Structural Basis of Oligomerization of N-Terminal Domain of Spider Aciniform Silk Protein

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Sequences of AcSpN and MaSpN from NA
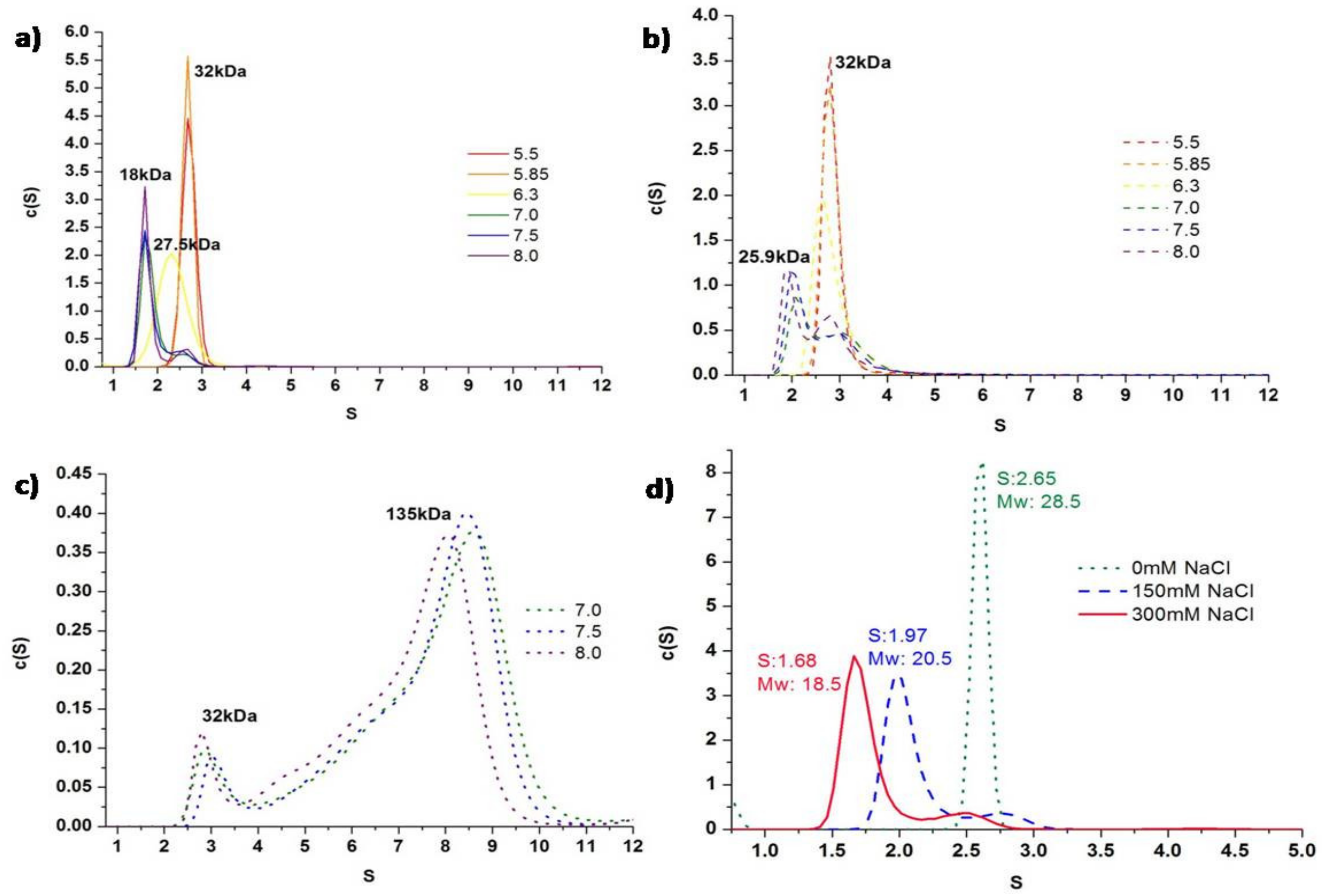
2.2. Effects of Salt Concentration and pH on Oligomerization of MaSpN and AcSpN
2.3. Structures of AcSpN and MaSpN Monomers from NA
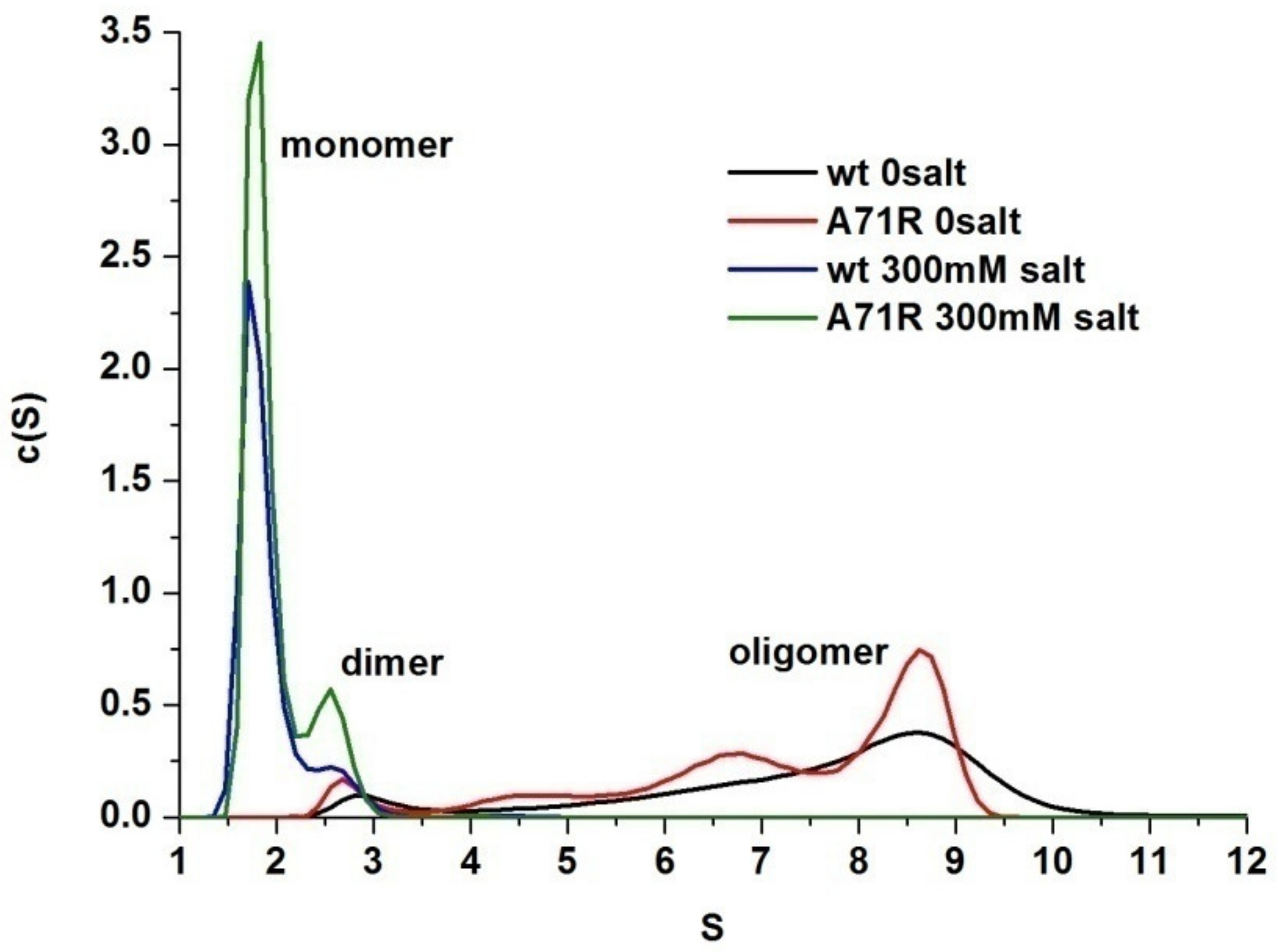
2.4. Identification of Oligomerization Interfaces for AcSpN
2.5. Structural Models of AcSpN Dimers and Oligomers
3. Materials and Methods
3.1. Sequence Identification, Cloning, Expression, and Purification
3.2. Size Exclusion Chromatography
3.3. Analytical Ultracentrifugation
3.4. NMR Spectroscopy and Structure Calculation
3.5. Hydrogen–Deuterium Exchange Mass Spectrometry (HDXMS)
3.6. Modelling of AcSpN Dimers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NMR | Nuclear magnetic resonance |
| HDXMS | Hydrogen–deuterium exchange mass spectrometry |
| CTD | C-terminal domain |
| NTD | N-terminal domain |
| RP | Repetitive domain |
| AcSp | Aciniform spidroin |
| MaSp | Major ampullate spidroin |
| AcSpN | NTD of AcSp |
| MaSpN | NTD of MaSp |
| AUC | Analytical ultra-centrifugation |
| HSQC | Heteronuclear single-quantum correlation |
References
- Humenik, M.; Pawar, K.; Scheibel, T. Nanostructured, Self-Assembled Spider Silk Materials for Biomedical Applications. Adv. Exp. Med. Biol. 2019, 1174, 187–221. [Google Scholar]
- Lewis, R.V. Spider silk: Ancient ideas for new biomaterials. Chem. Rev. 2006, 106, 3762–3774. [Google Scholar]
- Omenetto, F.G.; Kaplan, D.L. New Opportunities for an Ancient Material. Science 2010, 329, 528–531. [Google Scholar]
- Romer, L.; Scheibel, T. The elaborate structure of spider silk Structure and function of a natural high performance fiber. Prion 2008, 2, 154–161. [Google Scholar]
- Greenwood, M.J.; McIntosh, A.R.; Harding, J.S. Disturbance across an ecosystem boundary drives cannibalism propensity in a riparian consumer. Behav. Ecol. 2010, 21, 1227–1235. [Google Scholar]
- Rising, A.; Widhe, M.; Johansson, J.; Hedhammar, M. Spider silk proteins: Recent advances in recombinant production, structure-function relationships and biomedical applications. Cell. Mol. Life Sci. 2011, 68, 169–184. [Google Scholar]
- Teule, F.; Cooper, A.R.; Furin, W.A.; Bittencourt, D.; Rech, E.L.; Brooks, A.; Lewis, R.V. A protocol for the production of recombinant spider silk-like proteins for artificial fiber spinning. Nat. Protoc. 2009, 4, 341–355. [Google Scholar]
- Spiess, K.; Lammel, A.; Scheibel, T. Recombinant Spider Silk Proteins for Applications in Biomaterials. Macromol. Biosci. 2010, 10, 998–1007. [Google Scholar]
- Lin, Z.; Deng, Q.; Liu, X.Y.; Yang, D. Engineered large spider eggcase silk protein for strong artificial fibers. Adv. Mater. 2013, 25, 1216–1220. [Google Scholar]
- Andersson, M.; Jia, Q.; Abella, A.; Lee, X.Y.; Landreh, M.; Purhonen, P.; Hebert, H.; Tenje, M.; Robinson, C.V.; Meng, Q.; et al. Biomimetic spinning of artificial spider silk from a chimeric minispidroin. Nat. Chem. Biol. 2017, 13, 262–264. [Google Scholar]
- Hayashi, C.Y.; Lewis, R.V. Molecular architecture and evolution of a modular spider silk protein gene. Science 2000, 287, 1477–1479. [Google Scholar]
- Hayashi, C.Y.; Blackledge, T.A.; Lewis, R.V. Molecular and mechanical characterization of aciniform silk: Uniformity of iterated sequence modules in a novel member of the spider silk fibroin gene family. Mol. Biol. Evol. 2004, 21, 1950–1959. [Google Scholar]
- Huang, W.; Lin, Z.; Sin, Y.M.; Li, D.; Gong, Z.; Yang, D. Characterization and expression of a cDNA encoding a tubuliform silk protein of the golden web spider Nephila antipodiana. Biochimie 2006, 88, 849–858. [Google Scholar]
- Ayoub, N.A.; Garb, J.E.; Kuelbs, A.; Hayashi, C.Y. Ancient properties of spider silks revealed by the complete gene sequence of the prey-wrapping silk protein (AcSp1). Mol. Biol. Evol. 2013, 30, 589–601. [Google Scholar]
- Ayoub, N.A.; Garb, J.E.; Tinghitella, R.M.; Collin, M.A.; Hayashi, C.Y. Blueprint for a High-Performance Biomaterial: Full-Length Spider Dragline Silk Genes. PLoS ONE 2007, 2, e514. [Google Scholar]
- Lin, Z.; Huang, W.D.; Zhang, J.F.; Fan, J.S.; Yang, D. Solution structure of eggcase silk protein and its implications for silk fiber formation. Proc. Natl. Acad. Sci. USA 2009, 106, 8906–8911. [Google Scholar]
- Challis, R.J.; Goodacre, S.L.; Hewitt, G.M. Evolution of spider silks: Conservation and diversification of the C-terminus. Insect Mol. Biol. 2006, 15, 45–56. [Google Scholar]
- Hagn, F.; Eisoldt, L.; Hardy, J.G.; Vendrely, C.; Coles, M.; Scheibel, T.; Kessler, H. A conserved spider silk domain acts as a molecular switch that controls fibre assembly. Nature 2010, 465, 239–242. [Google Scholar]
- Gao, Z.; Lin, Z.; Huang, W.; Lai, C.C.; Fan, J.S.; Yang, D. Structural characterization of minor ampullate spidroin domains and their distinct roles in fibroin solubility and fiber formation. PLoS ONE 2013, 8, e56142. [Google Scholar]
- Wang, S.; Huang, W.; Yang, D. Structure and function of C-terminal domain of aciniform spidroin. Biomacromolecules 2014, 15, 468–477. [Google Scholar]
- Askarieh, G.; Hedhammar, M.; Nordling, K.; Saenz, A.; Casals, C.; Rising, A.; Johansson, J.; Knight, S.D. Self-assembly of spider silk proteins is controlled by a pH-sensitive relay. Nature 2010, 465, 236–238. [Google Scholar]
- Hijirida, D.H.; Do, K.G.; Michal, C.; Wong, S.; Zax, D.; Jelinski, L.W. C-13 NMR of Nephila clavipes major ampullate silk gland. Biophys. J. 1996, 71, 3442–3447. [Google Scholar]
- Landreh, M.; Askarieh, G.; Nordling, K.; Hedhammar, M.; Rising, A.; Casals, C.; Astorga-Wells, J.; Alvelius, G.; Knight, S.D.; Johansson, J.; et al. A pH-Dependent Dimer Lock in Spider Silk Protein. J. Mol. Biol. 2010, 404, 328–336. [Google Scholar]
- Jaudzems, K.; Askarieh, G.; Landreh, M.; Nordling, K.; Hedhammar, M.; Jörnvall, H.; Rising, A.; Knight, S.D.; Johansson, J. pH-dependent dimerization of spider silk N-terminal domain requires relocation of a wedged tryptophan side chain. J. Mol. Biol. 2012, 422, 477–487. [Google Scholar]
- Hagn, F.; Thamm, C.; Scheibel, T.; Kessler, H. pH-Dependent Dimerization and Salt-Dependent Stabilization of the N-terminal Domain of Spider Dragline Silk-Implications for Fiber Formation. Angew. Chem. Int. Ed. 2011, 50, 310–313. [Google Scholar]
- Bauer, J.; Schaal, D.; Eisoldt, L.; Schweimer, K.; Schwarzinger, S.; Scheibel, T. Acidic Residues Control the Dimerization of the N-terminal Domain of Black Widow Spiders’ Major Ampullate Spidroin 1. Sci. Rep. 2016, 6, 34442. [Google Scholar]
- Gaines, W.A.; Sehorn, M.G.; Marcotte, W.R.J. Spidroin N-terminal domain promotes a pH-dependent association of silk proteins during self-assembly. J. Biol. Chem. 2010, 285, 40745–40753. [Google Scholar]
- Kronqvist, N.; Otikovs, M.; Chmyrov, V.; Chen, G.; Andersson, M.; Nordling, K.; Landreh, M.; Sarr, M.; Jörnvall, H.; Wennmalm, S.; et al. Sequential pH-driven dimerization and stabilization of the N-terminal domain enables rapid spider silk formation. Nat. Commun. 2014, 5, 3254. [Google Scholar]
- Andersson, M.; Chen, G.; Otikovs, M.; Landreh, M.; Nordling, K.; Kronqvist, N.; Westermark, P.; Jörnvall, H.; Knight, S.; Ridderstråle, Y.; et al. Carbonic anhydrase generates CO2 and H+ that drive spider silk formation via opposite effects on the terminal domains. PLoS. Biol. 2014, 12, e1001921. [Google Scholar]
- Otikovs, M.; Chen, G.; Nordling, K.; Landreh, M.; Meng, Q.; Jörnvall, H.; Kronqvist, N.; Rising, A.; Johansson, J.; Jaudzems, K. Diversified Structural Basis of a Conserved Molecular Mechanism for pH-Dependent Dimerization in Spider Silk N-Terminal Domains. Chembiochem 2015, 16, 1720–1724. [Google Scholar]
- Rising, A.; Hjalm, G.; Engstrom, W.; Johansson, J. N-terminal nonrepetitive domain common to dragline, flagelliform, and cylindriform spider silk proteins. Biomacromolecules 2006, 7, 3120–3124. [Google Scholar]
- Tsumoto, K.; Ejima, D.; Senczuk, A.M.; Kita, Y.; Arakawa, T. Effects of salts on protein-surface interactions: Applications for column chromatography. J. Pharm. Sci. 2007, 96, 1677–1690. [Google Scholar]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar]
- Wales, T.E.; Engen, J.R. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom. Rev. 2006, 25, 158–170. [Google Scholar]
- Lim, X.X.; Chandramohan, A.; Lim, X.E.; Crowe, J.E.J.; Lok, S.M.; Anand, G.S. Epitope and Paratope Mapping Reveals Temperature-Dependent Alterations in the Dengue-Antibody Interface. Structure 2017, 25, 1391–1402. [Google Scholar]
- Li, G.Z.; Vissers, J.P.; Silva, J.C.; Golick, D.; Gorenstein, M.V.; Geromanos, S.J. Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics 2009, 9, 1696–1719. [Google Scholar]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar]
- Bauer, J.; Scheibel, T. Dimerization of the Conserved N-Terminal Domain of a Spider Silk Protein Controls the Self-Assembly of the Repetitive Core Domain. Biomacromolecules 2017, 18, 2521–2528. [Google Scholar]
- Wang, S.; Huang, W.; Yang, D. NMR structure note: Repetitive domain of aciniform spidroin 1 from Nephila antipodiana. J. Biomol. NMR 2012, 54, 415–420. [Google Scholar]
- Motriuk-Smith, D.; Smith, A.; Hayashi, C.Y.; Lewis, R.V. Analysis of the conserved N-terminal domains in major ampullate spider silk proteins. Biomacromolecules 2005, 6, 3152–3159. [Google Scholar]
- Brown, P.H.; Schuck, P. Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 2006, 90, 4651–4661. [Google Scholar]
- Kanelis, V.; Forman-Kay, J.D.; Kay, L.E. Multidimensional NMR methods for protein structure determination. IUBMB Life 2001, 52, 291–302. [Google Scholar]
- Yang, D.; Zheng, Y.; Liu, D.J.; Wyss, D.F. Sequence-specific assignments of methyl groups in high-molecular weight proteins. J. Am. Chem. Soc. 2004, 126, 3710–3711. [Google Scholar]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A.D. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar]
- Lin, Z.; Xu, Y.Q.; Yang, S.; Yang, D. Sequence-specific assignment of aromatic resonances of uniformly C-13,N-15-labeled proteins by using C-13- and N-15-edited NOESY spectra. Angew. Chem. Int. Ed. Engl. 2006, 45, 1960–1963. [Google Scholar]
- Xu, Y.; Zheng, Y.; Fan, J.; Yang, D. A new strategy for structure determination of large proteins in solution without deuteration. Nat. Methods 2006, 3, 931–937. [Google Scholar]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [Google Scholar]
- Schwieters, C.D.; Kuszewski, J.J.; Clore, G.M. Using Xplor-NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc. 2006, 48, 47–62. [Google Scholar]
- Tian, Y.; Schwieters, C.D.; Opella, S.J.; Marassi, F.M. A Practical Implicit Solvent Potential for NMR Structure Calculation. J. Magn. Res. 2014, 243, 54–64. [Google Scholar]
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar]
- Laskowski, R.A.; Rullmann, J.A.C.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakraborty, R.; Fan, J.-s.; Lai, C.C.; Raghuvamsi, P.V.; Chee, P.X.; Anand, G.S.; Yang, D. Structural Basis of Oligomerization of N-Terminal Domain of Spider Aciniform Silk Protein. Int. J. Mol. Sci. 2020, 21, 4466. https://doi.org/10.3390/ijms21124466
Chakraborty R, Fan J-s, Lai CC, Raghuvamsi PV, Chee PX, Anand GS, Yang D. Structural Basis of Oligomerization of N-Terminal Domain of Spider Aciniform Silk Protein. International Journal of Molecular Sciences. 2020; 21(12):4466. https://doi.org/10.3390/ijms21124466
Chicago/Turabian StyleChakraborty, Rusha, Jing-song Fan, Chong Cheong Lai, Palur Venkata Raghuvamsi, Pin Xuan Chee, Ganesh Srinivasan Anand, and Daiwen Yang. 2020. "Structural Basis of Oligomerization of N-Terminal Domain of Spider Aciniform Silk Protein" International Journal of Molecular Sciences 21, no. 12: 4466. https://doi.org/10.3390/ijms21124466
APA StyleChakraborty, R., Fan, J.-s., Lai, C. C., Raghuvamsi, P. V., Chee, P. X., Anand, G. S., & Yang, D. (2020). Structural Basis of Oligomerization of N-Terminal Domain of Spider Aciniform Silk Protein. International Journal of Molecular Sciences, 21(12), 4466. https://doi.org/10.3390/ijms21124466