Integrated RNA-seq Analysis and Meta-QTLs Mapping Provide Insights into Cold Stress Response in Rice Seedling Roots




Abstract
:1. Introduction
2. Results
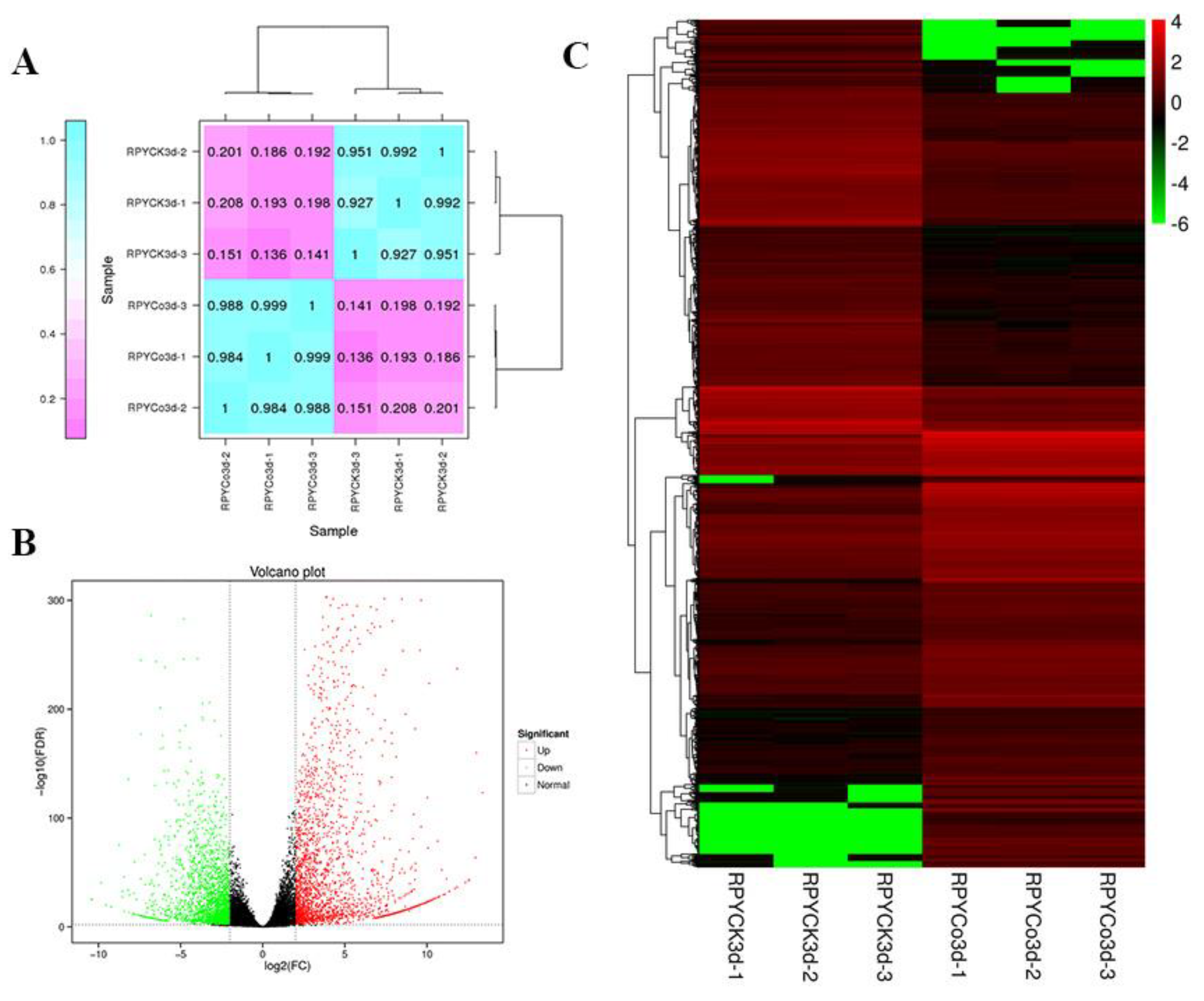
2.1. Transcriptome Sequencing and DEGs Analysis
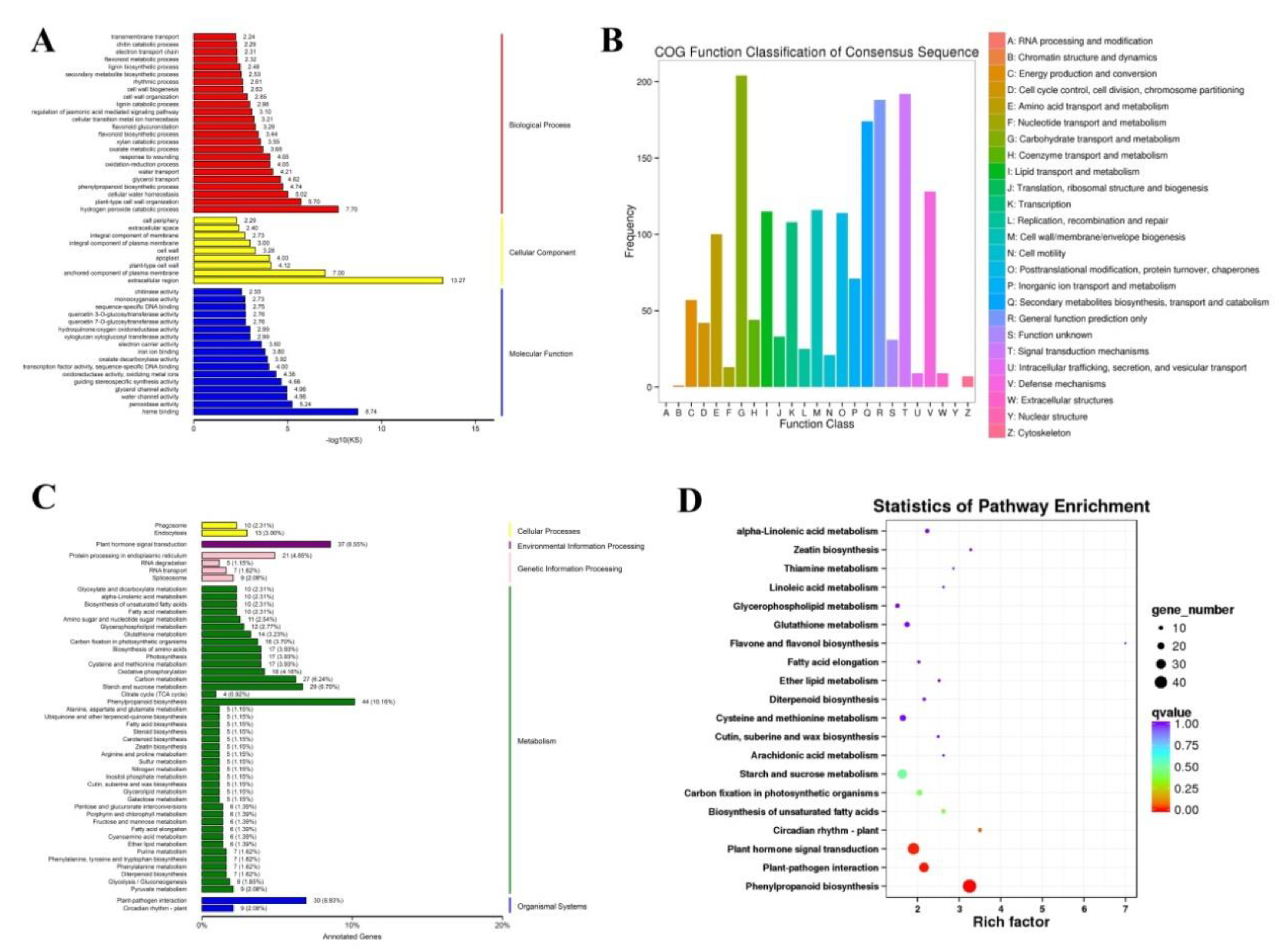
2.2. GO, COG, KEGG Enrichment Analysis of the DEGs
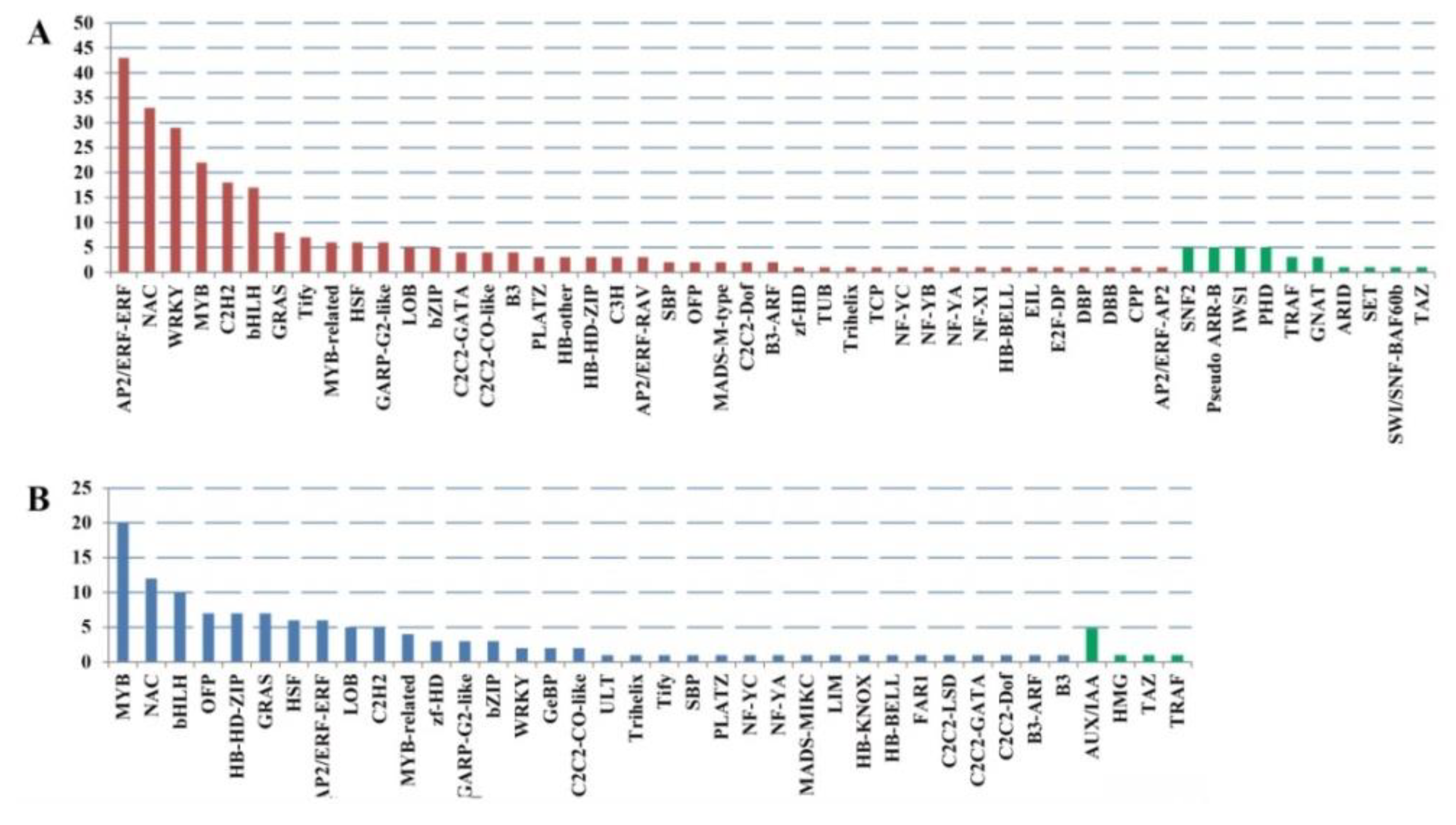
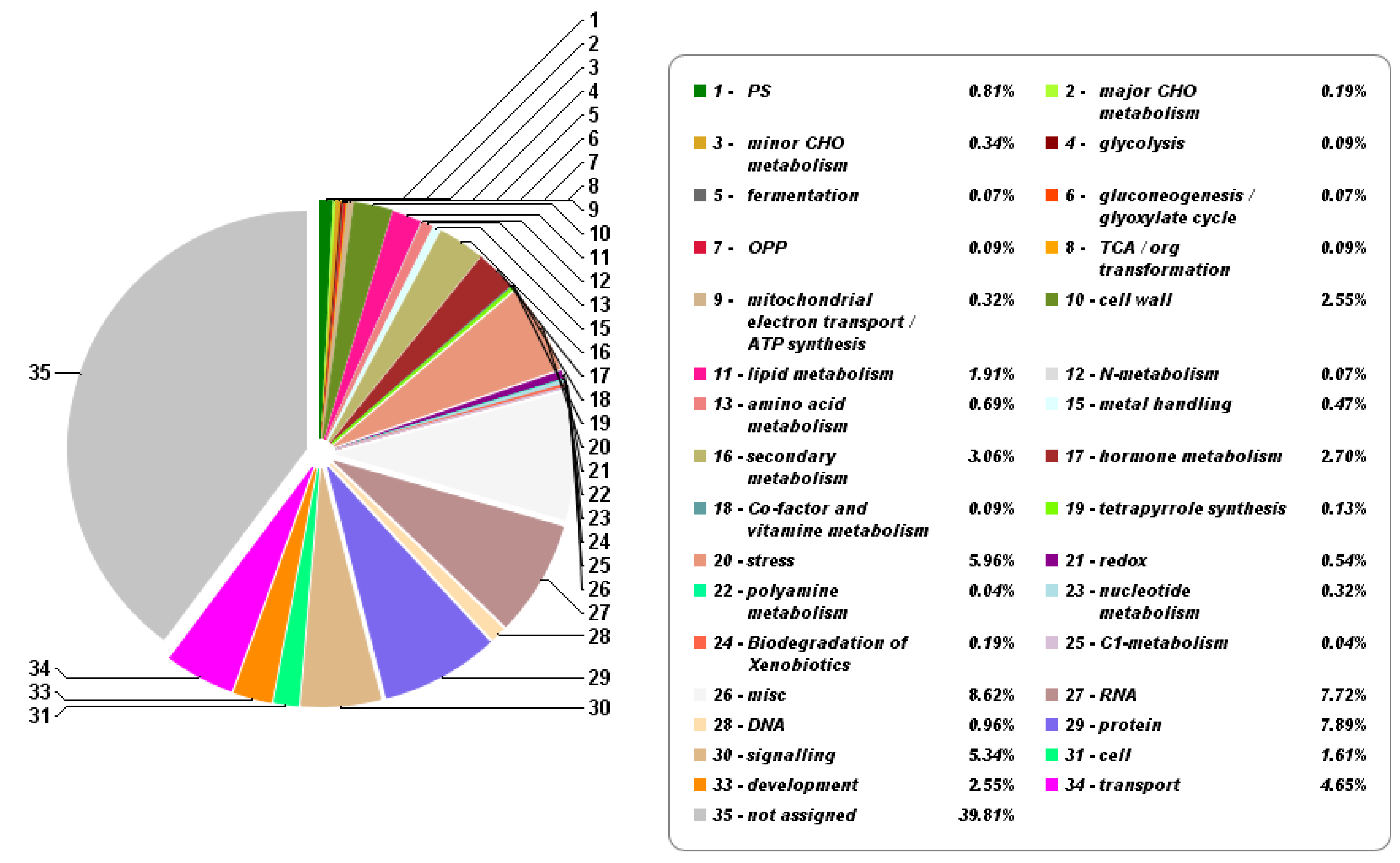
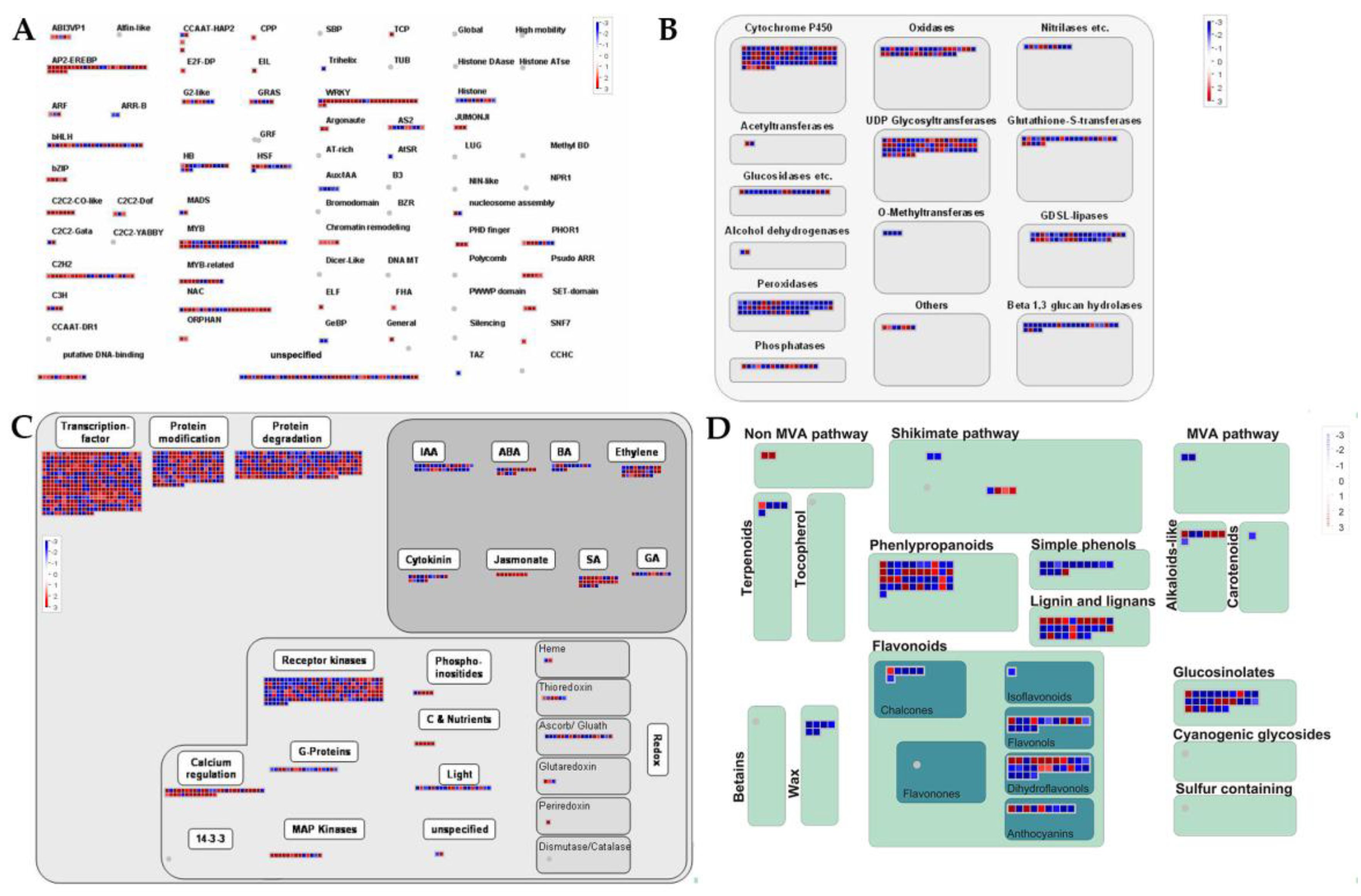
2.3. Cold Stress-Related TFs and Salt-Stress-Affected Pathways
2.4. Key DEGs locking Involved in the Response to Cold Stress
3. Discussion
3.1. Cold-Stress-Responsive DEGs in Rice Seedling Roots Associated with Plant Stress/Hormone Signal Transduction, Secondary Metabolites Mechanisms, TFs, and Function Proteins
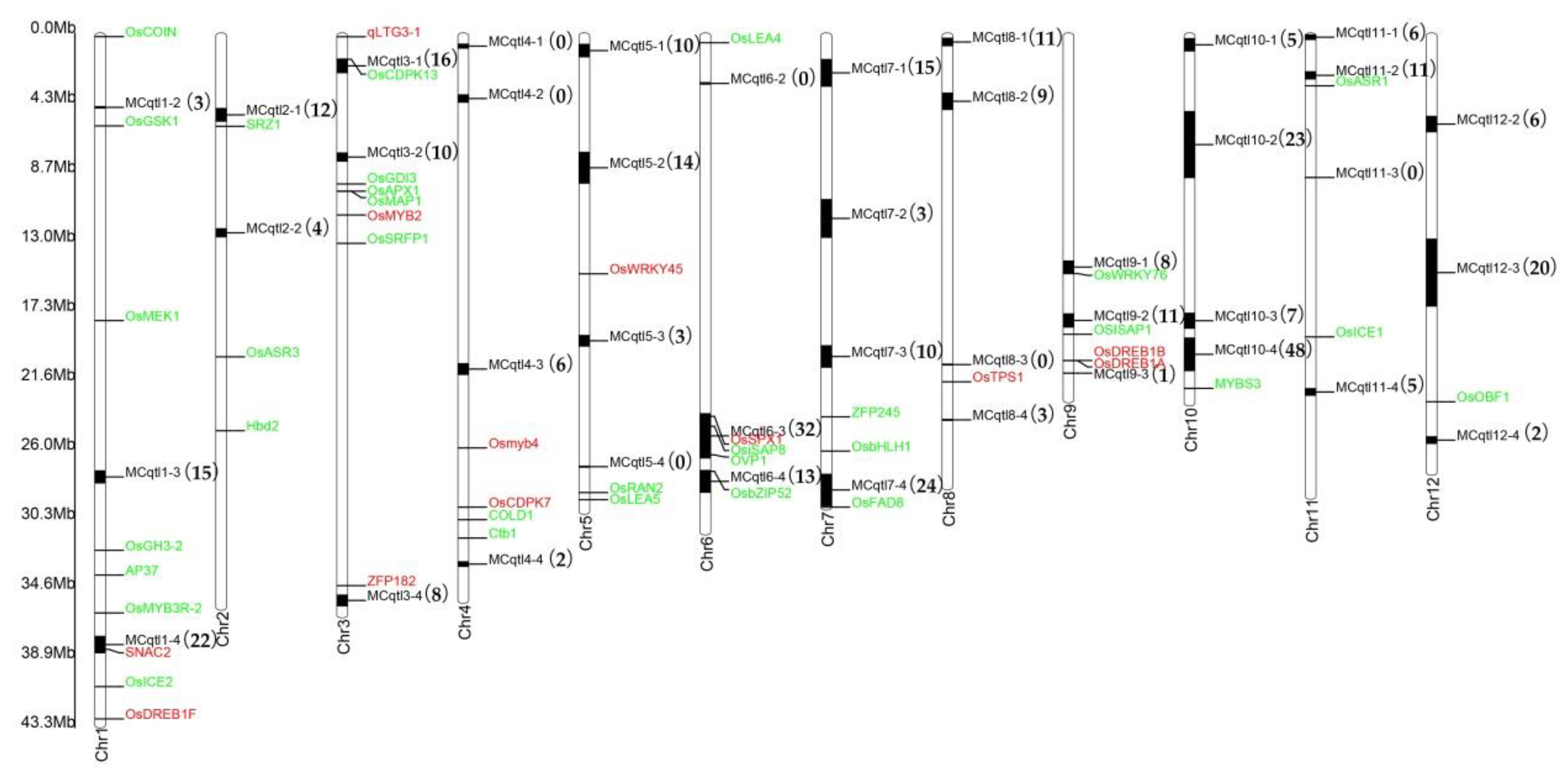
3.2. Meta-QTLs Mapping of DEGs Can Effectively Lock Main Effect Candidate Genes Related to Cold Stress Tolerance in Rice
4. Materials and Methods
4.1. Plant Materials and Treatments
4.2. Transcriptome Sequencing and RNA-seq Analysis
4.3. Functional Annotation and GO, KEGG Enrichment Analysis of the DEGs
4.4. Transcription Factors (TFs) and Transcriptional Regulators (TRs) Identification and Meta-QTLs Mapping.
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Song, Y.Q.; Jie, G.; Di, H.W.; Xin, D.T.; Jie, D.L. Comparative transcriptomics analysis reveals difference of key gene expression between banana and plantain in response to cold stress. BMC Genom. 2015, 16, 446. [Google Scholar]
- Guan, S.X.; Xu, Q.; Ma, D.R.; Zhang, W.Z.; Xu, Z.J.; Zhao, M.H.; Guo, Z.F. Transcriptomics profiling in response to cold stress in cultivated rice and weedy rice. Gene 2019, 685, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.W.; Chen, H.C.; Jen, W.F.; Liu, L.Y.; Chang, M.C. Comparative transcriptome analysis of shoots and roots of TNG67 and TCN1 rice seedlings under cold stress and following subsequent recovery: insights into metabolic pathways, phytohormones, and transcription factors. PLoS ONE 2015, 10, e0131391. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Hwang, S.; Kim, D.S.; Jang, C.S. Genome-wide transcriptome analysis of rice genes responsive to chilling stress. Can. J. Plant Sci. 2012, 92, 447–460. [Google Scholar] [CrossRef] [Green Version]
- Pradhan, S.K.; Nayak, D.K.; Guru, M.; Pandit, E.; Das, S.; Barik, S.R.; Mohanty, S.P.; Anandan, A. Screening and classification of genotypes for seedling-stage chilling stress tolerance in rice and validation of the trait using SSR markers. Plant Genet. Resour. 2016, 14, 173–182. [Google Scholar] [CrossRef]
- Pandit, E.; Swaleha, T.; Barik, S.R.; Mohanty, D.P.; Nayak, D.K.; Mohanty, S.P.; Das, S.; Pradhan, S.K. Genome-wide association mapping reveals multiple QTLs governing tolerance response for seedling stage chilling stress in Indica rice. Front. Plant Sci. 2017, 8, 552. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Liu, S.J.; Hou, M.Y.; Tang, J.Y.; Chen, L.M.; Zhai, H.Q.; Wan, J.M. Analysis of QTLs for seed low temperature germinability and anoxia germinability in rice (Oryza sativa L.). Field Crop. Res. 2006, 98, 68–75. [Google Scholar] [CrossRef]
- Yang, L.M.; Wang, J.X.; Lei, L.; Wang, J.G.; Muhammad, J.S.; Liu, H.L.; Sun, J.; Zheng, H.L.; Zhao, H.W.; Zou, D.t. QTL mapping for heading date, leaf area and chlorophyll content under cold and drought stress in two related recombinant inbred line populations (Japonica rice) and meta-analysis. Plant Breed. 2016, 2018, 1–19. [Google Scholar] [CrossRef]
- Mao, D.H.; Xin, Y.Y.; Tan, Y.J.; Hu, X.J.; Bai, J.J.; Liu, Z.Y.; Yu, Y.L.; Li, L.Y.; Peng, C.; Fan, T. Natural variation in the HAN1 gene confers chilling tolerance in rice and allowed adaptation to a temperate climate. Proc. Natl. Acad. Sci. USA 2019, 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Yun, M.; Dai, X.; Xu, Y.; Wei, L.; Zheng, X.; Zeng, D.; Pan, Y.; Lin, X.; Liu, H.; Zhang, D. COLD1 confers chilling tolerance in rice. Cell 2015, 6, 1209–1221. [Google Scholar]
- Liu, C.T.; Ou, S.J.; Mao, B.G.; Tang, J.Y.; Wang, W.; Wang, H.R.; Cao, S.Y.; Schlappi, M.; Zhao, B.R.; Xiao, G.Y. Early selection of bZIP73 facilitated adaptation of japonica rice to cold climates. Nat. Commun. 2018, 9, 3302. [Google Scholar] [CrossRef] [PubMed]
- Glass, G.V. Primary, secondary, and Meta-analysis of research. Educ. Res. 1976, 5, 3–8. [Google Scholar] [CrossRef]
- Yang, L.M. Genetic Analysis of Cold Tolerance QTL in Rice at Booting Stage; Northeast Agricultural University: Harbin, China, 2018. [Google Scholar]
- Ito, Y.; Katsura, K.; Maruyama, K.; Taji, T.; Kobayashi, M.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, a.K. Functional analysis of rice DREB1/CBF-type transcription factors involved in cold-responsive gene expression in transgenic rice. Plant Cell Physiol. 2006, 47, 141–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnusamy, V.; Zhu, J.; Zhu, J.K. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007, 12, 444–451. [Google Scholar] [CrossRef]
- Su, C.; Wang, Y.; Hsieh, T.; Lu, C.; Tseng, T.; Yu, S. A novel MYBS3-dependent pathway confers cold tolerance in rice. Plant Physiol. 2010, 153, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Chinnusamy, V.; Ohta, M.; Kanrar, S.; Lee, B.H.; Hong, X.; Agarwal, M.; Zhu, J.K. ICE1, a regulator of cold induced transcriptome and freezing tolerance in plants. Genes Dev. 2003, 17, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- Fursova, O.V.; Pogorelko, G.V.; Tarasov, V.A. Identification of ICE2, a gene involved in cold acclimation which determines freezing tolerance in Arabidopsis thaliana. Gene 2009, 429, 98–103. [Google Scholar] [CrossRef]
- Nakashima, K.; Takasaki, H.; Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. NAC transcription factors in plant abiotic stress responses. Bba Gene Regul. Mech. 2012, 1819, 97–103. [Google Scholar] [CrossRef]
- Maia, L.C.D.; Cadore, P.R.B.; Benitez, L.C.; Danielowski, R.; Braga, E.J.B.; Fagundes, P.R.R.; MagalhãEs, A.M.; Antonio, C.D.O. Transcriptome profiling of rice seedlings under cold stress. Funct. Plant Biol. 2017, 44, 419. [Google Scholar] [CrossRef]
- Zhang, T.; Huang, L.; Wang, Y.; Wang, W.; Zhao, X.; Zhang, S.; Jing, Z.; Hu, F.; Fu, B.; Li, Z. Differential transcriptome profiling of chilling stress response between shoots and rhizomes of Oryza longistaminata using RNA sequencing. PLoS ONE 2017, 12, e0188625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.Z.; Jin, Y.N.; Ding, X.H.; Wang, W.J.; Zhai, S.S.; Bai, L.P.; Guo, Z.F. Gene regulation and signal transduction in the ICE–CBF–COR signaling pathway during cold stress in plants. Biochemistry 2017, 82, 1103–1117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Zhao, X.Q.; Wang, W.S.; Pan, Y.J.; Huang, L.Y.; Liu, X.Y.; Zong, Y.; Zhu, L.H.; Yang, D.C.; Fu, B.Y. Comparative transcriptome profiling of chilling stress responsiveness in two contrasting rice genotypes. PLoS ONE 2012, 7, e43274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, C.; Li, D.; He, R.; Fang, Z.; Xia, Y.; Gao, J.; Shen, H.; Cao, M. Comparative transcriptome analysis of RNA-seq data for cold-tolerant and cold-sensitive rice genotypes under cold stress. J. Plant Biol. 2014, 57, 337–348. [Google Scholar] [CrossRef]
- Pradhan, S.K.; Pandit, E.; Nayak, D.K.; Behera, L.; Mohapatra, T. Genes, pathways and transcription factors involved in seedling stage chilling stress tolerance in indica rice through RNA-Seq analysis. BMC Plant Biol. 2019, 19, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Ma, X.D.; Cui, D.; Wang, Y.J.; Geng, L.Y.; Cao, G.L.; Zhang, H.; Han, L.Z. Comprehensive evaluation and analysis of the mechanism of cold tolerance based on the transcriptome of weedy rice seedlings. Rice 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Ouyang, B.; Zhang, J.H.; Wang, T.T.; Li, H.X.; Zhang, Y.Y.; Yu, C.Y.; Ye, Z.B. Differential modulation of photosynthesis, signaling, and transcriptional regulation between tolerant and sensitive tomato genotypes under cold stress. PLoS ONE 2012, 7, e50785. [Google Scholar] [CrossRef]
- Xin, H.P.; Zhu, W.; Wang, L.N.; Xiang, Y.; Fang, L.C.; Li, J.T.; Sun, X.M.; Wang, N.; Londo, J.P.; Li, S.H. Genome wide transcriptional profile analysis of Vitis amurensis and Vitis vinifera in response to cold stress. PLoS ONE 2013, 8, e58740. [Google Scholar] [CrossRef]
- Saijo, Y.; Hata, S.; Kyozuka, J.; Shimamoto, K.; Izui, K. Over-expression of a single Ca2+-dependent protein kinase confers both cold and salt/drought tolerance on rice plants. Plant J. 2000, 23, 319–327. [Google Scholar] [CrossRef]
- Tao, Z.; Kou, Y.; Liu, H.; Li, X.; Xiao, J.; Wang, S. OsWRKY45 alleles play different roles in abscisic acid signalling and salt stress tolerance but similar roles in drought and cold tolerance in rice. J. Exp. Bot. 2011, 62, 4863–4874. [Google Scholar] [CrossRef]
- Vannini, C.; Locatelli, F.; Bracale, M.; Magnani, E.; Coraggio, I. Overexpression of the rice OsMYB4 gene increases chilling and freezing tolerance of Arabidopsis thaliana plants. Plant J. 2004, 37, 115–127. [Google Scholar] [CrossRef]
- Ma, Q.B.; Dai, X.Y.; Xu, Y.Y.; Guo, J.; Liu, Y.J.; Chen, N.; Xiao, J.; Zhang, D.J.; Xu, Z.H.; Zhang, X.S. , et al. Enhanced tolerance to chilling stress in OsMYB3R-2 transgenic rice is mediated by alteration in cell cycle and ectopic expression of stress genes. Plant Physiol. 2009, 150, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Xu, Y.Y.; Ma, Q.B.; Xu, W.Y.; Wang, T.; Xue, Y.B.; Chong, K. Overexpression of an R1R2R3 MYB gene, OsMYB3R-2, increases tolerance to freezing, drought, and salt stress in transgenic Arabidopsis. Plant Physiol. 2007, 143, 1739–1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, W.L.; Zhong, H.; Gong, Z.Y.; Fang, X.Y.; Sun, T.; Deng, X.X.; Li, Y.S. Meta-analysis of salt stress transcriptome responses in different rice genotypes at the seedling stage. Plants 2019, 8, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86. [Google Scholar] [CrossRef] [Green Version]
- Kohli, A.; Sreenivasulu, N.; Lakshmanan, P.; Kumar, P.P. The phytohormone crosstalk paradigm takes center stage in understanding how plants respond to abiotic stresses. Plant Cell Rep. 2013, 32, 945–957. [Google Scholar] [CrossRef]
- Hu, Y.; Jiang, L.; Wang, F.; Yu, D. Jasmonate regulates the INDUCER OF CBF EXPRESSION–C-REPEAT BINDING FACTOR/DRE BINDING FACTOR1 cascade and freezing tolerance in Arabidopsis. Plant Cell 2013, 25, 2907–2924. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhang, Y.; Yin, L.; Qu, J.; Lu, J. Linkage of cold acclimation and disease resistance through plant–pathogen interaction pathway in Vitis amurensis grapevine. Funct. Integr. Genom. 2014, 14, 741–755. [Google Scholar] [CrossRef]
- Qian, X.; Xue-ren, Y.; Jiao-ke, Z.; Hang, G.; Min, S.; Chang-jie, X.; Xian, L.; Ferguson, I.B.; Kun-song, C. Activator- and repressor-type MYB transcription factors are involved in chilling injury induced flesh lignification in loquat via their interactions with the phenylpropanoid pathway. J. Exp. Bot. 2014, 15. [Google Scholar]
- Colquhoun, T.A.; Kim, J.; Wedde, A.E.; Levin, L.A.; Schmitt, K.; Schuurink, R.C.; Clark, D.G. PhMYB4 fine-tunes the floral volatile signature of Petunia×hybrida through PhC4H. J. Exp. Bot. 2011, 62, 1133–1143. [Google Scholar] [CrossRef]
- Yang, A.; Dai, X.; Zhang, W. A R2R3-type MYB gene, OsMYB2, is involved in salt, cold, and dehydration tolerance in rice. J. Exp. Bot. 2012, 63, 2541–2556. [Google Scholar] [CrossRef]
- Pasquali, G.; Biricolti, S.; Locatelli, F.; Baldoni, E.; Mattana, M. Osmyb4 expression improves adaptive responses to drought and cold stress in transgenic apples. Plant Cell Rep. 2008, 27, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; You, J.; Fang, Y.; Zhu, X.; Qi, Z.; Xiong, L. Characterization of transcription factor gene SNAC2 conferring cold and salt tolerance in rice. Plant Mol. Biol. 2008, 67, 169–181. [Google Scholar] [CrossRef]
- Wang, C.; Wei, Q.; Zhang, K.; Wang, L.; Liu, F.; Zhao, L.; Tan, Y.; Di, C.; Yan, H.; Yu, J. Down-regulation of OsSPX1 causes high sensitivity to cold and oxidative stresses in rice seedlings. PLoS ONE 2013, 8, e081849. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Liu, F.; Xu, W.; Di, C.; Zhou, S.; Xue, Y.; Yu, J.; Su, Z. Increased expression of OsSPX1 enhances cold/subfreezing tolerance in tobacco and Arabidopsis thaliana. Plant Biotechnol. J. 2009, 7, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.H.; Lin, L.Y.; Chen, M.L.; Lin, L.L.; Chen, X.F.; Lin, Y.H.; Chen, X.; Wang, Z.H.; Norvienyeku, J.; Zheng, H.K. Expression divergence as an evolutionary alternative mechanism adopted by two rice subspecies against rice blast infection. Rice 2019, 12. [Google Scholar] [CrossRef]
- Zhou, Y.; Yang, P.; Cui, F.; Zhang, F.; Luo, X.; Xie, J. Transcriptome analysis of salt stress responsiveness in the seedlings of Dongxiang wild rice (Oryza rufipogon Griff.). PLoS ONE 2016, 11, e0146242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, H.; Lou, Q.; Liu, H.; Han, H.; Wang, Q.; Tang, Z.; Ma, Y.; Wang, H. Differential regulation of anthocyanins in green and purple turnips revealed by combined de novo transcriptome and metabolome analysis. Int. J. Mol. Sci. 2019, 20, 4387. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Lin, F.; Li, B.; Zheng, Y. Hybrid-transcriptome sequencing and associated metabolite analysis reveal putative genes involved in flower color difference in rose mutants. Plants 2019, 8, 267. [Google Scholar] [CrossRef] [Green Version]
- Wei, T.; Deng, K.; Liu, D.; Gao, Y.; Liu, Y.; Yang, M.; Zhang, L.; Zheng, X.; Wang, C.; Song, W. Ectopic expression of DREB transcription factor, AtDREB1A, confers tolerance to drought in transgenic Salvia miltiorrhiza. Plant Cell Physiol. 2016, 57, 1593–1609. [Google Scholar] [CrossRef] [Green Version]
- Raymond, O.; Gouzy, J.; Just, J.; Badouin, H.; Verdenaud, M.; Lemainque, A.; Vergne, P.; Moja, S.; Choisne, N.; Pont, C. The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 2018, 50, 772. [Google Scholar] [CrossRef]
- Kong, W.L.; Bendahmane, M.; Fu, X.P. Genome-wide identification and characterization of aquaporins and their role in the flower opening processes in carnation (Dianthus caryophyllus). Molecules 2018, 23, 1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650. [Google Scholar] [CrossRef]
- Kong, W.L.; Gong, Z.Y.; Zhong, H.; Zhang, Y.; Zhao, G.Q.; Gautam, M.; Deng, X.X.; Liu, C.; Zhang, C.H.; Li, Y.S. Expansion and evolutionary patterns of glycosyltransferase family 8 in Gramineae crop genomes and their expression under salt and cold stresses in Oryza sativa ssp. japonica. Biomolecules 2019, 9, 188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, W.L.; Yang, S.Z.; Wang, Y.L.; Bendahmane, M.; Fu, X.P. Genome-wide identification and characterization of aquaporin gene family in Beta vulgaris. PeerJ 2017, 5, e3747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Tao, C.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914. [Google Scholar] [CrossRef]
- Jin, J.; Tian, F.; Yang, D.C.; Meng, Y.Q.; Kong, L.; Luo, J.; Gao, G. PlantTFDB 4.0: Toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017, 45, D1040–D1045. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Total Reads | GC Content | % ≥ Q30 | Mapped Reads |
|---|---|---|---|---|
| RPYCK3d-1 | 42,173,470 | 53.07% | 93.34% | 34,945,260 (82.86%) |
| RPYCK3d-2 | 42,574,242 | 52.95% | 93.55% | 35,593,632 (83.60%) |
| RPYCK3d-3 | 40,767,720 | 53.41% | 94.41% | 33,216,467 (81.48%) |
| RPYCo3d-1 | 45,230,168 | 52.24% | 93.31% | 36,277,084 (80.21%) |
| RPYCo3d-2 | 42,541,038 | 52.12% | 93.27% | 33,553,268 (78.87%) |
| RPYCo3d-3 | 42,812,246 | 52.36% | 92.86% | 34,405,870 (80.36%) |
| Gene IDs | Published Genes | FDR | Log2FC | Regulated |
|---|---|---|---|---|
| EVM0041152 | OsCDPK7 | 1.48E-188 | 3.005073 | up |
| EVM0023819 | OsDREB1B | 9.80E-161 | 12.99874 | up |
| EVM0037161 | OsDREB1F | 2.69E-14 | 8.265282 | up |
| EVM0000436 | OsMYB2 | 0 | 7.485158 | up |
| EVM0001849 | OsDREB1A | 0 | 9.69025 | up |
| EVM0001397 | SNAC2 | 0 | 7.397628 | up |
| EVM0005553 | OsWRKY45 | 1.28E-239 | 5.073813 | up |
| EVM0042736 | OsTPS1 | 1.18E-189 | 4.569165 | up |
| EVM0031981 | OsSPX1 | 1.48E-128 | 3.062169 | up |
| EVM0041750 | ZFP182 | 0 | 8.455205 | up |
| EVM0018891 | Osmyb4 | 0 | 5.400831 | up |
| EVM0018509 | qLTG3-1 | 8.43E-43 | -4.27137 | down |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, W.; Zhang, C.; Qiang, Y.; Zhong, H.; Zhao, G.; Li, Y. Integrated RNA-seq Analysis and Meta-QTLs Mapping Provide Insights into Cold Stress Response in Rice Seedling Roots. Int. J. Mol. Sci. 2020, 21, 4615. https://doi.org/10.3390/ijms21134615
Kong W, Zhang C, Qiang Y, Zhong H, Zhao G, Li Y. Integrated RNA-seq Analysis and Meta-QTLs Mapping Provide Insights into Cold Stress Response in Rice Seedling Roots. International Journal of Molecular Sciences. 2020; 21(13):4615. https://doi.org/10.3390/ijms21134615
Chicago/Turabian StyleKong, Weilong, Chenhao Zhang, Yalin Qiang, Hua Zhong, Gangqing Zhao, and Yangsheng Li. 2020. "Integrated RNA-seq Analysis and Meta-QTLs Mapping Provide Insights into Cold Stress Response in Rice Seedling Roots" International Journal of Molecular Sciences 21, no. 13: 4615. https://doi.org/10.3390/ijms21134615
APA StyleKong, W., Zhang, C., Qiang, Y., Zhong, H., Zhao, G., & Li, Y. (2020). Integrated RNA-seq Analysis and Meta-QTLs Mapping Provide Insights into Cold Stress Response in Rice Seedling Roots. International Journal of Molecular Sciences, 21(13), 4615. https://doi.org/10.3390/ijms21134615