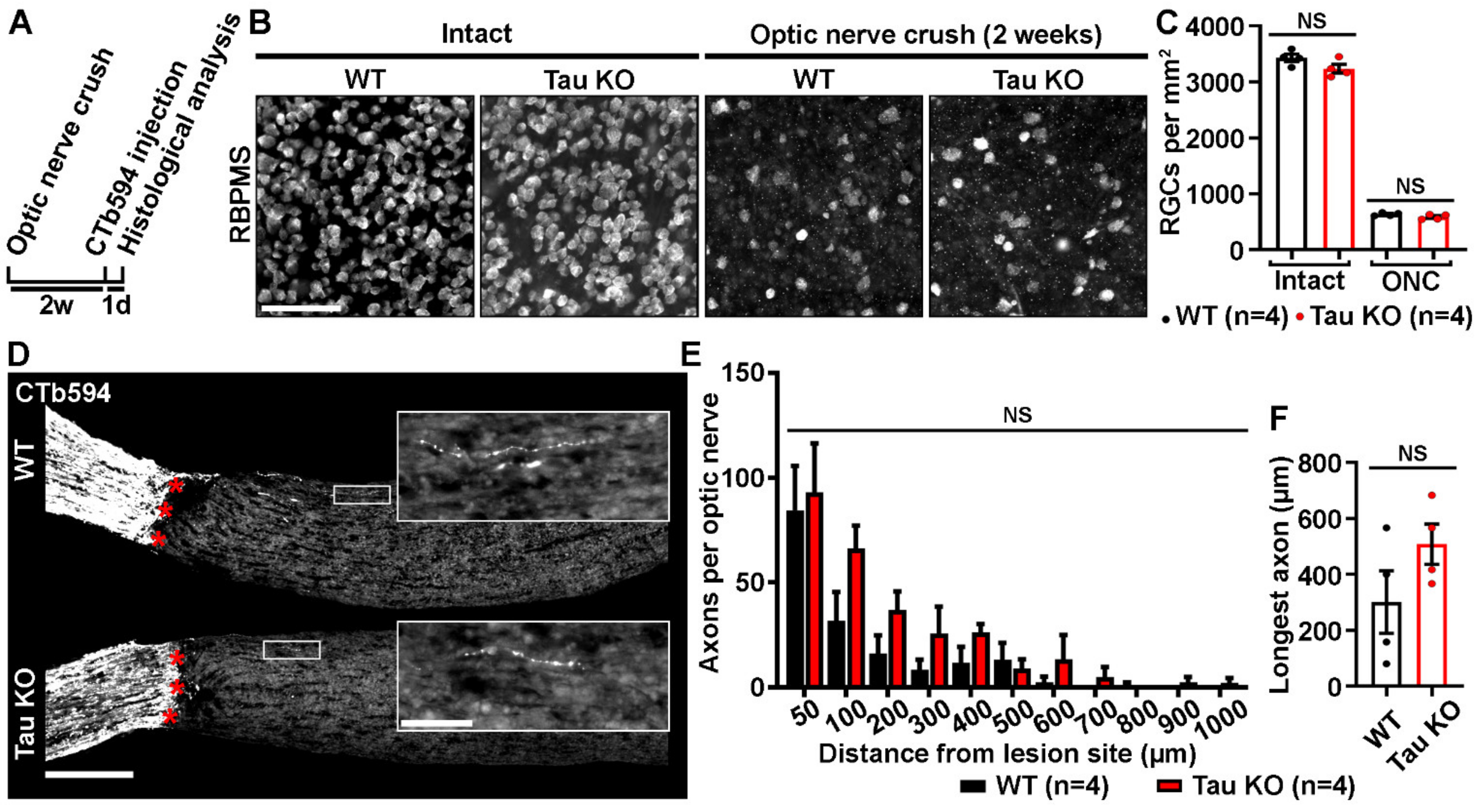
2.2. Tau Gene Deletion Does Not Influence Neuronal Survival and Axonal Growth after Optic Nerve Injury
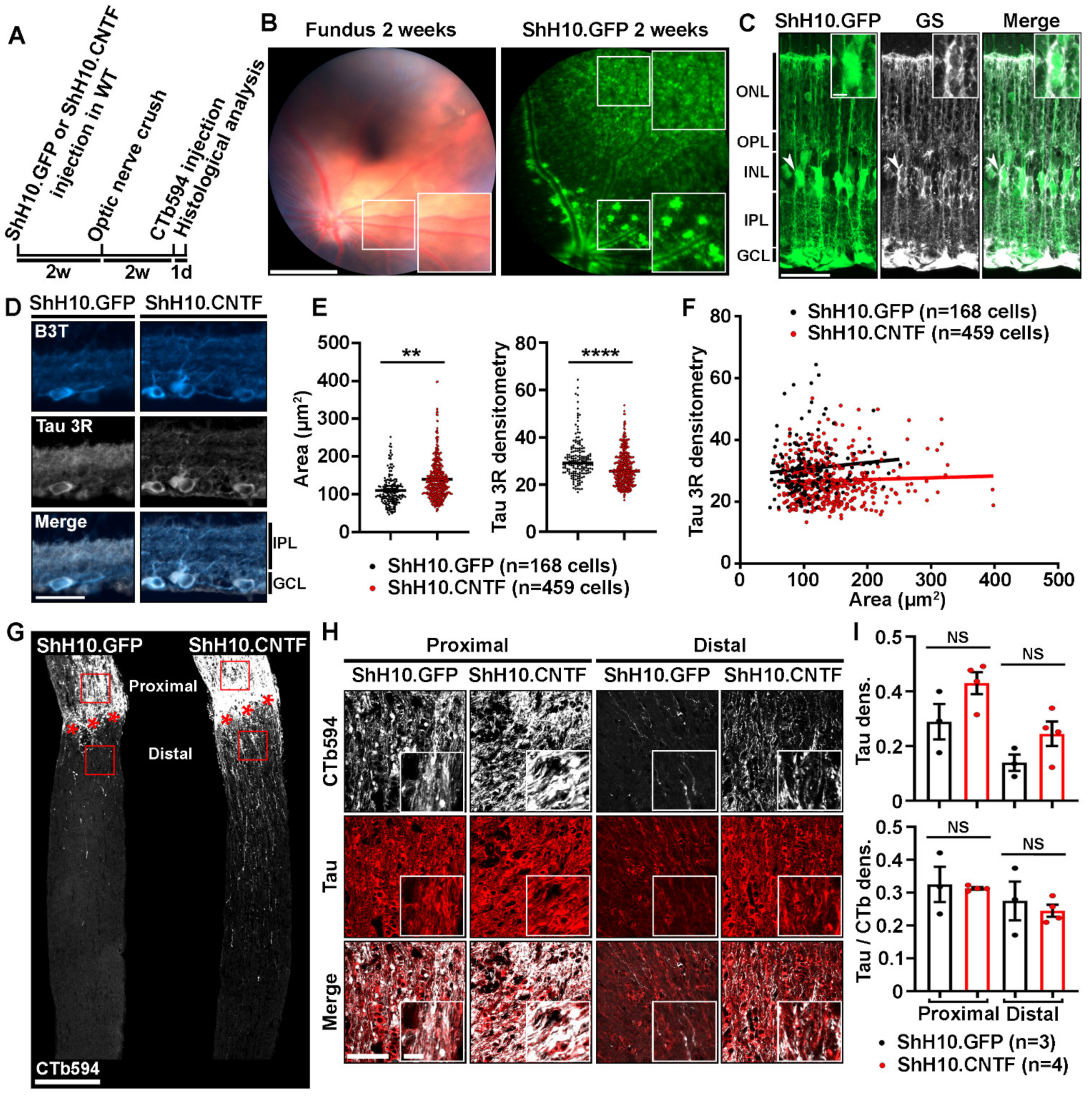
2.3. Analysis of Tau Expression in Retinal Ganglion Cells during CNTF-Induced Survival and Regeneration
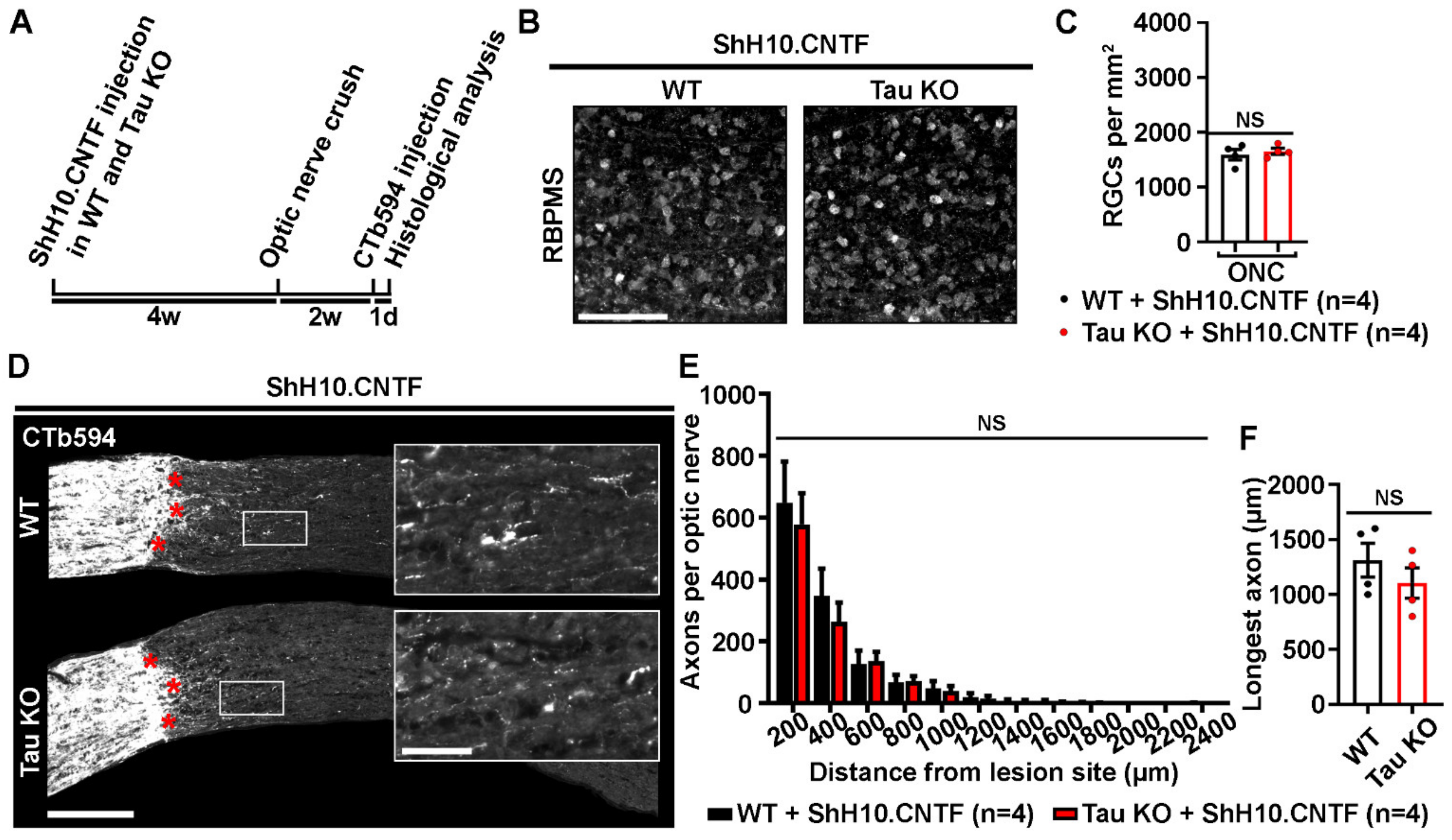
2.4. Tau Deletion Does Not Influence CNTF-Induced Neuronal Survival and Axonal Outgrowth in the Injured Visual System
3. Discussion
3.1. Tau Expression Changes in Injured and Regenerating RGCs
3.2. Tau is Not Required for Optic Nerve Axon Development and Regeneration after Crush Lesion
3.3. Tau Gene Deletion Does Not Rescue Injured RGCs from Optic Nerve Injury-Induced Cell Death
3.4. Differential Regenerative and Surviving Abilities of RGC Subtypes: Possible Implication for Tau
4. Material and Methods
4.1. Animals
4.2. Optic Nerve Injuries
4.3. Intravitreal Injections
4.4. Fundoscopy
4.5. Western Blot Analysis
4.6. Retinal Ganglion Cell (RGC) Survival
4.7. Immunofluorescence on Retinal and Optic Nerve Cryosections
4.8. Axonal Regeneration Analysis on Optic Nerve Sections
4.9. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | linear dichroism |
| AAV | Adeno-associated virus |
| CRMP2 | Collapsin response mediator protein-2 |
| CNS | Central nervous system |
| CNTF | Ciliary neurotrophic factor |
| CTb594 | Cholera toxin β subunit conjugated to Alexa 594 |
| GSK3β | Glycogen synthase kinase 3 β |
| ipRGCs | intrinsically photosensitive RGCs |
| KLF4 | Kruppel-like factor 4 |
| MAG | Myelin-associated glycoprotein |
| MAP 1A/1B | Microtubule-associated protein 1A/1B |
| mTOR | mammalian target of rapamycin |
| Nogo-A | Neurite outgrowth inhibitor A |
| OMgp | Oligodendrocyte myelin glycoprotein |
| ONC | Optic nerve crush |
| PBS | Phosphate buffered solution |
| PFA | Paraformaldehyde |
| PHF1 | Paired helical filaments 1 |
| RBPMS | RNA-binding protein with multiple splicing |
| RGC | Retinal ganglion cell |
| siRNA | small interfering RNA |
| Stat3 | Signal transducer and activator of transcription 3 |
| Tau KO | Tau knock-out |
| WT | Wild-type |
References
- Rodriguez, L.; Mdzomba, J.B.; Joly, S.; Boudreau-Laprise, M.; Planel, E.; Pernet, V. Human Tau Expression Does Not Induce Mouse Retina Neurodegeneration, Suggesting Differential Toxicity of Tau in Brain vs. Retinal Neurons. Front. Mol. Neurosci. 2018, 11, 293. [Google Scholar] [CrossRef] [PubMed]
- Chiasseu, M.; Cueva Vargas, J.L.; Destroismaisons, L.; Vande Velde, C.; Leclerc, N.; Di Polo, A. Tau Accumulation, Altered Phosphorylation, and Missorting Promote Neurodegeneration in Glaucoma. J. Neurosci. 2016, 36, 5785–5798. [Google Scholar] [CrossRef]
- Mey, J.; Thanos, S. Intravitreal injections of neurotrophic factors support the survival of axotomized retinal ganglion cells in adult rats in vivo. Brain Res. 1993, 602, 304–317. [Google Scholar] [CrossRef]
- Cheng, L.; Sapieha, P.; Kittlerova, P.; Hauswirth, W.W.; Di Polo, A. TrkB gene transfer protects retinal ganglion cells from axotomy-induced death in vivo. J. Neurosci. 2002, 22, 3977–3986. [Google Scholar] [CrossRef]
- Garcia-Sierra, F.; Wischik, C.M.; Harrington, C.R.; Luna-Munoz, J.; Mena, R. Accumulation of C-terminally truncated tau protein associated with vulnerability of the perforant pathway in early stages of neurofibrillary pathology in Alzheimer’s disease. J. Chem. Neuroanat. 2001, 22, 65–77. [Google Scholar] [CrossRef]
- Chiasseu, M.; Alarcon-Martinez, L.; Belforte, N.; Quintero, H.; Dotigny, F.; Destroismaisons, L.; Vande Velde, C.; Panayi, F.; Louis, C.; Di Polo, A. Tau accumulation in the retina promotes early neuronal dysfunction and precedes brain pathology in a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Biernat, J.; Wu, Y.Z.; Timm, T.; Zheng-Fischhofer, Q.; Mandelkow, E.; Meijer, L.; Mandelkow, E.M. Protein kinase MARK/PAR-1 is required for neurite outgrowth and establishment of neuronal polarity. Mol. Biol. Cell 2002, 13, 4013–4028. [Google Scholar] [CrossRef]
- Boyne, L.J.; Tessler, A.; Murray, M.; Fischer, I. Distribution of Big tau in the central nervous system of the adult and developing rat. J. Comp. Neurol. 1995, 358, 279–293. [Google Scholar] [CrossRef]
- Leibinger, M.; Andreadaki, A.; Golla, R.; Levin, E.; Hilla, A.M.; Diekmann, H.; Fischer, D. Boosting CNS axon regeneration by harnessing antagonistic effects of GSK3 activity. Proc. Natl. Acad. Sci. USA 2017, 114, E5454–E5463. [Google Scholar] [CrossRef] [PubMed]
- Sengottuvel, V.; Leibinger, M.; Pfreimer, M.; Andreadaki, A.; Fischer, D. Taxol facilitates axon regeneration in the mature CNS. J. Neurosci. 2011, 31, 2688–2699. [Google Scholar] [CrossRef]
- Berkelaar, M.; Clarke, D.B.; Wang, Y.C.; Bray, G.M.; Aguayo, A.J. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J. Neurosci. 1994, 14, 4368–4374. [Google Scholar] [CrossRef] [PubMed]
- Pernet, V.; Joly, S.; Dalkara, D.; Jordi, N.; Schwarz, O.; Christ, F.; Schaffer, D.V.; Flannery, J.G.; Schwab, M.E. Long-distance axonal regeneration induced by CNTF gene transfer is impaired by axonal misguidance in the injured adult optic nerve. Neurobiol. Dis. 2013, 51, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Park, K.K.; Liu, K.; Hu, Y.; Smith, P.D.; Wang, C.; Cai, B.; Xu, B.; Connolly, L.; Kramvis, I.; Sahin, M.; et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 2008, 322, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Sapieha, P.S.; Duplan, L.; Uetani, N.; Joly, S.; Tremblay, M.L.; Kennedy, T.E.; Di Polo, A. Receptor protein tyrosine phosphatase sigma inhibits axon regrowth in the adult injured CNS. Mol. Cell Neurosci. 2005, 28, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Kwong, J.M.; Caprioli, J.; Piri, N. RNA binding protein with multiple splicing: A new marker for retinal ganglion cells. Invest. Ophthalmol. Vis. Sci. 2010, 51, 1052–1058. [Google Scholar] [CrossRef] [PubMed]
- Leaver, S.G.; Cui, Q.; Plant, G.W.; Arulpragasam, A.; Hisheh, S.; Verhaagen, J.; Harvey, A.R. AAV-mediated expression of CNTF promotes long-term survival and regeneration of adult rat retinal ganglion cells. Gene Ther 2006, 13, 1328–1341. [Google Scholar] [CrossRef]
- Leaver, S.G.; Cui, Q.; Bernard, O.; Harvey, A.R. Cooperative effects of bcl-2 and AAV-mediated expression of CNTF on retinal ganglion cell survival and axonal regeneration in adult transgenic mice. Eur. J. Neurosci. 2006, 24, 3323–3332. [Google Scholar] [CrossRef]
- Muller, A.; Hauk, T.G.; Leibinger, M.; Marienfeld, R.; Fischer, D. Exogenous CNTF stimulates axon regeneration of retinal ganglion cells partially via endogenous CNTF. Mol. Cell Neurosci. 2009, 41, 233–246. [Google Scholar] [CrossRef]
- Leibinger, M.; Muller, A.; Andreadaki, A.; Hauk, T.G.; Kirsch, M.; Fischer, D. Neuroprotective and axon growth-promoting effects following inflammatory stimulation on mature retinal ganglion cells in mice depend on ciliary neurotrophic factor and leukemia inhibitory factor. J. Neurosci. 2009, 29, 14334–14341. [Google Scholar] [CrossRef]
- Hauk, T.G.; Leibinger, M.; Muller, A.; Andreadaki, A.; Knippschild, U.; Fischer, D. Stimulation of axon regeneration in the mature optic nerve by intravitreal application of the toll-like receptor 2 agonist Pam3Cys. Invest. Ophthalmol. Vis. Sci. 2010, 51, 459–464. [Google Scholar] [CrossRef]
- Leibinger, M.; Andreadaki, A.; Diekmann, H.; Fischer, D. Neuronal STAT3 activation is essential for CNTF- and inflammatory stimulation-induced CNS axon regeneration. Cell Death Dis. 2013, 4, e805. [Google Scholar] [CrossRef] [PubMed]
- Joly, S.; Dalkara, D.; Pernet, V. Sphingosine 1-Phosphate Receptor 1 Modulates CNTF-Induced Axonal Growth and Neuroprotection in the Mouse Visual System. Neural. Plast 2017, 2017, 6818970. [Google Scholar] [CrossRef] [PubMed]
- Klimczak, R.R.; Koerber, J.T.; Dalkara, D.; Flannery, J.G.; Schaffer, D.V. A novel adeno-associated viral variant for efficient and selective intravitreal transduction of rat Muller cells. PLoS ONE 2009, 4, e7467. [Google Scholar] [CrossRef]
- Lu, M.; Kosik, K.S. Competition for microtubule-binding with dual expression of tau missense and splice isoforms. Mol. Biol Cell 2001, 12, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Goode, B.L.; Feinstein, S.C. Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J. Cell Biol. 1994, 124, 769–782. [Google Scholar] [CrossRef]
- Butner, K.A.; Kirschner, M.W. Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 1991, 115, 717–730. [Google Scholar] [CrossRef]
- Pernet, V.; Joly, S.; Jordi, N.; Dalkara, D.; Guzik-Kornacka, A.; Flannery, J.G.; Schwab, M.E. Misguidance and modulation of axonal regeneration by Stat3 and Rho/ROCK signaling in the transparent optic nerve. Cell Death Dis. 2013, 4, e734. [Google Scholar] [CrossRef]
- Biswas, S.; Kalil, K. The Microtubule-Associated Protein Tau Mediates the Organization of Microtubules and Their Dynamic Exploration of Actin-Rich Lamellipodia and Filopodia of Cortical Growth Cones. J. Neurosci. 2018, 38, 291–307. [Google Scholar] [CrossRef]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef]
- Oku, H.; Kida, T.; Horie, T.; Taki, K.; Mimura, M.; Kojima, S.; Ikeda, T. Tau Is Involved in Death of Retinal Ganglion Cells of Rats From Optic Nerve Crush. Invest. Ophthalmol Vis. Sci. 2019, 60, 2380–2387. [Google Scholar] [CrossRef]
- Selles-Navarro, I.; Ellezam, B.; Fajardo, R.; Latour, M.; McKerracher, L. Retinal ganglion cell and nonneuronal cell responses to a microcrush lesion of adult rat optic nerve. Exp. Neurol. 2001, 167, 282–289. [Google Scholar] [CrossRef]
- Yin, Y.; Cui, Q.; Li, Y.; Irwin, N.; Fischer, D.; Harvey, A.R.; Benowitz, L.I. Macrophage-derived factors stimulate optic nerve regeneration. J. Neurosci. 2003, 23, 2284–2293. [Google Scholar] [CrossRef] [PubMed]
- Pease, M.E.; McKinnon, S.J.; Quigley, H.A.; Kerrigan-Baumrind, L.A.; Zack, D.J. Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma. Invest. Ophthalmol Vis. Sci. 2000, 41, 764–774. [Google Scholar] [PubMed]
- McKerracher, L.; Vidal-Sanz, M.; Essagian, C.; Aguayo, A.J. Selective impairment of slow axonal transport after optic nerve injury in adult rats. J. Neurosci. 1990, 10, 2834–2841. [Google Scholar] [CrossRef] [PubMed]
- Leon, S.; Yin, Y.; Nguyen, J.; Irwin, N.; Benowitz, L.I. Lens injury stimulates axon regeneration in the mature rat optic nerve. J. Neurosci. 2000, 20, 4615–4626. [Google Scholar] [CrossRef]
- Sun, F.; Park, K.K.; Belin, S.; Wang, D.; Lu, T.; Chen, G.; Zhang, K.; Yeung, C.; Feng, G.; Yankner, B.A.; et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature 2011, 480, 372–375. [Google Scholar] [CrossRef]
- Erturk, A.; Hellal, F.; Enes, J.; Bradke, F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J. Neurosci. 2007, 27, 9169–9180. [Google Scholar] [CrossRef]
- Sengottuvel, V.; Fischer, D. Facilitating axon regeneration in the injured CNS by microtubules stabilization. Commun. Integr. Biol. 2011, 4, 391–393. [Google Scholar] [CrossRef]
- Erturk, A.; Mauch, C.P.; Hellal, F.; Forstner, F.; Keck, T.; Becker, K.; Jahrling, N.; Steffens, H.; Richter, M.; Hubener, M.; et al. Three-dimensional imaging of the unsectioned adult spinal cord to assess axon regeneration and glial responses after injury. Nat. Med. 2011, 18, 166–171. [Google Scholar] [CrossRef]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef]
- Takei, Y.; Teng, J.; Harada, A.; Hirokawa, N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J. Cell Biol. 2000, 150, 989–1000. [Google Scholar] [CrossRef] [PubMed]
- Takei, Y.; Kondo, S.; Harada, A.; Inomata, S.; Noda, T.; Hirokawa, N. Delayed development of nervous system in mice homozygous for disrupted microtubule-associated protein 1B (MAP1B) gene. J. Cell Biol. 1997, 137, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Meixner, A.; Haverkamp, S.; Wassle, H.; Fuhrer, S.; Thalhammer, J.; Kropf, N.; Bittner, R.E.; Lassmann, H.; Wiche, G.; Propst, F. MAP1B is required for axon guidance and Is involved in the development of the central and peripheral nervous system. J. Cell Biol. 2000, 151, 1169–1178. [Google Scholar] [CrossRef]
- Edelmann, W.; Zervas, M.; Costello, P.; Roback, L.; Fischer, I.; Hammarback, J.A.; Cowan, N.; Davies, P.; Wainer, B.; Kucherlapati, R. Neuronal abnormalities in microtubule-associated protein 1B mutant mice. Proc. Natl. Acad. Sci. USA 1996, 93, 1270–1275. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, L.; Joly, S.; Zine-Eddine, F.; Mdzomba, J.B.; Pernet, V. Tau modulates visual plasticity in adult and aging mice. Neurobiol. Aging 2020. under review. [Google Scholar]
- Yuan, A.; Kumar, A.; Peterhoff, C.; Duff, K.; Nixon, R.A. Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J. Neurosci. 2008, 28, 1682–1687. [Google Scholar] [CrossRef]
- Dieterich, D.C.; Trivedi, N.; Engelmann, R.; Gundelfinger, E.D.; Gordon-Weeks, P.R.; Kreutz, M.R. Partial regeneration and long-term survival of rat retinal ganglion cells after optic nerve crush is accompanied by altered expression, phosphorylation and distribution of cytoskeletal proteins. Eur J. Neurosci. 2002, 15, 1433–1443. [Google Scholar] [CrossRef] [PubMed]
- Badea, T.C.; Nathans, J. Quantitative analysis of neuronal morphologies in the mouse retina visualized by using a genetically directed reporter. J. Comp. Neurol. 2004, 480, 331–351. [Google Scholar] [CrossRef]
- Li, R.S.; Chen, B.Y.; Tay, D.K.; Chan, H.H.; Pu, M.L.; So, K.F. Melanopsin-expressing retinal ganglion cells are more injury-resistant in a chronic ocular hypertension model. Invest. Ophthalmol. Vis. Sci. 2006, 47, 2951–2958. [Google Scholar] [CrossRef]
- Duan, X.; Qiao, M.; Bei, F.; Kim, I.J.; He, Z.; Sanes, J.R. Subtype-specific regeneration of retinal ganglion cells following axotomy: Effects of osteopontin and mTOR signaling. Neuron 2015, 85, 1244–1256. [Google Scholar] [CrossRef]
- Bray, E.R.; Yungher, B.J.; Levay, K.; Ribeiro, M.; Dvoryanchikov, G.; Ayupe, A.C.; Thakor, K.; Marks, V.; Randolph, M.; Danzi, M.C.; et al. Thrombospondin-1 Mediates Axon Regeneration in Retinal Ganglion Cells. Neuron 2019, 103, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Hattar, S.; Liao, H.W.; Takao, M.; Berson, D.M.; Yau, K.W. Melanopsin-containing retinal ganglion cells: Architecture, projections, and intrinsic photosensitivity. Science 2002, 295, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Tucker, K.L.; Meyer, M.; Barde, Y.A. Neurotrophins are required for nerve growth during development. Nat. Neurosci. 2001, 4, 29–37. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, L.; Joly, S.; Mdzomba, J.B.; Pernet, V. Tau Gene Deletion Does Not Influence Axonal Regeneration and Retinal Neuron Survival in the Injured Mouse Visual System. Int. J. Mol. Sci. 2020, 21, 4100. https://doi.org/10.3390/ijms21114100
Rodriguez L, Joly S, Mdzomba JB, Pernet V. Tau Gene Deletion Does Not Influence Axonal Regeneration and Retinal Neuron Survival in the Injured Mouse Visual System. International Journal of Molecular Sciences. 2020; 21(11):4100. https://doi.org/10.3390/ijms21114100
Chicago/Turabian StyleRodriguez, Léa, Sandrine Joly, Julius Baya Mdzomba, and Vincent Pernet. 2020. "Tau Gene Deletion Does Not Influence Axonal Regeneration and Retinal Neuron Survival in the Injured Mouse Visual System" International Journal of Molecular Sciences 21, no. 11: 4100. https://doi.org/10.3390/ijms21114100
APA StyleRodriguez, L., Joly, S., Mdzomba, J. B., & Pernet, V. (2020). Tau Gene Deletion Does Not Influence Axonal Regeneration and Retinal Neuron Survival in the Injured Mouse Visual System. International Journal of Molecular Sciences, 21(11), 4100. https://doi.org/10.3390/ijms21114100