Abstract
The vascular system is critical infrastructure that transports oxygen and nutrients around the body, and dynamically adapts its function to an array of environmental changes. To fulfil the demands of diverse organs, each with unique functions and requirements, the vascular system displays vast regional heterogeneity as well as specialized cell types. Our understanding of the heterogeneity of vascular cells and the molecular mechanisms that regulate their function is beginning to benefit greatly from the rapid development of single cell technologies. Recent studies have started to analyze and map vascular beds in a range of organs in healthy and diseased states at single cell resolution. The current review focuses on recent biological insights on the vascular system garnered from single cell analyses. We cover the themes of vascular heterogeneity, phenotypic plasticity of vascular cells in pathologies such as atherosclerosis and cardiovascular disease, as well as the contribution of defective microvasculature to the development of neurodegenerative disorders such as Alzheimer’s disease. Further adaptation of single cell technologies to study the vascular system will be pivotal in uncovering the mechanisms that drive the array of diseases underpinned by vascular dysfunction.
1. Introduction
A healthy and functioning vascular system is critical for ensuring that sufficient nutrients and oxygen reach the ~37 trillion cells in our bodies and the cellular waste products are efficiently removed. To service such a colossal quantity of cells, there is over 100,000 km of vasculature present within our bodies [1], which forms a continuum of vessels from arteries to arterioles, capillaries, venules and veins (Figure 1A) [2]. Differences in vascular cell phenotypes along the artery to vein axis are referred to as vascular zonation and correspond to unique cellular subtypes in each section of the vasculature (Figure 1A) [3]. Blood vessels are generally composed of up to three distinct regions or layers: (i) the tunica intima, a monolayer of flat squamous endothelial cells (ECs) that are in direct contact with the blood stream and mounted on a fibro-elastic basement membrane filled with extracellular matrix; (ii) the tunica media, composed of vascular smooth muscle cells (SMCs) and pericytes; and (iii) the tunica adventitia, composed of connective tissue, including fibroblasts and mesenchymal stem cells (MSCs), as well as lymphatic and neural plexi (Figure 1B,C) [2].
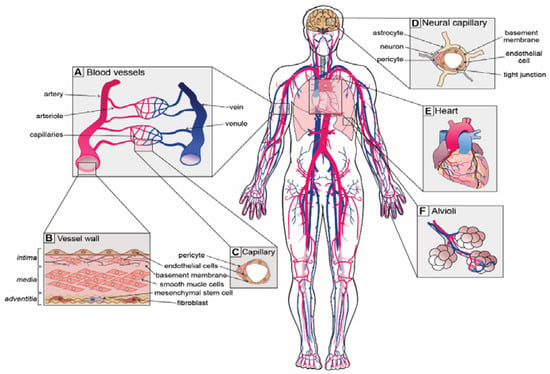
Figure 1.
Unique vascular beds in the human body. (A) Blood vessels are zonated and display unique cellular phenotypes and functionality. The 5 major zonation states of vessels are arteries, arterioles, capillaries, venules and veins. (B) Walls of arterial vessels are typically composed of 3 layers: tunica intima, tunica media and tunica adventitia. The intima is the innermost layer formed by endothelial cells that are in direct contact with the blood. The intima layer is mounted on the basement membrane, which is filled with fibro-elastic extracellular matrix, pericytes and smooth muscle cells. Media, the middle contractile layer, is composed of smooth muscle cells that provide support and flexibility to the vessel. Adventitia, the outmost layer of connective tissue surrounding the vessel, contains fibroblasts, a few mesenchymal stem cells and neurons. (C) Capillaries, the smallest blood vessels, are involved in direct solute exchange with the tissue. Capillaries possess a single layer of ECs that is surrounded by basement membrane and contains extracellular matrix and pericytes. Pericytes regulate the permeability of capillaries and their precise density varies from organ to organ. (D) Neural capillaries are characterized by an unfenestrated structure and ECs with tight junctions. Neural capillaries are densely populated by pericytes and are often contacted by astrocytes and microglia. (E) The heart is the central organ in the cardiovascular system that pumps blood through the whole body, and its function is supported by coronary arteries. (F) Lungs possess specialized vasculature that enables oxygen and carbon dioxide exchange between alveoli and pulmonary capillaries.
Although the general structure of blood vessels is somewhat conserved throughout the body, each organ has unique functions and demands on the vascular system. For instance, arteries and arterioles, the so-called resistance vessels, endure high pressure and shear stress [4]. The pressure and shear stress gradually reduce towards the veins, which endure up to 70-fold less pressure than arteries [4]. Due to the necessity of withstanding such high forces, arteries and arterioles possess a thick media layer with numerous SMCs that provide elastic support to the vessel walls. Capillaries, on the other hand, are the smallest and thinnest vessels. They only possess an intima layer covered with basement membrane and are supported by pericytes (Figure 1C). Together, SMCs and pericytes constitute the mural cell population that promotes EC differentiation, maintains vascular tone and regulates the permeability of capillaries [5,6]. Consistent with their morphology, capillaries are the major sites of nutrient and gas exchange.
Blood vessels display specializations that are closely associated with organ function. For instance, vessels of the kidney glomerulus need to interact with podocytes and allow filtration of blood and reabsorption of fluids. Therefore, capillary ECs in the kidney are highly fenestrated and permeable [7]. In contrast, the brain has a highly vulnerable environment and is thus afforded protection via the blood–brain barrier (BBB) that restricts the movement of cells, metabolites, infectious agents and proteins in and out of the brain [8,9]. Consistently, ECs of the BBB possess highly specialized non-fenestrated vasculature with specialized tight junctions to maintain the selective nature of the BBB [8,9]. To achieve unique organ-specific functions, vascular cells from distinct organs display unique gene expression programs and phenotypic heterogeneity [10], which will be discussed further in this review.
Given the critical role of blood vessels in maintaining homeostasis, deregulation of vascular function is associated with numerous disorders such as atherosclerosis [11] and associated complications such as ischemia, thrombosis, stroke and myocardial infarction [12], as well as neurodegeneration [13], age-related cognitive decline [14] and cancer [15]. However, due to technical limitations, many of the molecular and subtle phenotypic changes in diseased vascular cells remained unknown. Recent advances in genomics and molecular biology techniques such as single cell sequencing have opened new avenues for researchers, allowing characterization of the precise cellular composition of various tissues, including the detection of rare vascular cell subtypes [3,10]. Importantly, single cell technologies have made it possible to identify cellular heterogeneity, as well as phenotypic and transcriptional changes in the context of healthy and diseased conditions. Through these analyses, researchers have begun to unravel novel molecular mechanisms that underlie the healthy development of the vascular system, as well as changes associated with pathological states.
Single cell technologies now span the analyses of RNA, chromatin accessibility, whole genome DNA sequencing and DNA methylation. In this review, we focus on the biological insights garnered from single cell studies, rather than their technical aspects, which have been recently reviewed elsewhere [16,17,18]. We cover studies collating the diversity of vascular cells through the use of single cell and complementary genomics technologies. We discuss the lessons learnt from these novel data in the context of vascular homeostasis. Given the involvement of vascular dysfunction in a range of diseases, we discuss pathological mechanisms underlying vascular dysfunction and inflammation in atherosclerosis, cardiac and pulmonary disease, as well as neurodegeneration. We highlight recent advances in our understanding of the inflammatory pathways in vascular cells that drive these pathologies and discuss important questions that will be critical towards progressing our understanding of vascular function and disease.
2. Overview of Single Cell Analyses
Although numerous methods for single cell analyses have now been established, all methods generally utilize a similar design [16]. Single cells are isolated by tissue dissociation and physically separated into individual wells of a collection plate through fluorescence-activated cell sorting (FACS), or encapsulated in droplets using droplet-based technologies (e.g., inDrop, 10x Genomics). For transcriptome analyses, RNA from each cell is isolated, reverse transcribed and barcoded with unique cell-specific oligonucleotides. Moreover, to overcome the PCR duplication artifacts of the extremely low RNA content of single cells, unique molecular identifiers (UMIs) are introduced into the cDNA. Barcoded cDNA from multiple cells is then mixed and libraries are produced for sequencing. Following sequencing, gene transcripts from each unique cell are identified based on the corresponding barcodes. The identity of cells is determined based on their gene-expression profile as well as the expression of known cell markers. The transcriptional profile of each cell type can then be compared between different conditions, for example between diseased and healthy states.
There are unique benefits of single cell approaches over traditional “bulk” analyses that investigate whole tissues or FACS-enriched cell populations [19]. Single cell analyses deconvolute the full complexity of tissues by resolving and characterizing rare cell populations, which would be otherwise masked by more abundant cell types when undertaking analyses of whole complex tissues. Furthermore, through established bioinformatic pipelines, single cell analyses can pick up subtle phenotypic shifts in diseased conditions as well as intermediate cell states during disease onset or during normal cellular differentiation [20]. This is particularly important when studying the molecular mechanisms that induce the transition from a healthy to a diseased cell state. Thus, while “bulk” analyses remain a powerful tool for understanding the average molecular changes in a tissue, single cell analyses allow the identification of cellular heterogeneity as well as phenotypic and transcriptional transitions in individual cell types.
3. Single Cell Technology towards Creation of a Comprehensive Vascular Cell Atlas
The first large-scale initiative to produce a reference single cell atlas of healthy wild type mice was pioneered by the Tabula Muris Consortium that sequenced more than 100,000 cells from 20 mouse organs [21]. An analogous Human Cell Atlas consortium [22,23], together with other datasets developed by independent laboratories [24,25,26,27,28], is already on the way to producing a comprehensive single cell atlas of all human tissues. While these single cell RNA (scRNA)-seq databases provide an extremely valuable resource, the vascular system remained to be covered in depth, as vascular cells represent only a minor proportion of an organ. Vascular cells are generally grouped together in global databases and consequently their heterogeneity is overlooked, as bioinformatic algorithms typically do not distinguish between the vascular subtypes amongst the huge diversity of other, more common cell types in whole organs. Thus, considerable effort has since been spent on generating a vascular cell atlas.
Kalucka and colleagues focused on characterizing EC heterogeneity by analyzing more than 32,000 ECs from 11 mouse tissues including the brain, spleen, testis, lung, kidney and heart [10]. They uncovered that EC gene expression profiles are highly tissue dependent. Nevertheless, there were strong similarities in ECs isolated from the small intestine and colon, skeletal muscle and heart, as well as the brain and testis. Moreover, the authors detected specialized EC subtypes in the intestine (Aqp7+, Madcam1+) and brain (Plvap-high, Esm1-high), as well as proliferative and activated EC populations in various organs. Although this is a valuable resource for the field, the heterogeneity of vascular cell types such as pericytes, SMCs and peri-vascular fibroblasts remains to be systematically compared between different organs using similar techniques.
Considerable attention has been paid to the brain due to its highly specialized vasculature that is critical for maintaining a homeostatic neural environment. Vanlandewijck and colleagues isolated ECs (Cldn+ cells), SMCs (Tagln+ cells), pericytes (Pdgfrb+, Cspg4+ cells) and fibroblasts (Pdgfra+ cells) from the adult brain by FACS and analyzed them via scRNA-seq [3]. By coupling their transcriptomics data with known EC and mural cell markers, they were able to expand vascular zonation markers using bioinformatic algorithms (Table 1). In addition, these analyses also uncovered zonation-specific transcription factors and transporters enriched in neural ECs and mural cells.

Table 1.
Cell type specific marker genes for vascular cell types from single cell data. Marker genes were derived from References [3,10,29]. Endothelial cells (ECs); smooth muscle cells (SMCs).
In addition to global EC databases, vascular cells in the adult human heart have been recently annotated [30]. Comprehensive single cell and single nuclei analyses of six distinct regions of the heart uncovered heterogeneity in all major cell types depending on their spatial location and function [30]. In addition to the expected differences between atrial and ventricular cardiomyocytes [31], vascular cells, which constitute approximately 7% of cells in this dataset, displayed zonation-based clustering with the highest proportion of ECs identified as capillary ECs (53%) [30]. Moreover, ECs, SMCs and pericytes exhibited chamber specificity, showing distinct gene expression profiles between atrial and ventricular counterparts. This study emphasizes the importance of single cell approaches to study vascular cells due to their heterogeneity and tissue-specific function.
Single cell technologies have started to uncover critical insights into numerous aspects of vascular function and homeostasis in healthy organisms. In addition, studies are now utilizing single technologies to interrogate phenotypic changes in vascular cells in diseased states. In the sections below, we focus specifically on the themes of chronic vascular dysfunction, which promotes and accompanies atherosclerosis, myocardial infarction and cognitive disorders such as Alzheimer’s disease.
4. Vascular Inflammation
The vascular system not only supplies our bodies with nutrients and oxygen, but is also directly involved in the execution of immune reactions to pro-inflammatory stimuli. There are a multitude of stimuli that lead to vascular inflammation including alterations in blood flow and shear stress, increased concentration of low-density lipoproteins (LDLs), metabolic changes such as increased fatty acids, hypertension, agiotensin II signaling, as well as pro-inflammatory cytokines and chemokines that are upregulated in response to viral or bacterial infections [11,32,33,34]. Vascular inflammation is accompanied by active attraction of circulating leukocytes to the site of injury and their transmigration into the intima of the vessel wall, to clear the tissue from the source of inflammation and dead cells and ultimately resolve the inflammation. However, if the inflammation transitions to a chronic state, it can lead to adverse outcomes through the development of vascular diseases like atherosclerosis. Here, we will discuss the role of vascular inflammation in atherosclerosis, myocardial infarction, pulmonary arterial hypertension and neurodegeneration, and focus on new insights obtained from single cell studies.
4.1. Atherosclerosis
Atherosclerosis is a common, multi-factorial disorder that typically underlies ischemic heart disease, stroke and peripheral artery disease. Atherosclerosis affects arteries and arterioles by narrowing and stiffening of the vessels due to the build-up of lipid-rich plaques. According to the well-established dogma, atherosclerosis starts with EC dysfunction in response to increased levels of oxidized low-density lipoproteins (LDLs) in the blood stream [35]. ECs sense and uptake toxic LDLs, which promote oxidative stress, a reduction in nitric oxide synthesis and increased apoptosis. LDL-loaded ECs upregulate adhesion molecules like ICAM1, VCAM1 and E- and P-selectins, as well as pro-inflammatory cytokines such as CCL2 (MCP1) and IL1β, and the complement system. These changes in EC phenotype promote attraction, attachment and slow rolling of circulating immune cells on the vessel wall [36]. An inflammatory state leads to opening of adherent junctions, promoting immune cell transmigration into intima of the vessel wall, where monocytes differentiate into macrophages and thus facilitate an inflammatory state and clearance of excessive lipids and dead cells [37]. Macrophages that uptake LDLs transform into foam cells and are typical of atherosclerotic plaques [37].
SMCs, on the other hand, contribute to vascular wall remodeling. SMCs migrate into the intima of the lesion area, where they undergo a phenotypic switch and start to excessively produce extracellular matrix (ECM) and proliferate. Some SMCs reportedly express macrophage markers in response to LDLs and pro-inflammatory stimuli, suggesting the possibility of trans-differentiation [38,39]. The opposite also seems to take place, where macrophages and myeloid cells begin to express a number of SMC markers [40,41,42]. Moreover, ECs have been shown to undergo endothelial-to-mesenchymal transition (EndMT) in atherosclerotic plaques in vivo, as well as in response to disturbed flow in vitro via activation of Yes-associated protein/transcriptional coactivator with PDZ-binding motif (YAP/TAZ) complex and TGF-β signaling [43,44]. ECs acquire a contractile phenotype with expression of SMC-specific genes like α-smooth muscle actin (ACTA2), connective tissue growth factor (CTGF) and fibroblast activation protein (FAP), while losing EC-specific gene expression (Figure 2) [43]. Mesenchymal stem cells (MSCs) normally reside in the adventitia of the vessel and are also found in atherosclerotic lesions, but it is unclear if they migrate from the adventitia or originate from trans-differentiation of ECs or SMCs [45,46]. Further plaque growth is accompanied by lipid accumulation, calcification, apoptosis and necrotic core formation. Infiltrating immune cells secrete metalloproteinases that digest and thin the fibrous cap, thus promoting plaque rupture. If the plaque is ruptured, it leads to thrombus formation and ultimately vessel occlusion that can result in adverse outcomes such as stroke and myocardial infarction.
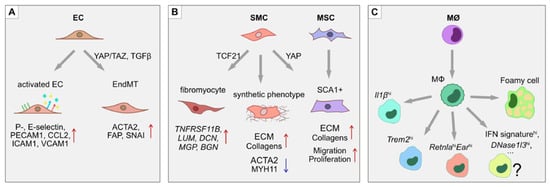
Figure 2.
Phenotypic switches during atherosclerosis. In atherosclerotic lesions, several processes of phenotypic modulation and trans-differentiation take place. (A) ECs upregulate adhesion molecules such as ICAM1, VCAM1, E- and P-selectins, as well as secreting pro-inflammatory cytokines CCL2 and IL1β, which help attract leukocytes. In atherosclerotic plaques, ECs also undergo endothelial-to-mesenchymal transition (EndMT) through the activation of YAP/TAZ- and TGFβ-driven pathways. The EndMT transitions are characterized by the loss of endothelial identity markers such as PECAM1, together with the upregulation of mesenchymal markers α-smooth muscle actin (ACTA2), fibroblast activation protein (FAP) and the SNAI transcription factors. (B) SMCs undergo a phenotypic switch from a contractile to a synthetic state by increasing production of ECM proteins and downregulating MYH11. Moreover, a subset of SMCs in the atherosclerotic lesion express the stem cell marker SCA1, suggesting either mesenchymal stem cell differentiation into SMCs, or de-differentiation of SMCs towards an MSC-like state. (C) Monocytes, upon transmigration into the intima of the lesion, differentiate into macrophages that display at least 3 unique subsets: (i) resident-like anti-inflammatory cells, (ii) pro-inflammatory Il1βhi cells, and (iii) Trem2hi cells. Macrophages that take up low-density lipoproteins (LDLs) upregulate lipid metabolism related genes and take on a “foamy macrophage” phenotype.
As it becomes clearer that atherosclerosis involves cellular plasticity and phenotypic transitions including EndMT, SMCs and immune cell trans-differentiation, single cell technologies are becoming increasingly instrumental in addressing heterogeneity of atherosclerotic plaques. A number of studies have begun to utilize scRNA-seq to investigate total diseased arterial wall tissue [47], samples enriched for SMCs [48,49], and purified cell populations of infiltrating immune cells [50,51] to gain insights into the pathophysiological mechanisms underlying atherosclerosis.
To understand atherosclerosis development in controlled animal models, Kalluri and co-workers performed a single cell analysis of more than 10,000 cells derived from aortas of 12-week-old wild type C57/BL6 mice on either chow or Western diets [47]. They detected all major cell types in the plaque including ECs, SMCs, fibroblasts, monocytes and macrophages. Moreover, they discovered three functionally distinct EC populations: (i) Vcam1-enriched population, (ii) EC population with higher lipid metabolism and angiogenesis related genes and (iii) a lymphatic population. In comparison to mice on the control diet, Western diet-fed mice developed changes in EC expression with increased transcription of contractile genes that might be related to EndMT.
Other studies have focused on the non-EC compartments in the atherosclerotic plaques. Working on the ApoE−/− atherosclerosis mouse model, Gu and co-workers found that non-immune cells also appear in a pro-inflammatory and stem-/progenitor-like state that is typified by the expression of stem cell antigen 1 (SCA1) in SMCs [52]. Moreover, isolated SCA1+ SMCs had high CCL2 expression in vitro, consistent with the pro-inflammatory signature. Other cell types such as resident macrophages were also in an activated pro-inflammatory state [52]. While Western diet or ApoE knockout mouse models do not result in formation of progressive atherosclerotic lesions, these models nevertheless provide important insights into its early development and first-wave responses of ECs and SMCs to atherosclerosis development.
4.1.1. Smooth Muscle Cells in Atherosclerosis
One important question in the field has been the origin of SMCs found in atherosclerotic plaques. In order to answer this question, several studies have combined scRNA-seq with Myh11-driven SMC lineage tracing in ApoE−/− animals [48,49]. Firstly, SMCs from the aortic arch and descending thoracic aorta show transcriptional differences suggesting the presence of SMC subtypes. Secondly, a rare subset of progenitor-like SCA1+ cells is present in plaques, which is characterized by low contractile function and increased expression of genes related to migration, proliferation and ECM production [48]. SCA1+ SMCs are also increased in atherosclerotic lesions of ApoE−/− mice, suggesting that regulation of Sca1 expression may contribute to the switch from contractile to the activated SMC phenotype. Consistently, SCA1+ SMCs are activated upon arterial injury and are crucial for vascular repair and regeneration mediated by YAP1, a member of the Hippo pathway [53]. Together, the presence of SCA1+ cells in atherosclerotic lesions might be viewed as an exaggerated repair response to chronic inflammation and injury.
Recently, attention has shifted towards identifying important regulatory molecules via scRNA-seq analysis. Transcription factor 21 (TCF21) is highly expressed within atherosclerosis lesions and coincides with SMC markers [54]. A recent scRNA-seq study found TCF21 to mediate the SMC phenotypic switch and fibrous cap formation [49]. The authors discovered a phenotypic switch of SMCs in atherosclerotic plaques towards a fibroblast-like transcriptional signature that they named “fibromyocyte”. Fibromyocytes were characterized by upregulation of fibroblast marker genes lumican (Lum), decorin (Dcn) and biglycan (Bgn) and increased expression of Tcf21. On the other hand, SMCs did not shift towards a macrophage phenotype. Despite expressing the Lgals3 macrophage marker, SMCs did not acquire expression of other macrophage markers including Cd68, Cd16, Cd32, Cd11b, Cd64 and Cd86. Thus, these results seemingly oppose the hypothesis of SMC-to-macrophage trans-differentiation [38]. The existence of “fibromyocytes” was further confirmed in four human atherosclerotic plaque samples by scRNA-seq and complimentary in situ RNA hybridization, where osteoprotegerin (TNFRSF11B) was found to mark the fibromyocyte cell population [49]. Moreover, SMC-specific Tcf21 knockout in mice led to a reduced fibromyocyte population at the fibrous cap and in the atherosclerotic lesion, while overexpression of TCF21 in the human coronary artery SMC cell line led to the upregulation of LUM, DCN and MGP fibromyocyte genes, suggesting a conserved role of TCF21 in mouse and human models [49]. Consistent with findings in atherosclerosis, the involvement of TCF21 in fibrosis has been reported in a number of other organs [55,56,57]. Together, these studies suggest that TCF21 is an important player in atherosclerosis-related fibrosis and might modulate plaque stability via fibrous cup support.
Taken together, SMCs found in atherosclerotic plaques undergo a phenotypic switch and acquire stem cell-like and/or fibroblast-like markers. However, future work is still required to determine the origin of these cells.
4.1.2. Immune Cells in Atherosclerosis
As atherosclerosis is a chronic inflammatory disease, there is a strong contribution from the immune cell compartment. To characterize the heterogeneity of immune cell infiltrates in atherosclerotic lesions, studies have undertaken FACS isolation of CD45+ hematopoietic cells followed by scRNA-seq [50,51]. These analyses have established that atherosclerotic plaques are rich in various immune cell types, including monocytes, macrophages, B-cells, T-cells, NK-cells and granulocytes [50,51]. In turn, each infiltrating immune cell population also displays significant heterogeneity. For instance, three distinct populations of macrophages in atherosclerotic plaques have been described [50]. Of these, two macrophage cell populations were specific to atherosclerotic lesions in Ldlr−/− mice maintained on a high fat diet (HFD), namely (i) inflammatory macrophages with high expression of Il1b and (ii) macrophages with high expression of triggered receptor expressed on myeloid cells 2 (Trem2hi). Importantly, these two macrophage populations were also independently detected in the ApoE−/− high fat diet atherosclerosis model [50], strengthening the claim that these populations may be a general characteristic of atherosclerotic plaques. Furthermore, increased numbers of foamy macrophages are also found in plaques and their number correlates with the size of the atherosclerotic lesion [51]. Consistent with their LDL-loaded composition, foamy macrophages display increased expression of fatty acid and cholesterol transport and uptake genes. In contrast, inflammation-related genes including Il1b are most highly expressed in the non-foamy macrophage populations [51].
Due to high numbers of immune cells in the atherosclerotic lesion, it is debatable if all immune cells originate from the blood or whether they might be derived from resident macrophages and proliferate in the lesion itself [58]. To address this question, Lin and colleagues examined the fate of infiltrating CX3CR1+ CD11b+ monocytes and macrophages during atherosclerosis progression [59]. Ten different subpopulations of myeloid lineage cells were detected by dataset clustering [59], including the three previously reported macrophage populations (resident-like, inflammatory, and Trem2hi) [50]. Novel macrophage populations including DNase1l3high, IFNhigh, as well as Retnlahi/Earhi, were strongly enriched in progressive plaques compared to regressive ones [59]. Interestingly, a highly proliferative “stem-like macrophage” population was detected in both progressing and regressing plaques [59], suggesting that macrophage precursors can proliferate within atherosclerotic plaques and give rise to distinct types of macrophages.
Taken together, scRNA-seq studies of atherosclerotic plaques provide new evidence of complex and intricate regulation of cell phenotypes and function in response to chronic inflammation (Figure 2). Despite the important insights gained from scRNA-seq studies on atherosclerosis models, there remain a number of outstanding questions. For instance, while the final diseased state in atherosclerotic animals has been well defined, the molecular changes leading to the development of the atherosclerotic plaque remain to be investigated in detail. It would be highly informative to produce a comprehensive scRNA-seq based timeline of atherosclerotic lesion development to uncover the precise sequence of molecular events that drive changes in cellular phenotypes. Moreover, it is of paramount importance to study how the vast number of cell types, each with unique disease-specific phenotypes, communicate with each other. These intercellular communication networks remain to be mapped. Further, there are only limited data available in human subjects. While the mouse models provide significant molecular insights, future studies utilizing primary human plaques, together with single cell RNA and chromatin analyses, will reveal whether molecular networks discovered in animal models also hold true in human samples.
4.2. Cardiac Vasculature in Disease
As a disease that impairs blood circulation, atherosclerosis has direct effects on the fitness of the cardiovascular system, and on the heart in particular. Vessel wall thickening and stiffening eventually results in increased blood pressure and potentially heart failure. Unstable atherosclerotic plaques may rupture and thereby increase the risk of thrombosis, acute coronary syndrome and myocardial infarction (MI) due to insufficient blood supply through coronary arteries [60]. MI leads to muscle damage and scarring, as well as the loss of contractile and conductive function of the heart, ultimately resulting in cardiac insufficiency and the need for heart support or replacement. As any scar tissue, cardiac scars after MI are characterized by increased migration and proliferation of fibroblasts, extensive ECM production and inflammation targeted to remove dead cells [61].
Changes in cell composition and phenotypic switches after MI have been addressed by a number of studies, where an influx of immune cells [62] and increased fibrosis [63,64] in the scar have been reported. Scar formation is propagated via fibroblast activation modulated by periostin (Postn) and TGFβ-WNT signaling pathways, as well as fibroblast trans-differentiation into myofibroblasts, which is accompanied by the acquisition of SMC markers [65,66]. There is also new evidence of an increase in the EC population starting as early as day 5 post-MI [64]. This change can be related to re-vascularization of the scar tissue, which is critical for the healing process. Indeed, increased EC proliferation and clonal expansion of EC into the border zone of MI was reported seven days post-MI in animal models [67]. ScRNA-seq analysis of FACS-sorted ECs from healthy and ischemic hearts at seven days post-MI detected 10 distinct EC subpopulations, including a small CD45+ population [67], which could represent either a phenotypic switch of EC into an inflammatory state, or infiltrating immune cells taking on some EC-like characteristics. Other EC subpopulations include proliferative, angiogenic and regenerative signatures, which represent a neovascularization response to injury. Moreover, ECs with high expression of cardiomyocyte genes were also detected, suggesting a mechanism to compensate for myocyte loss. These ECs display strong expression of inflammatory mediators to attract immune cells and facilitate clearance of debris in the scar tissue.
Taken together, cardiac injury is characterized by dynamic changes in the EC population and multiple events of trans-differentiation that are mostly related to fibroblasts. However, new data on scar neovascularization promise novel opportunities for post-MI treatment and recovery, emphasizing the importance of vascular cells in the period following MI.
4.3. Pulmonary Arterial Hypertension
Pulmonary vasculature undertakes the essential function of oxygen and carbon dioxide exchange between the blood and air at the interface of alveoli and alveolar capillaries. However, a number of factors, including genetics, side effects of some drugs and pre-existing conditions such as heart or lung disease, can lead to the development of pulmonary arterial hypertension (PAH) [68]. PAH is characterized by a chronic increase in pulmonary artery pressure and vascular resistance despite normal heart function. It is accompanied by oxidative stress and vascular inflammation affecting all three layers of the vascular wall, which leads to irreversible vascular remodeling and obliteration of pulmonary arterioles, and ultimately to lung and left ventricle dysfunction [69]. Consistently, PAH development is associated with vasoconstriction, increased ECM production, EndMT, upregulation of inflammatory modulators, immune cell infiltration and extensive EC, SMC and pericyte migration and proliferation that is mediated by BMP-TGFβ signaling, as well as growth factors such as PDGF and FGF [70].
Due to recent efforts in assembling single cell lung atlases, the precise tissue composition of the lungs in humans, mice, rats and pigs has been described [71,72,73]. More recent studies have also begun to focus on diseased states such as PAH. ScRNA-Seq analyses in human PAH patients have uncovered the upregulation of angiogenesis-associated genes in ECs such as endoglin (ENG), NOTCH1 and KDR, together with genes involved in SMC migration and proliferation such as PDGFB and FGFR1, as well as vasoconstrictor endothelin 1 (EDN1) [74]. Interestingly, apelin (APLN) and apelin cleaving enzyme lysosomal pro-X carboxypeptidase (PRCP) were elevated in PAH samples [74], contradicting the previously reported reduction in APLN and PRCP expression in pulmonary artery ECs isolated from PAH patients in vitro and the regression of PAH in mice after APLN administration [75,76]. Genes upregulated in SMCs and pericytes were related to ECM components that support vessel wall remodeling and increased ECM stiffness [74]. Furthermore, bioinformatic analyses of regulatory networks derived a number of transcription factors such as SOX18, STRA13, LYL1 and TFDP1 [74], which are likely to play an important role in ECs in PAH.
Given the cellular complexity of the lungs, a number of scRNA-seq studies have also focused on intercellular communication [71,72,73,77], which is of great importance for understanding the contribution of individual cell types to complex diseases. For instance, the interaction between alveolar type I epithelial cells and capillary ECs through VEGF and semaphorin signaling is strongly conserved across species [77], suggesting the importance of interactions between these two cell types. Additionally, bioinformatic analyses have confirmed that lung ECs are able to detect and process major circulating hormones [73]; Lung ECs express the angiotensin-converting enzyme (ACE) and endothelin-converting enzyme (ECE1), which are utilized for the production of biologically active peptides that can in turn be sensed by pericytes. Furthermore, acetylcholine receptors are expressed by lung ECs, which stimulate the production of the vasodilatory nitric oxide and prostacyclin upon activation [73].
In addition to transcriptional changes, there is growing evidence that epigenetics may also play an important role in ECs during PAH. The histone acetylation mark, histone H3 lysine 27 acetylation (H3K27ac), normally found on active regulatory elements such as enhancers and promoters, has been recently reported to differ in small pulmonary artery ECs isolated from healthy and PAH patients [78]. Intriguingly, genes associated with differentially acetylated enhancers in PAH versus control samples were related to cell proliferation, migration and angiogenesis processes [78]. Moreover, a number of transcription factors including members of the AP1 complex, STAT1, STAT4, RFX3, RFX4, GATA4 and GATA6 showed increased activity in PAH samples based on H3K27ac signal, whereas the VEGF target NOS3, the serotonin receptor HTR1BRC, and YAP1 enhancers showed a reduction in H3K27ac signal [78]. Interestingly, the mechanosensitive YAP/TAZ complex responds to increased ECM stiffness in PAH by promoting glycolysis and reducing mitochondrial oxidative phosphorylation in pulmonary arterial ECs and SMCs [79]. Together, these data suggest the involvement of AP1, TGFβ, YAP/TAZ, serotonin and VEGF signaling in PAH, resulting in inflammation, EndMT, vascular stiffness and aberrant angiogenesis. To support the notion of epigenetic modulation in PAH, changes in the expression of chromatin regulators FAM60A and HDAC7 in ECs derived from PAH patients have also been reported [74]. The transcriptional repressor complex member FAM60A responds to hypoxia via recruitment of HDAC7 to the HIF2A promoter [80,81], whereas the histone deacetylases HDAC6 and HDAC7 play an important role in regulating sprouting, angiogenesis and endothelial barrier function [82,83,84]. Thus, further investigation of epigenetic mechanisms in EC subtypes through single cell technologies (discussed below) would be important for a better understanding of molecular events underlying the development of PAH.
4.4. Vascular Dysfunction in Neural Disorders
Compared to the blood vessels of other organs, the vasculature in the nervous system is unique and underpinned by the blood–brain barrier (BBB). In particular, brain ECs are deficient in fenestrations and possess tight junctions that strongly limit the transport of macromolecules, metabolites and toxins into the brain parenchyma [85,86]. In addition to specialized ECs, neural vasculature possesses particularly high numbers of pericytes, with estimates ranging from one pericyte to every one to three ECs [5]. In comparison, the vessels of skeletal muscle, for example, are thought to have only around one pericyte per 100 ECs. Pericytes are critical for the formation of the BBB during development [87,88,89], and their ongoing functionality is critical for modulating its permeability [90,91]. In addition to the two main vascular cell types, numerous brain cells including astrocytes and microglia have close contacts with the blood vessels and are able to directly modulate their activity (Figure 1D). Consistent with this, changes in the neural metabolic environment [34], neural activity [92], inflammatory signals [93], buildup of toxic products such as β-amyloid [94], and infection [95], can all modulate vascular properties such as blood flow and permeability.
Transcriptional analyses [3,19], together with imaging techniques such as light-sheet microscopy [96,97], are starting to shed important light on the makeup of brain vasculature. Single cell transcriptomics focusing on the mouse brain have revealed that neural EC subtypes exist in a zonated manner [3]. Indeed, there is a clear transcriptomic and phenotypic distinction between arterial ECs, capillary ECs and venous ECs, with 1798 transcripts showing differential expression amongst the major neural EC subtypes. For instance, Bmx, Efnb2, Vegfc and Sema3g are highly enriched in arterial ECs, Mfsd2a and Trfc in capillary ECs, whereas Nr2f2 and Slc38a5 are enriched in venous ECs [3]. While there are numerous genes reportedly expressed in a zonated fashion in neural ECs, the precise function of the majority of identified genes in the specification or functionality of EC subtypes remains to be ascertained. In contrast to ECs, mural cells do not show zonation, but rather display two major clusters based on their transcriptional profiles [3]. The major mural cluster consists of pericytes and SMCs associated with capillaries and venous vessels that display high levels of Pdgfrb, Vtn and Kcnj8 mRNA levels. In contrast, arteriole and arterial smooth muscle cells make up the second mural cell cluster and express high levels of Myh11, Acta2 and Myl9 [3]. These data are consistent with unique recruitment mechanisms of SMCs and pericytes to different parts of the zonated vasculature. For instance, depletion of Pdgfrb primarily results in the loss of pericytes from capillaries, while mural cell recruitment to arterioles is less affected [89]. While the neural vasculature scRNA-seq dataset provides great insights into neural vascular cells at steady states, precisely which subtype of vascular cells change in pathological conditions and through which molecular mechanisms remains to be identified. This is particularly important given the range of neural pathologies impacted by defective vascular function.
The importance of the neural vasculature in a range of neurological disorders, including vascular dementia [98], Alzheimer’s disease [99,100,101,102], Parkinson’s disease [103], Huntington’s disease [104], amyotrophic lateral sclerosis [105] and age-related cognitive decline [90,100], is starting to become clear. Amongst these disorders, the role of vascular dysfunction in Alzheimer’s disease (AD) has been widely studied. Defects in neural vasculature, including increased permeability are thought to be amongst the earliest changes observed and are thought to precede the occurrence of β-amyloid fibrils [100,102]. Indeed, reduced pericyte functionality in mouse models of AD accelerates AD development [99], while higher levels of pericyte stress, evident through increased CSF concentrations of PDGFRβ, strongly predicts accelerated cognitive decline in humans in the following 4.5 years [102].
While a number of scRNA-seq databases have been developed for human AD [106,107], vascular cells only represent a miniscule proportion of cells analyzed and the underlying changes in neural vascular cells remain to be identified. Nevertheless, there is some evidence from scRNA-seq analyses of brain ECs from aging mouse models that capillary ECs maybe the most vulnerable to age-related decline in vascular function [108]. However, given the recent recognition of the key role that pericytes play in the development of neurodegenerative disorders [102,109], it would be of paramount importance to map pericyte transition from a healthy to a diseased state through single cell analyses.
Although there is now increasing appreciation that vascular defects are key parts of the pathology of neurodegenerative disorders, whether environmental factors could also contribute to neurovascular defects remains unclear. Recent work has suggested that a defective metabolic environment could induce vascular inflammation and increased vascular permeability in the developing brain [34]. Through scRNA-seq and transcriptomic analyses of FACS-isolated neural and vascular cells, it was established that increased free fatty acids can trigger a toll-like receptor 4 (TLR4)-NFkB-dependent pro-inflammatory response in brain pericytes. This ultimately results in morphological changes in brain pericytes, associated with vascular dilation, breakdown of the extracellular matrix and increased vascular permeability [34]. This finding is compelling, as metabolic changes are thought to be a core component of neurodegenerative and neurodevelopmental disorders [110,111] and APOE, a key genetic risk factor for AD, plays a central role in lipid metabolism and transport [112,113]. However, whether metabolic changes induce defects in vascular cells during the onset of neurodegeneration in human remains to be determined.
Despite our ever-increasing appreciation of neural vasculature dysfunction in a range of neurological disorders, transcriptomic analysis of vascular phenotypes in neural pathologies is in its infancy. Future studies utilizing single cell technologies will provide invaluable insights into molecular mechanisms that drive vascular phenotypic changes associated with neural pathologies. Given that many of the vascular-associated neural pathologies are related to aging, the importance of environmental factors and epigenetic changes in mediating vascular breakdown will be an additional important line of study.
5. Outlook and Perspectives
5.1. Intercellular Communication
Inflammation is a process that involves multiple intercellular interactions and communication by means of secreted factors and chemokines on one side and receiving receptors on the other side. In order to address the heterocellular interactions within the context of complex tissues and inflammation, considerable effort has been spent on creating a comprehensive ligand-receptor database for humans [114]. Studies have utilized this Ramilowski database to uncover inter-cellular interactions in the developing mouse brain [19], heart [115], kidney [116] and tumors [117]. Furthermore, a number of bioinformatic tools [118,119,120] utilizing ligand-receptor databases such as the Ramilowski database [114] or REACTOME [121] are making intercellular communication analysis more accessible to single cell studies.
Bioinformatic tools have now also been employed to interrogate cellular interactions in the context of atherosclerosis [52]. In atherosclerosis, increased interactions between mesenchymal-like cells and immune cells have been reported in the ApoE−/− mouse model [52]. Increased interacting ligand-receptor pairs were enriched in gene ontologies related to inflammation, leukocyte chemotaxis and ECM interactions, consistent with the important roles of these cellular processes in the context of atherosclerosis. In contrast, ligand-receptor interaction analysis of human heart vascular cells revealed that differences in cellular communication modules were dependent on vascular zonation, including specific patterning of Notch ligands. Arterial ECs mainly expressed DLL4, JAG1 and JAG2 that are recognized by NOTCH2 in arterial SMCs, whereas venous EC expressed DLL1 that is recognized by NOTCH3 of venous SMCs [30]. Furthermore, neural vascular cells, microglia and neural cells of the developing mouse brain also display a complex intercellular communication network [19]. The authors reported that APOE, a protein implicated in Alzheimer’s disease and in atherosclerosis [122,123], is expressed by microglia during development and has reciprocal receptors in neural cells [19], suggesting APOE may play an important and currently undefined role during neural development.
In addition to the use of bioinformatic tools to identify potential cell–cell communication modules, a recently developed method, physically interacting cells (PIC-seq) attempts to directly probe inter-cellular interactions [124]. PIC-seq involves sorting single cells and physically interacting cell aggregates (PICs) into 384-well plates. Transcripts originating in single cells and PICs are computationally deconvoluted and putative ligand-receptor interactions are predicted. This method has been successfully applied to murine dendritic and T-cells both in vitro and in vivo and has the potential to further our knowledge of physically interacting cells in vascular inflammation models.
Together, these examples show the importance of cellular communication in homeostasis and pathogenesis of disease. Given that vascular cells show unique organ-dependent phenotypes and are involved in complex pathologies inflicting whole organs, future studies focusing on communication between different cell types will be critical for a better understanding of the contribution of vascular cells to disease.
5.2. Single Cell Epigenetics
Single cell technologies are rapidly developing. New adaptations of single cell technologies to epigenomic methods such as chromatin immunoprecipitation (ChIP)-seq [125,126], assay for transposase-accessible chromatin (ATAC)-seq [127,128] and chromosomal conformation capture for studying the 3D chromatin structure [129,130,131,132] are increasingly improving. Thus far, single cell ATAC-seq is the only widely employed single cell epigenomic technique [127,128]. This technique detects accessible open chromatin regions in the genome [133] and is available as a kit from 10x Genomics, making it a popular option. In addition, new elegant methods probing a combination of the epigenetic state and transcriptomics on a single cell level are being developed [134,135,136,137]. For instance, NOME-seq [138] is now being utilized to simultaneously probe DNA methylation and open chromatin in single cells [134,135]. Surprisingly, neither scATAC-seq, nor single cell methylation profiles have been generated for vascular cells. On the other hand, single cell ChIP-seq has already been successfully applied to vascular development. Mapping of H3K27ac in Cdh5-traced EC lineages showed unique profiles of active promoters and enhancers in the ECs isolated from 10 different tissues of E16.5 embryos [125]. This is consistent with scRNA-seq studies that identify organ-dependent EC heterogeneity [10], suggesting that unique epigenetic factors are likely to drive the identity of unique EC-subtypes. These epigenetic mechanisms remain to be identified.
Future utilization of the single cell epigenomic technologies in the context of vascular dysfunction and inflammation will help unravel the underlying chromatin changes that modulate transcription and drive pathological changes in cellular phenotype.
6. Concluding Remarks
Vascular diseases have been studied for decades, but we are only now starting to understand the full complexity and cellular heterogeneity underlying disease-associated pathophysiological conditions. A range of phenotypic switches and trans-differentiation of major vascular and immune cells have been reported with the help of single cell technologies (Table 2). It is now important to harvest the unprecedented amount of data and try to integrate them with the previously gathered knowledge on signaling pathways and cell interactions in the context of diseases such as atherosclerosis and neurodegenerative disorders. Intercellular communication studies are of particular interest as human diseases involve a multitude of cell types and the root cause(s) is often difficult to ascertain. Future studies focusing not only on transcriptomic changes, but also on underlying epigenomic alterations, will provide us with a more comprehensive understanding of cellular and molecular mechanisms that drive changes in vascular cells associated with a multitude of pathologies.
Table 2.
Major single cell studies focusing on the vascular system.
Author Contributions
All authors wrote, edited and finalized the text. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the free-state of Saxony and Helmholtz Center Munich.
Acknowledgments
The authors thank Maria Shvedunova, Leila Varghese and Jesús Rodriguez-Aguilera for the thorough reading of this manuscript and helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Aird, W.C. Spatial and temporal dynamics of the endothelium. J. Thromb. Haemost. 2005, 3, 1392–1406. [Google Scholar] [CrossRef] [PubMed]
- Pugsley, M.K.; Tabrizchi, R. The vascular system. An overview of structure and function. J. Pharmacol. Toxicol. Methods 2000, 44, 333–340. [Google Scholar] [CrossRef]
- Vanlandewijck, M.; He, L.; Mae, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Lavina, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, T.G.; Stefanadis, C. Vascular wall shear stress: Basic principles and methods. Hellenic J. Cardiol. 2005, 46, 9–15. [Google Scholar] [PubMed]
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef]
- Holm, A.; Heumann, T.; Augustin, H.G. Microvascular Mural Cell Organotypic Heterogeneity and Functional Plasticity. Trends Cell Biol. 2018, 28, 302–316. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef]
- Kalucka, J.; de Rooij, L.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Hadi, H.A.; Carr, C.S.; Al Suwaidi, J. Endothelial dysfunction: Cardiovascular risk factors, therapy, and outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar] [PubMed]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The pathobiology of vascular dementia. Neuron 2013, 80, 844–866. [Google Scholar] [CrossRef] [PubMed]
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the Role of Angiogenesis in Cancer Ecosystems. Front. Oncol. 2018, 8, 248. [Google Scholar] [CrossRef]
- Kalisky, T.; Oriel, S.; Bar-Lev, T.H.; Ben-Haim, N.; Trink, A.; Wineberg, Y.; Kanter, I.; Gilad, S.; Pyne, S. A brief review of single-cell transcriptomic technologies. Brief. Funct. Genom. 2018, 17, 64–76. [Google Scholar] [CrossRef]
- Kelsey, G.; Stegle, O.; Reik, W. Single-cell epigenomics: Recording the past and predicting the future. Science 2017, 358, 69–75. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Bondareva, O.; Guhathakurta, S.; Tsang, T.H.; Sikora, K.; Aizarani, N.; Sagar Holz, H.; Grun, D.; Hein, L.; Akhtar, A.; et al. Systematic Identification of Cell-Cell Communication Networks in the Developing Brain. iScience 2019, 21, 273–287. [Google Scholar] [CrossRef]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef]
- Tabula Muris, C.; Overall, C.; Logistical, C.; Organ, C.P.; Library, P.S.; Computational, D.A.; Cell, T.A.; Writing, G. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef]
- Regev, A.; Teichmann, S.A.; Lander, E.S.; Amit, I.; Benoist, C.; Birney, E.; Bodenmiller, B.; Campbell, P.; Carninci, P.; Clatworthy, M.; et al. The Human Cell Atlas. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Rozenblatt-Rosen, O.; Stubbington, M.J.T.; Regev, A.; Teichmann, S.A. The Human Cell Atlas: From vision to reality. Nature 2017, 550, 451–453. [Google Scholar] [CrossRef] [PubMed]
- Reyfman, P.A.; Walter, J.M.; Joshi, N.; Anekalla, K.R.; McQuattie-Pimentel, A.C.; Chiu, S.; Fernandez, R.; Akbarpour, M.; Chen, C.I.; Ren, Z.; et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 199, 1517–1536. [Google Scholar] [CrossRef]
- Aizarani, N.; Saviano, A.; Sagar Mailly, L.; Durand, S.; Herman, J.S.; Pessaux, P.; Baumert, T.F.; Grun, D. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 2019, 572, 199–204. [Google Scholar] [CrossRef]
- Tucker, N.R.; Chaffin, M.; Fleming, S.J.; Hall, A.W.; Parsons, V.A.; Bedi, K.; Akkad, A.-D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and Cellular Diversity of the Human Heart. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef]
- Cui, Y.; Zheng, Y.; Liu, X.; Yan, L.; Fan, X.; Yong, J.; Hu, Y.; Dong, J.; Li, Q.; Wu, X.; et al. Single-Cell Transcriptome Analysis Maps the Developmental Track of the Human Heart. Cell Rep. 2019, 26, 1934–1950. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Eichten, A.; Parveen, A.; Adler, C.; Huang, Y.; Wang, W.; Ding, Y.; Adler, A.; Nevins, T.; Ni, M.; et al. Single-Cell Transcriptome Analyses Reveal Endothelial Cell Heterogeneity in Tumors and Changes following Antiangiogenic Treatment. Cancer Res. 2018, 78, 2370–2382. [Google Scholar] [CrossRef]
- Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Fasouli, E.S.; Samari, S.; et al. Cells and gene expression programs in the adult human heart. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wu, S.P.; Cheng, C.M.; Lanz, R.B.; Wang, T.; Respress, J.L.; Ather, S.; Chen, W.; Tsai, S.J.; Wehrens, X.H.; Tsai, M.J.; et al. Atrial identity is determined by a COUP-TFII regulatory network. Dev. Cell 2013, 25, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P. Endothelial dysfunction and hypertension. Hypertension 2014, 64, 924–928. [Google Scholar] [CrossRef]
- Higashi, Y.; Kihara, Y.; Noma, K. Endothelial dysfunction and hypertension in aging. Hypertens. Res. 2012, 35, 1039–1047. [Google Scholar] [CrossRef]
- Sheikh, B.N.; Guhathakurta, S.; Tsang, T.H.; Schwabenland, M.; Renschler, G.; Herquel, B.; Bhardwaj, V.; Holz, H.; Stehle, T.; Bondareva, O.; et al. Neural metabolic imbalance induced by MOF dysfunction triggers pericyte activation and breakdown of vasculature. Nat. Cell Biol. 2020. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Schnitzler, J.G.; Hoogeveen, R.M.; Ali, L.; Prange, K.H.; Waissi, F.; van Weeghel, M.; Bachmann, J.C.; Versloot, M.; Borrelli, M.J.; Yeang, C.; et al. Atherogenic Lipoprotein(a) Increases Vascular Glycolysis, Thereby Facilitating Inflammation and Leukocyte Extravasation. Circ. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef]
- Martin, K.; Weiss, S.; Metharom, P.; Schmeckpeper, J.; Hynes, B.; O’Sullivan, J.; Caplice, N. Thrombin stimulates smooth muscle cell differentiation from peripheral blood mononuclear cells via protease-activated receptor-1, RhoA, and myocardin. Circ. Res. 2009, 105, 214–218. [Google Scholar] [CrossRef]
- Stewart, H.J.; Guildford, A.L.; Lawrence-Watt, D.J.; Santin, M. Substrate-induced phenotypical change of monocytes/macrophages into myofibroblast-like cells: A new insight into the mechanism of in-stent restenosis. J. Biomed. Mater. Res. Part A 2009, 90, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res. 2012, 95, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.R.; d’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2016, 7, 11853. [Google Scholar] [CrossRef] [PubMed]
- Bondareva, O.; Tsaryk, R.; Bojovic, V.; Odenthal-Schnittler, M.; Siekmann, A.F.; Schnittler, H.J. Identification of atheroprone shear stress responsive regulatory elements in endothelial cells. Cardiovasc. Res. 2019, 115, 1487–1499. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, Z.; Torsney, E.; Afzal, A.R.; Davison, F.; Metzler, B.; Xu, Q. Abundant progenitor cells in the adventitia contribute to atherosclerosis of vein grafts in ApoE-deficient mice. J. Clin. Investig. 2004, 113, 1258–1265. [Google Scholar] [CrossRef]
- Kramann, R.; Goettsch, C.; Wongboonsin, J.; Iwata, H.; Schneider, R.K.; Kuppe, C.; Kaesler, N.; Chang-Panesso, M.; Machado, F.G.; Gratwohl, S.; et al. Adventitial MSC-like Cells Are Progenitors of Vascular Smooth Muscle Cells and Drive Vascular Calcification in Chronic Kidney Disease. Cell Stem Cell 2016, 19, 628–642. [Google Scholar] [CrossRef]
- Kalluri, A.S.; Vellarikkal, S.K.; Edelman, E.R.; Nguyen, L.; Subramanian, A.; Ellinor, P.T.; Regev, A.; Kathiresan, S.; Gupta, R.M. Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation 2019, 140, 147–163. [Google Scholar] [CrossRef]
- Dobnikar, L.; Taylor, A.L.; Chappell, J.; Oldach, P.; Harman, J.L.; Oerton, E.; Dzierzak, E.; Bennett, M.R.; Spivakov, M.; Jorgensen, H.F. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat. Commun. 2018, 9, 4567. [Google Scholar] [CrossRef]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.E.; Zernecke, A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef]
- Kim, K.; Shim, D.; Lee, J.S.; Zaitsev, K.; Williams, J.W.; Kim, K.W.; Jang, M.Y.; Seok Jang, H.; Yun, T.J.; Lee, S.H.; et al. Transcriptome Analysis Reveals Nonfoamy Rather Than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ. Res. 2018, 123, 1127–1142. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Ni, Z.; Tan, Y.Q.; Deng, J.; Zhang, S.J.; Lv, Z.C.; Wang, X.J.; Chen, T.; Zhang, Z.; Hu, Y.; et al. Adventitial Cell Atlas of wt (Wild Type) and ApoE (Apolipoprotein E)-Deficient Mice Defined by Single-Cell RNA Sequencing. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1055–1071. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wang, H.; Huang, X.; Li, F.; Zhu, H.; Li, Y.; He, L.; Zhang, H.; Pu, W.; Liu, K.; et al. Arterial Sca1(+) Vascular Stem Cells Generate De Novo Smooth Muscle for Artery Repair and Regeneration. Cell Stem Cell 2020, 26, 81–96. [Google Scholar] [CrossRef] [PubMed]
- Nurnberg, S.T.; Cheng, K.; Raiesdana, A.; Kundu, R.; Miller, C.L.; Kim, J.B.; Arora, K.; Carcamo-Oribe, I.; Xiong, Y.; Tellakula, N.; et al. Coronary Artery Disease Associated Transcription Factor TCF21 Regulates Smooth Muscle Precursor Cells That Contribute to the Fibrous Cap. PLoS Genet. 2015, 11, e1005155. [Google Scholar] [CrossRef] [PubMed]
- Braitsch, C.M.; Kanisicak, O.; van Berlo, J.H.; Molkentin, J.D.; Yutzey, K.E. Differential expression of embryonic epicardial progenitor markers and localization of cardiac fibrosis in adult ischemic injury and hypertensive heart disease. J. Mol. Cell Cardiol. 2013, 65, 108–119. [Google Scholar] [CrossRef]
- Ganieva, U.; Nakamura, T.; Osuka, S.; Bayasula Nakanishi, N.; Kasahara, Y.; Takasaki, N.; Muraoka, A.; Hayashi, S.; Nagai, T.; Murase, T.; et al. Involvement of Transcription Factor 21 in the Pathogenesis of Fibrosis in Endometriosis. Am. J. Pathol. 2020, 190, 145–157. [Google Scholar] [CrossRef]
- Nakano, Y.; Kamiya, A.; Sumiyoshi, H.; Tsuruya, K.; Kagawa, T.; Inagaki, Y. A Deactivation Factor of Fibrogenic Hepatic Stellate Cells Induces Regression of Liver Fibrosis in Mice. Hepatology 2019. [Google Scholar] [CrossRef]
- Rosenfeld, M.E. Macrophage proliferation in atherosclerosis: An historical perspective. Arterioscler. Thromb. Vasc. Biol. 2014, 34, e21–e22. [Google Scholar] [CrossRef]
- Lin, J.D.; Nishi, H.; Poles, J.; Niu, X.; McCauley, C.; Rahman, K.; Brown, E.J.; Yeung, S.T.; Vozhilla, N.; Weinstock, A.; et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef] [PubMed]
- King, K.R.; Aguirre, A.D.; Ye, Y.X.; Sun, Y.; Roh, J.D.; Ng, R.P., Jr.; Kohler, R.H.; Arlauckas, S.P.; Iwamoto, Y.; Savol, A.; et al. IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef]
- Farbehi, N.; Patrick, R.; Dorison, A.; Xaymardan, M.; Janbandhu, V.; Wystub-Lis, K.; Ho, J.W.; Nordon, R.E.; Harvey, R.P. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Skelly, D.A.; Chen, M.; Daigle, S.; Morelli, K.A.; Hon, O.; Philip, V.M.; Costa, M.W.; Rosenthal, N.A.; Furtado, M.B. Dynamic Interstitial Cell Response during Myocardial Infarction Predicts Resilience to Rupture in Genetically Diverse Mice. Cell Rep. 2020, 30, 3149–3163. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef]
- Li, Z.; Solomonidis, E.G.; Meloni, M.; Taylor, R.S.; Duffin, R.; Dobie, R.; Magalhaes, M.S.; Henderson, B.E.P.; Louwe, P.A.; D’Amico, G.; et al. Single-cell transcriptome analyses reveal novel targets modulating cardiac neovascularization by resident endothelial cells following myocardial infarction. Eur. Heart J. 2019, 40, 2507–2520. [Google Scholar] [CrossRef]
- Simonneau, G.; Gatzoulis, M.A.; Adatia, I.; Celermajer, D.; Denton, C.; Ghofrani, A.; Gomez Sanchez, M.A.; Krishna Kumar, R.; Landzberg, M.; Machado, R.F.; et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 2013, 62, D34–D41. [Google Scholar] [CrossRef]
- Huertas, A.; Tu, L.; Humbert, M.; Guignabert, C. Chronic inflammation within the vascular wall in pulmonary arterial hypertension: More than a spectator. Cardiovasc. Res. 2020, 116, 885–893. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Humbert, M. Pathobiology of pulmonary arterial hypertension: Understanding the roads less travelled. Eur. Respir. Rev. 2017, 26. [Google Scholar] [CrossRef]
- Cohen, M.; Giladi, A.; Gorki, A.D.; Solodkin, D.G.; Zada, M.; Hladik, A.; Miklosi, A.; Salame, T.M.; Halpern, K.B.; David, E.; et al. Lung Single-Cell Signaling Interaction Map Reveals Basophil Role in Macrophage Imprinting. Cell 2018, 175, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Angelidis, I.; Simon, L.M.; Fernandez, I.E.; Strunz, M.; Mayr, C.H.; Greiffo, F.R.; Tsitsiridis, G.; Ansari, M.; Graf, E.; Strom, T.M.; et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat. Commun. 2019, 10, 963. [Google Scholar] [CrossRef]
- Travaglini, K.J.; Nabhan, A.N.; Penland, L.; Sinha, R.; Gillich, A.; Sit, R.V.; Chang, S.; Conley, S.D.; Mori, Y.; Seita, J.; et al. A molecular cell atlas of the human lung from single cell RNA sequencing. bioRxiv 2020. [Google Scholar] [CrossRef]
- Saygin, D.; Tabib, T.; Bittar, H.E.T.; Valenzi, E.; Sembrat, J.; Chan, S.Y.; Rojas, M.; Lafyatis, R. Transcriptional profiling of lung cell populations in idiopathic pulmonary arterial hypertension. Pulm. Circ. 2020, 10. [Google Scholar] [CrossRef]
- Kim, J.; Kang, Y.; Kojima, Y.; Lighthouse, J.K.; Hu, X.; Aldred, M.A.; McLean, D.L.; Park, H.; Comhair, S.A.; Greif, D.M.; et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat. Med. 2013, 19, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Alastalo, T.P.; Li, M.; Perez Vde, J.; Pham, D.; Sawada, H.; Wang, J.K.; Koskenvuo, M.; Wang, L.; Freeman, B.A.; Chang, H.Y.; et al. Disruption of PPARgamma/beta-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J. Clin. Investig. 2011, 121, 3735–3746. [Google Scholar] [CrossRef]
- Raredon, M.S.B.; Adams, T.S.; Suhail, Y.; Schupp, J.C.; Poli, S.; Neumark, N.; Leiby, K.L.; Greaney, A.M.; Yuan, Y.; Horien, C.; et al. Single-cell connectomic analysis of adult mammalian lungs. Sci. Adv. 2019, 5, eaaw3851. [Google Scholar] [CrossRef]
- Reyes-Palomares, A.; Gu, M.; Grubert, F.; Berest, I.; Sa, S.; Kasowski, M.; Arnold, C.; Shuai, M.; Srivas, R.; Miao, S.; et al. Remodeling of active endothelial enhancers is associated with aberrant gene-regulatory networks in pulmonary arterial hypertension. Nat. Commun. 2020, 11, 1673. [Google Scholar] [CrossRef]
- Bertero, T.; Oldham, W.M.; Cottrill, K.A.; Pisano, S.; Vanderpool, R.R.; Yu, Q.; Zhao, J.; Tai, Y.; Tang, Y.; Zhang, Y.Y.; et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Investig. 2016, 126, 3313–3335. [Google Scholar] [CrossRef]
- Tiana, M.; Acosta-Iborra, B.; Puente-Santamaria, L.; Hernansanz-Agustin, P.; Worsley-Hunt, R.; Masson, N.; Garcia-Rio, F.; Mole, D.; Ratcliffe, P.; Wasserman, W.W.; et al. The SIN3A histone deacetylase complex is required for a complete transcriptional response to hypoxia. Nucleic Acids Res. 2018, 46, 120–133. [Google Scholar] [CrossRef]
- Biddlestone, J.; Batie, M.; Bandarra, D.; Munoz, I.; Rocha, S. SINHCAF/FAM60A and SIN3A specifically repress HIF-2alpha expression. Biochem. J. 2018, 475, 2073–2090. [Google Scholar] [CrossRef] [PubMed]
- Mottet, D.; Bellahcene, A.; Pirotte, S.; Waltregny, D.; Deroanne, C.; Lamour, V.; Lidereau, R.; Castronovo, V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ. Res. 2007, 101, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Turtoi, A.; Mottet, D.; Matheus, N.; Dumont, B.; Peixoto, P.; Hennequiere, V.; Deroanne, C.; Colige, A.; De Pauw, E.; Bellahcene, A.; et al. The angiogenesis suppressor gene AKAP12 is under the epigenetic control of HDAC7 in endothelial cells. Angiogenesis 2012, 15, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Kaluza, D.; Kroll, J.; Gesierich, S.; Yao, T.P.; Boon, R.A.; Hergenreider, E.; Tjwa, M.; Rossig, L.; Seto, E.; Augustin, H.G.; et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 2011, 30, 4142–4156. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and Dysfunction of the Blood-Brain Barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef]
- Hellstrom, M.; Kalen, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar]
- Lindahl, P.; Johansson, B.R.; Leveen, P.; Betsholtz, C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Pericyte-specific expression of PDGF beta receptor in mouse models with normal and deficient PDGF beta receptor signaling. Mol. Neurodegener. 2010, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef]
- Nishioku, T.; Dohgu, S.; Takata, F.; Eto, T.; Ishikawa, N.; Kodama, K.B.; Nakagawa, S.; Yamauchi, A.; Kataoka, Y. Detachment of brain pericytes from the basal lamina is involved in disruption of the blood-brain barrier caused by lipopolysaccharide-induced sepsis in mice. Cell. Mol. Neurobiol. 2009, 29, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid beta oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 2019, 365. [Google Scholar] [CrossRef] [PubMed]
- Bonney, S.; Seitz, S.; Ryan, C.A.; Jones, K.L.; Clarke, P.; Tyler, K.L.; Siegenthaler, J.A. Gamma Interferon Alters Junctional Integrity via Rho Kinase, Resulting in Blood-Brain Barrier Leakage in Experimental Viral Encephalitis. mBio 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Todorov, M.I.; Paetzold, J.C.; Schoppe, O.; Tetteh, G.; Shit, S.; Efremov, V.; Todorov-Volgyi, K.; During, M.; Dichgans, M.; Piraud, M.; et al. Machine learning analysis of whole mouse brain vasculature. Nat. Methods 2020, 17, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Di Giovanna, A.P.; Tibo, A.; Silvestri, L.; Mullenbroich, M.C.; Costantini, I.; Allegra Mascaro, A.L.; Sacconi, L.; Frasconi, P.; Pavone, F.S. Whole-Brain Vasculature Reconstruction at the Single Capillary Level. Sci. Rep. 2018, 8, 12573. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, W.M.; Skoog, I.; Schneider, J.A.; Pantoni, L.; Mok, V.; Chen, C.L.H.; Scheltens, P. Vascular cognitive impairment. Nat. Rev. Dis. Primers 2018, 4, 18003. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Korczyn, A.D. Vascular parkinsonism--characteristics, pathogenesis and treatment. Nat. Rev. Neurol. 2015, 11, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Drouin-Ouellet, J.; Sawiak, S.J.; Cisbani, G.; Lagace, M.; Kuan, W.L.; Saint-Pierre, M.; Dury, R.J.; Alata, W.; St-Amour, I.; Mason, S.L.; et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann. Neurol. 2015, 78, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Grubman, A.; Chew, G.; Ouyang, J.F.; Sun, G.; Choo, X.Y.; McLean, C.; Simmons, R.K.; Buckberry, S.; Vargas-Landin, D.B.; Poppe, D.; et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 2019, 22, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Zhao, L.; Li, Z.; Vong, J.S.L.; Chen, X.; Lai, H.-M.; Yan, L.Y.C.; Huang, J.; Sy, S.K.H.; Tian, X.; Huang, Y.; et al. Zonation-dependent single-endothelial cell transcriptomic changes in the aged brain. bioRxiv 2019. [Google Scholar] [CrossRef]
- Blanchard, J.W.; Bula, M.; Davila-Velderrain, J.; Akay, L.A.; Zhu, L.; Frank, A.; Victor, M.B.; Bonner, J.M.; Mathys, H.; Lin, Y.T.; et al. Reconstruction of the human blood-brain barrier in vitro reveals a pathogenic mechanism of APOE4 in pericytes. Nat. Med. 2020, 26, 952–963. [Google Scholar] [CrossRef]
- van Karnebeek, C.D.; Shevell, M.; Zschocke, J.; Moeschler, J.B.; Stockler, S. The metabolic evaluation of the child with an intellectual developmental disorder: Diagnostic algorithm for identification of treatable causes and new digital resource. Mol. Genet. Metab. 2014, 111, 428–438. [Google Scholar] [CrossRef]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; de Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metabolism 2016, 65, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Rebeck, G.W. The role of APOE on lipid homeostasis and inflammation in normal brains. J. Lipid Res. 2017, 58, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Farmer, B.C.; Kluemper, J.; Johnson, L.A. Apolipoprotein E4 Alters Astrocyte Fatty Acid Metabolism and Lipid Droplet Formation. Cells 2019, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Ramilowski, J.A.; Goldberg, T.; Harshbarger, J.; Kloppmann, E.; Lizio, M.; Satagopam, V.P.; Itoh, M.; Kawaji, H.; Carninci, P.; Rost, B.; et al. A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2015, 6, 7866. [Google Scholar] [CrossRef]
- Skelly, D.A.; Squiers, G.T.; McLellan, M.A.; Bolisetty, M.T.; Robson, P.; Rosenthal, N.A.; Pinto, A.R. Single-Cell Transcriptional Profiling Reveals Cellular Diversity and Intercommunication in the Mouse Heart. Cell Rep. 2018, 22, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Kirita, Y.; Donnelly, E.L.; Humphreys, B.D. Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis. J. Am. Soc. Nephrol. 2019, 30, 23–32. [Google Scholar] [CrossRef]
- Kumar, M.P.; Du, J.; Lagoudas, G.; Jiao, Y.; Sawyer, A.; Drummond, D.C.; Lauffenburger, D.A.; Raue, A. Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep. 2018, 25, 1458–1468. [Google Scholar] [CrossRef]
- Cabello-Aguilar, S.; Tack, F.K.S.; Alame, M.; Fau, C.; Lacroix, M.; Colinge, J. SingleCellSignalR: Inference of intercellular networks from single-cell transcriptomics. bioRxiv 2019. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.; Zhang, S.; Song, S.; Jiang, C.; Han, G.; Wang, M.; Ajani, J.; Futreal, A.; Wang, L. iTALK: An R Package to Characterize and Illustrate Intercellular Communication. bioRxiv 2019. [Google Scholar] [CrossRef]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; PericakVance, M.A.; Risch, N.; vanDuijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease—A meta-analysis. JAMA-J. Am. Med. Assoc. 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Giladi, A.; Cohen, M.; Medaglia, C.; Baran, Y.; Li, B.; Zada, M.; Bost, P.; Blecher-Gonen, R.; Salame, T.M.; Mayer, J.U.; et al. Dissecting cellular crosstalk by sequencing physically interacting cells. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Xiong, H.; Ai, S.; Yu, X.; Liu, Y.; Zhang, J.; He, A. CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Mol. Cell 2019, 76, 206–216. [Google Scholar] [CrossRef]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O.; et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490. [Google Scholar] [CrossRef]
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348, 910–914. [Google Scholar] [CrossRef]
- Nagano, T.; Lubling, Y.; Yaffe, E.; Wingett, S.W.; Dean, W.; Tanay, A.; Fraser, P. Single-cell Hi-C for genome-wide detection of chromatin interactions that occur simultaneously in a single cell. Nat. Protoc. 2015, 10, 1986–2003. [Google Scholar] [CrossRef]
- Flyamer, I.M.; Gassler, J.; Imakaev, M.; Brandao, H.B.; Ulianov, S.V.; Abdennur, N.; Razin, S.V.; Mirny, L.A.; Tachibana-Konwalski, K. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 2017, 544, 110–114. [Google Scholar] [CrossRef]
- Stevens, T.J.; Lando, D.; Basu, S.; Atkinson, L.P.; Cao, Y.; Lee, S.F.; Leeb, M.; Wohlfahrt, K.J.; Boucher, W.; O’Shaughnessy-Kirwan, A.; et al. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 2017, 544, 59–64. [Google Scholar] [CrossRef]
- Beagrie, R.A.; Scialdone, A.; Schueler, M.; Kraemer, D.C.; Chotalia, M.; Xie, S.Q.; Barbieri, M.; de Santiago, I.; Lavitas, L.M.; Branco, M.R.; et al. Complex multi-enhancer contacts captured by genome architecture mapping. Nature 2017, 543, 519–524. [Google Scholar] [CrossRef]
- Satpathy, A.T.; Granja, J.M.; Yost, K.E.; Qi, Y.; Meschi, F.; McDermott, G.P.; Olsen, B.N.; Mumbach, M.R.; Pierce, S.E.; Corces, M.R.; et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat. Biotechnol. 2019, 37, 925–936. [Google Scholar] [CrossRef]
- Pott, S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. Elife 2017, 6. [Google Scholar] [CrossRef]
- Clark, S.J.; Argelaguet, R.; Kapourani, C.A.; Stubbs, T.M.; Lee, H.J.; Alda-Catalinas, C.; Krueger, F.; Sanguinetti, G.; Kelsey, G.; Marioni, J.C.; et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat. Commun. 2018, 9, 781. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Cusanovich, D.A.; Ramani, V.; Aghamirzaie, D.; Pliner, H.A.; Hill, A.J.; Daza, R.M.; McFaline-Figueroa, J.L.; Packer, J.S.; Christiansen, L.; et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 2018, 361, 1380–1385. [Google Scholar] [CrossRef] [PubMed]
- Barnett, K.R.; Decato, B.E.; Scott, T.J.; Hansen, T.J.; Chen, B.; Attalla, J.; Smith, A.D.; Hodges, E. ATAC-Me Captures Prolonged DNA Methylation of Dynamic Chromatin Accessibility Loci during Cell Fate Transitions. Mol. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.K.; Liu, Y.; Lay, F.D.; Liang, G.; Berman, B.P.; Jones, P.A. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012, 22, 2497–2506. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).