Comparative Analysis, Structural Insights, and Substrate/Drug Interaction of CYP128A1 in Mycobacterium tuberculosis

 , ,
, ,  , ,
, , 

Abstract
:1. Introduction
2. Results and Discussion
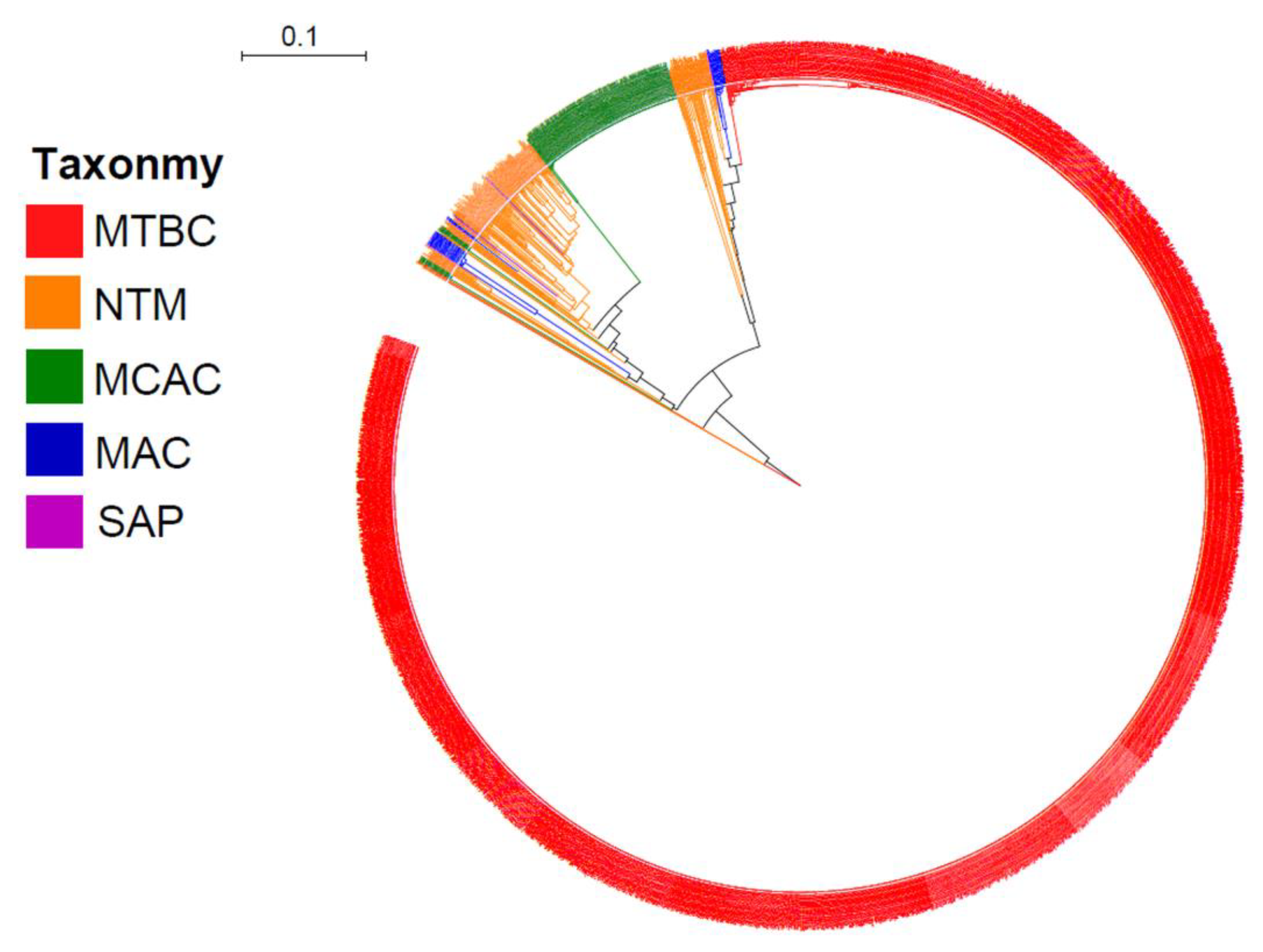
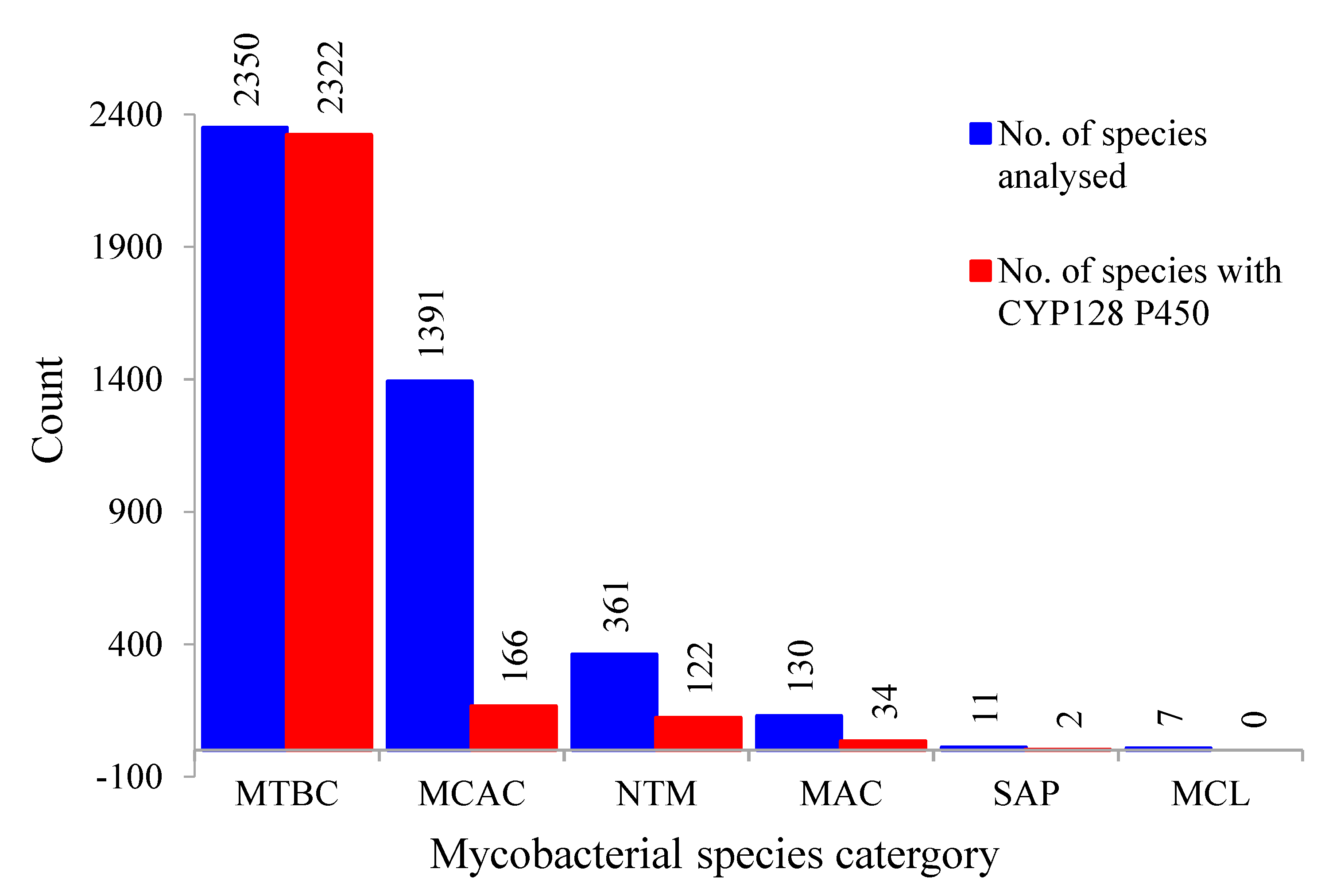
2.1. Presence of CYP128 P450s in Five of the Six Mycobacterial Category Species
2.2. Different Mycobacterial Category Species Have Different CYP128 Subfamily Preferences
2.3. CYP128 Family Ranked Fifth among P450 Families
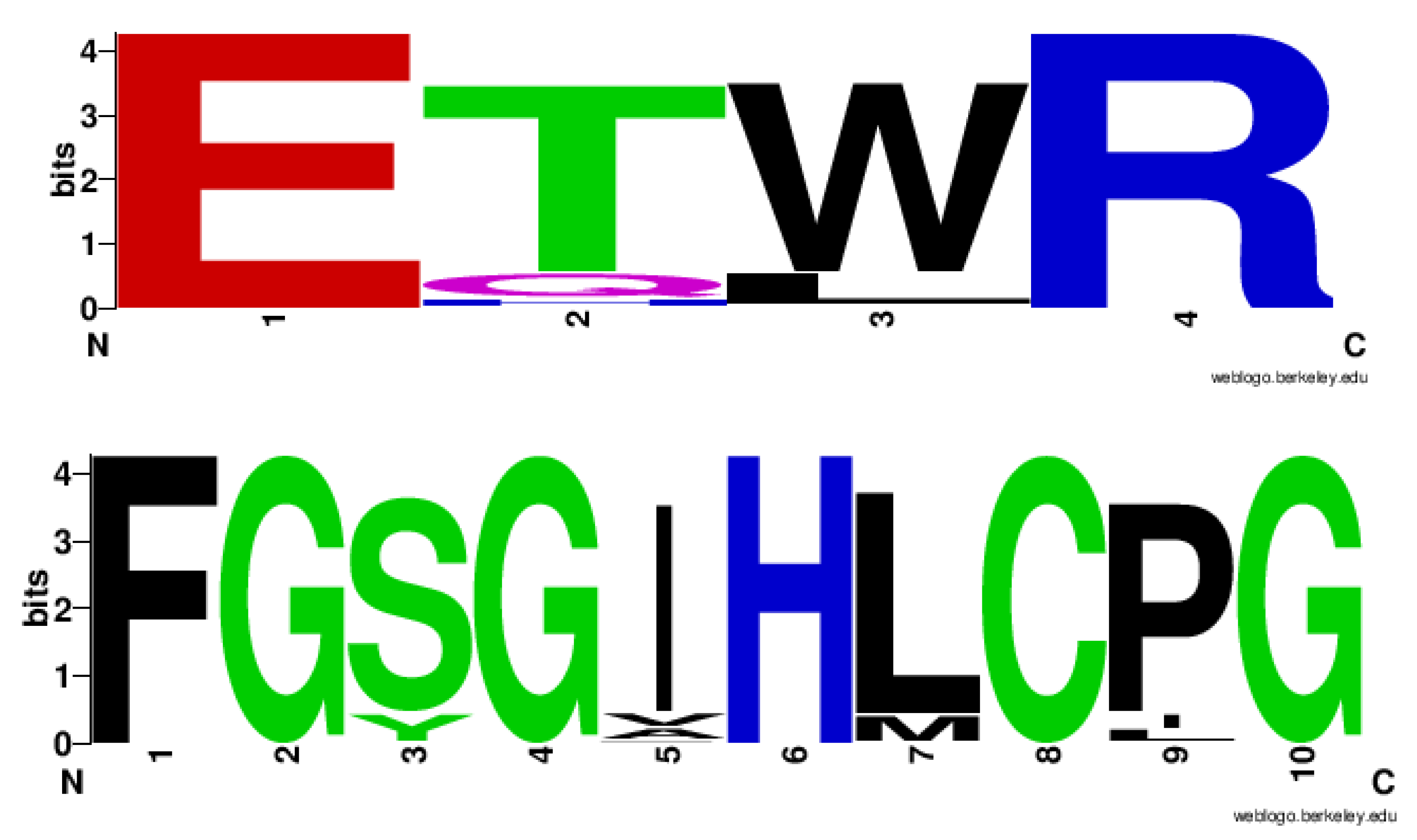
2.4. CYP128 Family Has Distinctive Amino Acid Patterns at EXXR and CXG Motif
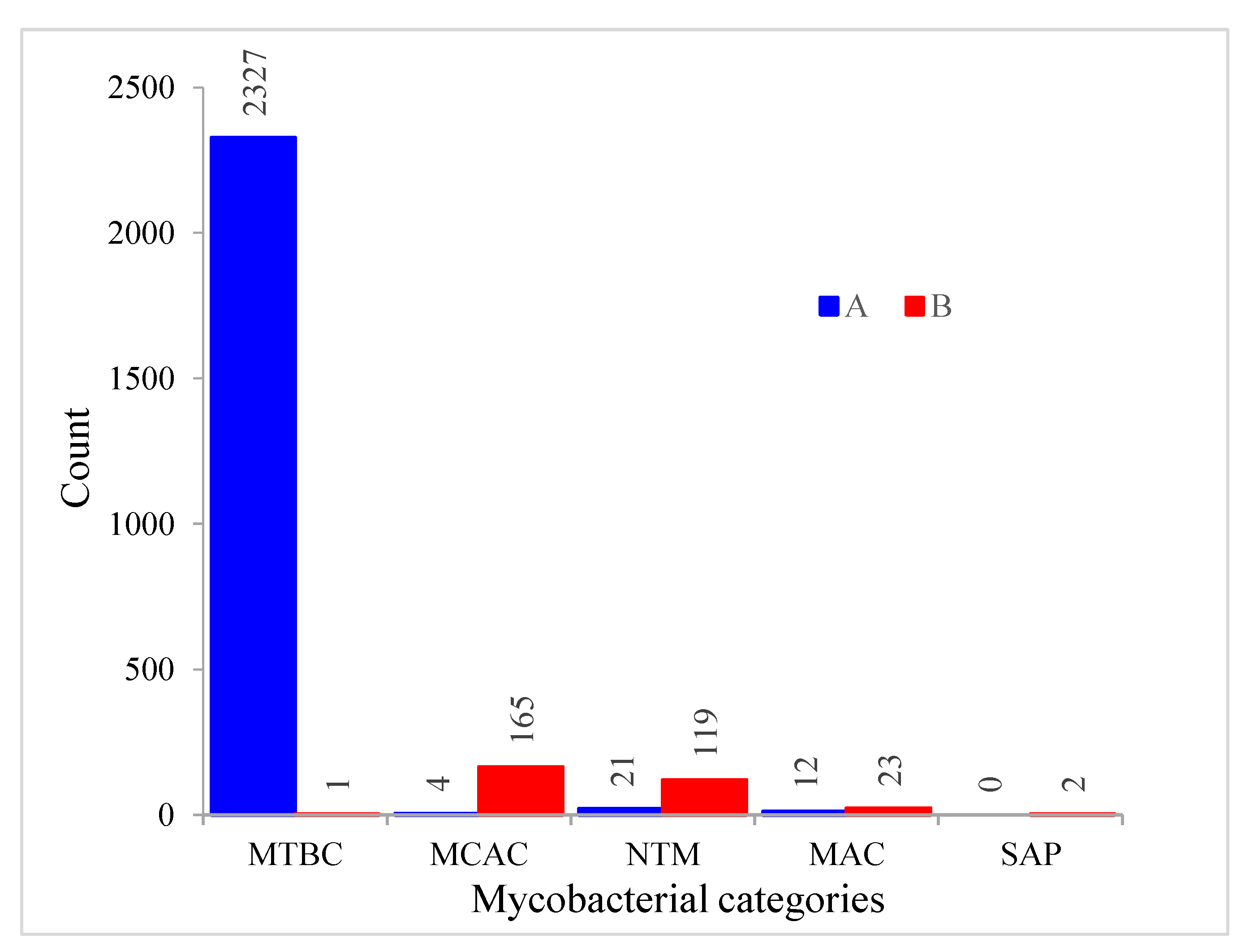
2.5. Most CYP128 P450s Exist in Secondary Metabolite Biosynthetic Gene Clusters
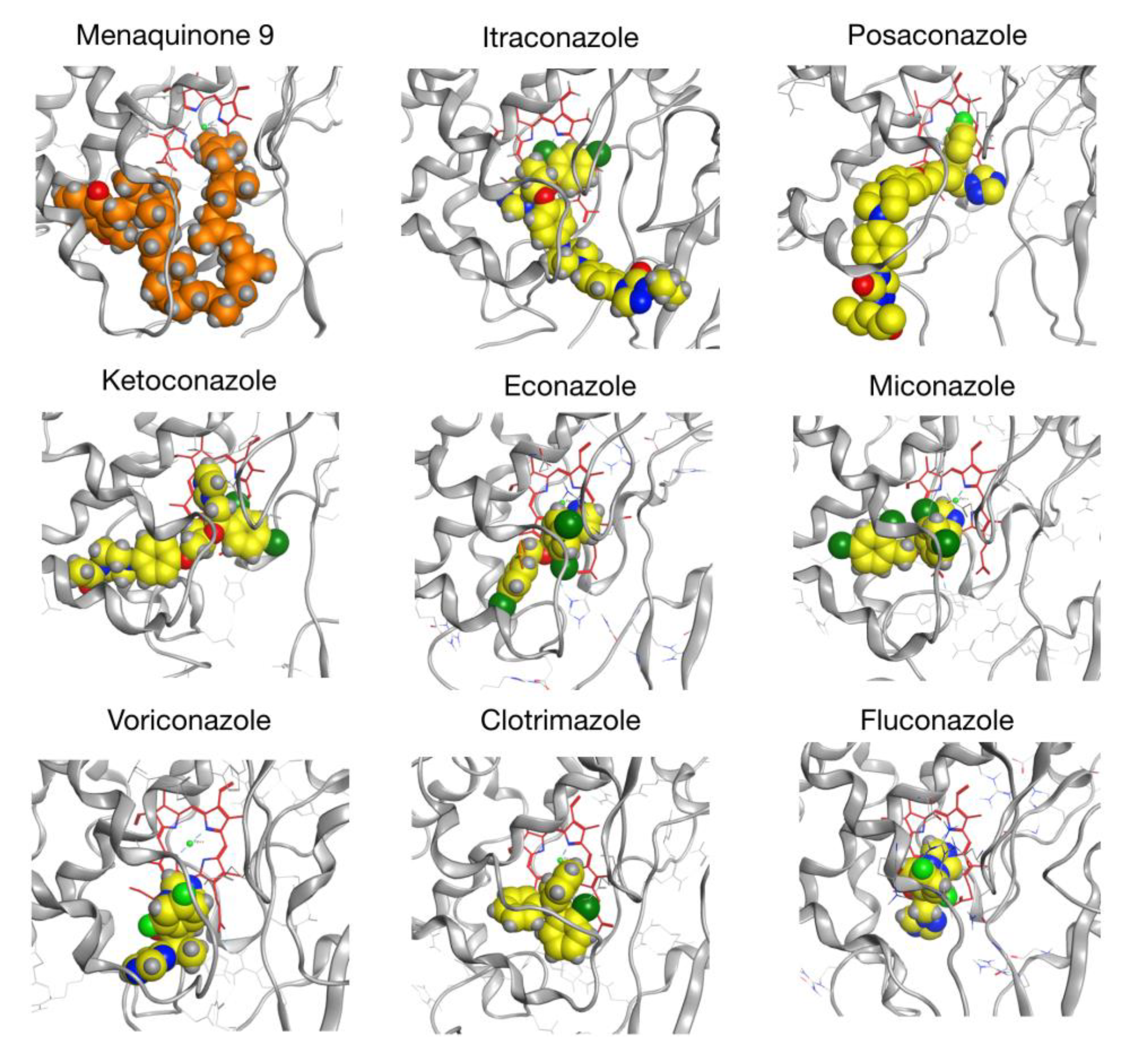
2.6. CYP128A1 Protein Model Has High Affinity to Menaquinone-9
3. Materials and Methods
3.1. Species and Databases
3.2. Genome Data Mining and Annotation of CYP128 P450s
3.3. Phylogenetic Analysis of CYP128 P450s
3.4. Amino Acid Conservation Analysis
3.5. Generation of EXXR and CXG Sequence Logos
3.6. Identification of CYP128 P450 Secondary Metabolite BGCs
3.7. CYP128 Homology Modeling
3.8. Molecular Docking of the CYP128 Model
3.9. Density Functional Theory
3.10. Molecular Dynamics
3.11. Redocking of Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Global Tuberculosis Report 2019. 2019. ISBN 978-92-4-156571-4. Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 7 December 2019).
- SSA. Mortality and Causes of Death in South Africa, 2016: Findings From Death Notification; Statistics South Africa. 2018. Available online: http://www.statssa.gov.za/publications/P03093/P030932016.pdf (accessed on 22 March 2019).
- Cole, S.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.; Eiglmeier, K.; Gas, S.; Barry Iii, C. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537. [Google Scholar] [CrossRef] [PubMed]
- Ghazaei, C. Mycobacterium tuberculosis and lipids: Insights into molecular mechanisms from persistence to virulence. J. Res. Med Sci. Off. J. Isfahan Univ. Med. Sci. 2018, 23, 63. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Potential drug targets in the Mycobacterium tuberculosis cytochrome P450 system. J. Inorg. Biochem. 2018, 180, 235–245. [Google Scholar] [CrossRef]
- Senate, L.M.; Tjatji, M.P.; Pillay, K.; Chen, W.; Zondo, N.M.; Syed, P.R.; Mnguni, F.C.; Chiliza, Z.E.; Bamal, H.D.; Karpoormath, R. Similarities, variations, and evolution of cytochrome P450s in Streptomyces versus Mycobacterium. Sci. Rep. 2019, 9, 3962. [Google Scholar] [CrossRef] [Green Version]
- Nelson, D.R. Cytochrome P450 diversity in the tree of life. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 141–154. [Google Scholar] [CrossRef]
- Van Wyk, R.; van Wyk, M.; Mashele, S.S.; Nelson, D.R.; Syed, K. Comprehensive comparative analysis of cholesterol catabolic genes/proteins in mycobacterial species. Int. J. Mol. Sci. 2019, 20, 1032. [Google Scholar] [CrossRef] [Green Version]
- Sassetti, C.M.; Rubin, E.J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 2003, 100, 12989–12994. [Google Scholar] [CrossRef] [Green Version]
- Lamichhane, G.; Zignol, M.; Blades, N.J.; Geiman, D.E.; Dougherty, A.; Grosset, J.; Broman, K.W.; Bishai, W.R. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: Application to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2003, 100, 7213–7218. [Google Scholar] [CrossRef] [Green Version]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002, 43, 717–731. [Google Scholar] [CrossRef]
- Rustad, T.R.; Harrell, M.I.; Liao, R.; Sherman, D.R. The enduring hypoxic response of Mycobacterium tuberculosis. PLoS ONE 2008, 3, e1502. [Google Scholar] [CrossRef]
- Sogi, K.M.; Holsclaw, C.M.; Fragiadakis, G.K.; Nomura, D.K.; Leary, J.A.; Bertozzi, C.R. Biosynthesis and Regulation of Sulfomenaquinone, a Metabolite Associated with Virulence in Mycobacterium tuberculosis. ACS Infect. Dis. 2016, 2, 800–806. [Google Scholar] [CrossRef] [Green Version]
- Ouellet, H.; Johnston, J.B.; de Montellano, P.R.O. The Mycobacterium tuberculosis cytochrome P450 system. Arch. Biochem. Biophys. 2010, 493, 82–95. [Google Scholar] [CrossRef] [Green Version]
- Driscoll, M. Investigating Orphan Cytochromes P450 from Mycobacterium tuberculosis: The Search for Potential Drug Targets. Ph.D. Thesis, University of Manchester, Manchester, UK, 2011. [Google Scholar]
- Holsclaw, C.M.; Sogi, K.M.; Gilmore, S.A.; Schelle, M.W.; Leavell, M.D.; Bertozzi, C.R.; Leary, J.A. Structural characterization of a novel sulfated menaquinone produced by stf3 from Mycobacterium tuberculosis. ACS Chem. Biol. 2008, 3, 619624. [Google Scholar] [CrossRef] [Green Version]
- Dhiman, R.K.; Mahapatra, S.; Slayden, R.A.; Boyne, M.E.; Lenaerts, A.; Hinshaw, J.C.; Angala, S.K.; Chatterjee, D.; Biswas, K.; Narayanasamy, P. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol. Microbiol. 2009, 72, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Zhang, Y. Antituberculosis activity of certain antifungal and antihelmintic drugs. Tuber. Lung Dis. 1999, 79, 319–320. [Google Scholar] [CrossRef]
- Ahmad, Z.; Sharma, S.; Khuller, G.K. The potential of azole antifungals against latent/persistent tuberculosis. FEMS Microbiol. Lett. 2006, 258, 200–203. [Google Scholar] [CrossRef]
- Ahmad, Z.; Sharma, S.; Khuller, G. In vitro and ex vivo antimycobacterial potential of azole drugs against Mycobacterium tuberculosis H37Rv. FEMS Microbiol. Lett. 2005, 251, 19–22. [Google Scholar] [CrossRef] [Green Version]
- Chenge, J.T.; Duyet, L.V.; Swami, S.; McLean, K.J.; Kavanagh, M.E.; Coyne, A.G.; Rigby, S.E.; Cheesman, M.R.; Girvan, H.M.; Levy, C.W.; et al. Structural Characterization and Ligand/Inhibitor Identification Provide Functional Insights into the Mycobacterium tuberculosis Cytochrome P450 CYP126A1. J. Biol. Chem. 2017, 292, 1310–1329. [Google Scholar]
- Parvez, M.; Qhanya, L.B.; Mthakathi, N.T.; Kgosiemang, I.K.; Bamal, H.D.; Pagadala, N.S.; Xie, T.; Yang, H.; Chen, H.; Theron, C.W.; et al. Molecular evolutionary dynamics of cytochrome P450 monooxygenases across kingdoms: Special focus on mycobacterial P450s. Sci. Rep. 2016, 6, 33099. [Google Scholar]
- Mthethwa, B.; Chen, W.; Ngwenya, M.; Kappo, A.; Syed, P.; Karpoormath, R.; Yu, J.-H.; Nelson, D.; Syed, K. Comparative analyses of cytochrome P450s and those associated with secondary metabolism in Bacillus species. Int. J. Mol. Sci. 2018, 19, 3623. [Google Scholar] [CrossRef] [Green Version]
- Syed, K.; Shale, K.; Pagadala, N.S.; Tuszynski, J. Systematic identification and evolutionary analysis of catalytically versatile cytochrome P450 monooxygenase families enriched in model basidiomycete fungi. PLoS ONE 2014, 9, e86683. [Google Scholar] [CrossRef] [PubMed]
- Jawallapersand, P.; Mashele, S.S.; Kovačič, L.; Stojan, J.; Komel, R.; Pakala, S.B.; Kraševec, N.; Syed, K. Cytochrome P450 monooxygenase CYP53 family in fungi: Comparative structural and evolutionary analysis and its role as a common alternative anti-fungal drug target. PLoS ONE 2014, 9, e107209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sello, M.M.; Jafta, N.; Nelson, D.R.; Chen, W.; Yu, J.-H.; Parvez, M.; Kgosiemang, I.K.R.; Monyaki, R.; Raselemane, S.C.; Qhanya, L.B. Diversity and evolution of cytochrome P450 monooxygenases in Oomycetes. Sci. Rep. 2015, 5, 11572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qhanya, L.B.; Matowane, G.; Chen, W.; Sun, Y.; Letsimo, E.M.; Parvez, M.; Yu, J.-H.; Mashele, S.S.; Syed, K. Genome-wide annotation and comparative analysis of cytochrome P450 monooxygenases in Basidiomycete biotrophic plant pathogens. PLoS ONE 2015, 10, e0142100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kgosiemang, I.K.R.; Syed, K.; Mashele, S.S. Comparative genomics and evolutionary analysis of cytochrome P450 monooxygenases in fungal subphylum Saccharomycotina. J. Pure Appl. Microbiol. 2014, 8, 291–302. [Google Scholar]
- Nelson, D.R. Cytochrome P450 and the individuality of species. Arch. Biochem Biophys 1999, 369, 1–10. [Google Scholar] [CrossRef]
- Yoshida, Y.; Aoyama, Y.; Noshiro, M.; Gotoh, O. Sterol 14-demethylase P450 (CYP51) provides a breakthrough for the discussion on the evolution of cytochrome P450 gene superfamily. Biochem. Biophys. Res. Commun. 2000, 273, 799–804. [Google Scholar] [CrossRef]
- Syed, P.R.; Chen, W.; Nelson, D.R.; Kappo, A.P.; Yu, J.-H.; Karpoormath, R.; Syed, K. Cytochrome P450 Monooxygenase CYP139 Family Involved in the Synthesis of Secondary Metabolites in 824 Mycobacterial Species. Int. J. Mol. Sci. 2019, 20, 2690. [Google Scholar] [CrossRef] [Green Version]
- Bamal, H.D.; Chen, W.; Mashele, S.S.; Nelson, D.R.; Kappo, A.P.; Mosa, R.A.; Yu, J.-H.; Tuszynski, J.A.; Syed, K. Comparative analyses and structural insights of the novel cytochrome P450 fusion protein family CYP5619 in Oomycetes. Sci. Rep. 2018, 8, 6597. [Google Scholar] [CrossRef] [Green Version]
- Pei, J.; Kim, B.-H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef]
- Syed, K.; Mashele, S.S. Comparative analysis of P450 signature motifs EXXR and CXG in the large and diverse kingdom of fungi: Identification of evolutionarily conserved amino acid patterns characteristic of P450 family. PLoS ONE 2014, 9, e95616. [Google Scholar] [CrossRef] [Green Version]
- Greule, A.; Stok, J.E.; De Voss, J.J.; Cryle, M.J. Unrivalled diversity: The many roles and reactions of bacterial cytochromes P450 in secondary metabolism. Nat. Prod. Rep. 2018, 35, 757–791. [Google Scholar] [CrossRef] [Green Version]
- Podust, L.M.; Sherman, D.H. Diversity of P450 enzymes in the biosynthesis of natural products. Nat. Prod. Rep. 2012, 29, 1251–1266. [Google Scholar] [CrossRef] [Green Version]
- Yasutake, Y.; Nishioka, T.; Imoto, N.; Tamura, T. A single mutation at the ferredoxin binding site of P450 Vdh enables efficient biocatalytic production of 25-hydroxyvitamin D (3). Chembiochem 2013, 14, 2284–2291. [Google Scholar] [CrossRef]
- Nagano, S.; Cupp-Vickery, J.R.; Poulos, T.L. Crystal Structures of the Ferrous Dioxygen Complex of Wild-type Cytochrome P450eryF and Its Mutants, A245S and A245T INVESTIGATION OF THE PROTON TRANSFER SYSTEM IN P450eryF. J. Biol. Chem. 2005, 280, 22102–22107. [Google Scholar] [CrossRef] [Green Version]
- Yasutake, Y.; Fujii, Y.; Nishioka, T.; Cheon, W.K.; Arisawa, A.; Tamura, T. Structural evidence for enhancement of sequential vitamin D3 hydroxylation activities by directed evolution of cytochrome P450 vitamin D3 hydroxylase. J. Biol. Chem. 2010, 285, 31193–31201. [Google Scholar] [CrossRef] [Green Version]
- Yasutake, Y.; Kameda, T.; Tamura, T. Structural insights into the mechanism of the drastic changes in enzymatic activity of the cytochrome P450 vitamin D3 hydroxylase (CYP107BR1) caused by a mutation distant from the active site. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 266–275. [Google Scholar] [CrossRef]
- Matowane, R.G.; Wieteska, L.; Bamal, H.D.; Kgosiemang, I.K.R.; Van Wyk, M.; Manume, N.A.; Abdalla, S.M.H.; Mashele, S.S.; Gront, D.; Syed, K. In silico analysis of cytochrome P450 monooxygenases in chronic granulomatous infectious fungus Sporothrix schenckii: Special focus on CYP51. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 166–177. [Google Scholar] [CrossRef]
- Chen, I.-M.A.; Chu, K.; Palaniappan, K.; Pillay, M.; Ratner, A.; Huang, J.; Huntemann, M.; Varghese, N.; White, J.R.; Seshadri, R. IMG/M v. 5.0: An integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 2018, 47, D666–D677. [Google Scholar] [CrossRef]
- Nelson, D.R. Cytochrome P450 Nomenclature, 2004. In Cytochrome P450 Protocols; Springer: Berlin, Germany, 2006; pp. 1–10. [Google Scholar]
- Nelson, D.R. Cytochrome P450 nomenclature. In Cytochrome P450 Protocols; Springer: Berlin, Germany, 1998; pp. 15–24. [Google Scholar]
- Nelson, D.R. The cytochrome p450 homepage. Hum. Genom. 2009, 4, 59. [Google Scholar] [CrossRef] [Green Version]
- Sirim, D.; Widmann, M.; Wagner, F.; Pleiss, J. Prediction and analysis of the modular structure of cytochrome P450 monooxygenases. BMC Struct. Biol. 2010, 10, 34. [Google Scholar] [CrossRef] [Green Version]
- Gotoh, O. Substrate recognition sites in cytochrome P450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J. Biol. Chem. 1992, 267, 83–90. [Google Scholar]
- Katoh, K.; Kuma, K.-i.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Boc, A.; Diallo, A.B.; Makarenkov, V. T-REX: A web server for inferring, validating and visualizing phylogenetic trees and networks. Nucleic Acids Res. 2012, 40, W573–W579. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucleic Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Pascal Andreu, V.; de los Santos, E.L.C.; Del Carratore, F.; Lee, S.Y.; Medema, M.H.; Weber, T. The antiSMASH database version 2: A comprehensive resource on secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2018, 47, D625–D630. [Google Scholar] [CrossRef] [Green Version]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Case, D.; Berryman, J.; Betz, R.; Cerutti, D.; Cheatham, T., III; Darden, T.; Duke, R.; Giese, T.; Gohlke, H.; Goetz, A. AMBER 2015; University of California: San Francisco, CA, USA, 2015. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P450 Family | Number of Member P450s | Kingdom | PROMALS3D Conservation Index | Rank (Highest to Lowest Conservation) | ||||
|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 7 | 8 | 9 | ||||
| CYP141 | 29 | Bacteria | 0 | 0 | 0 | 0 | 389 | 1 |
| CYP51 | 50 | Bacteria | 11 | 102 | 0 | 0 | 264 | 2 |
| CYP137 | 38 | Bacteria | 145 | 0 | 0 | 0 | 251 | 3 |
| CYP121 | 34 | Bacteria | 0 | 0 | 0 | 0 | 233 | 4 |
| CYP128 | 2191 | Bacteria | 118 | 25 | 0 | 0 | 217 | 5 (previously 23rd) |
| CYP132 | 39 | Bacteria | 175 | 0 | 0 | 0 | 217 | 5 |
| CYP5619 | 23 | Stramenopila | 118 | 38 | 170 | 0 | 199 | 6 |
| CYP124 | 71 | Bacteria | 52 | 35 | 59 | 0 | 170 | 7 |
| CYP139 | 894 | Bacteria | 0 | 127 | 0 | 0 | 165 | 8 |
| CYP188 | 67 | Bacteria | 62 | 0 | 100 | 0 | 141 | 9 |
| CYP123 | 74 | Bacteria | 62 | 0 | 82 | 0 | 137 | 10 |
| Data type | Mycobacterial Category | ||||
|---|---|---|---|---|---|
| MTBC | MCAC | NTM | MAC | SAP | |
| Total number of CYP128 P450s | 2328 | 169 | 140 | 35 | 2 |
| Number of CYP128 P450s not part of BGCs | 282 | 147 | 120 | 34 | 1 |
| Number of CYP128 P450s part of BGCs | 2046 | 22 | 20 | 1 | 1 |
| Number of BGCs with CYP128, CYP121 and CYP124 P450s | 1908 | 0 | 0 | 0 | 0 |
| Number of BGCs with CYP128 P450 | 136 | 22 | 21 | 1 | 1 |
| Number of BGCs with CYP128 and CYP121 P450s | 0 | 0 | 0 | 0 | 0 |
| Number of BGCs with CYP128 and CYP124 P450s | 2 | 0 | 0 | 0 | 0 |
| Compound | Top Score (kcal/mol) | Score of First Well-Oriented Pose (kcal/mol) | Rank of First Well-Oriented Pose |
|---|---|---|---|
| Menaquinone-9 (MK9) | −13.48 | −12.83 (1) | 7 |
| Itraconazole | −12.57 | −10.72 (3) | 10 |
| Posaconazole | −11.81 | −11.81 (2) | 1 |
| Ketoconazole | −10.47 | −9.84 (4) | 9 |
| Econazole | −8.24 | −7.56 (7) | 6 |
| Miconazole | −8.10 | −8.10 (5) | 1 |
| Voriconazole | −7.63 | −7.57 (6) | 2 |
| Clotrimazole | −7.46 | −7.21 (9) | 5 |
| Fluconazole | −7.38 | −7.38 (8) | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngcobo, N.S.; Chiliza, Z.E.; Chen, W.; Yu, J.-H.; Nelson, D.R.; Tuszynski, J.A.; Preto, J.; Syed, K. Comparative Analysis, Structural Insights, and Substrate/Drug Interaction of CYP128A1 in Mycobacterium tuberculosis. Int. J. Mol. Sci. 2020, 21, 4816. https://doi.org/10.3390/ijms21144816
Ngcobo NS, Chiliza ZE, Chen W, Yu J-H, Nelson DR, Tuszynski JA, Preto J, Syed K. Comparative Analysis, Structural Insights, and Substrate/Drug Interaction of CYP128A1 in Mycobacterium tuberculosis. International Journal of Molecular Sciences. 2020; 21(14):4816. https://doi.org/10.3390/ijms21144816
Chicago/Turabian StyleNgcobo, Nokwanda Samantha, Zinhle Edith Chiliza, Wanping Chen, Jae-Hyuk Yu, David R. Nelson, Jack A. Tuszynski, Jordane Preto, and Khajamohiddin Syed. 2020. "Comparative Analysis, Structural Insights, and Substrate/Drug Interaction of CYP128A1 in Mycobacterium tuberculosis" International Journal of Molecular Sciences 21, no. 14: 4816. https://doi.org/10.3390/ijms21144816
APA StyleNgcobo, N. S., Chiliza, Z. E., Chen, W., Yu, J.-H., Nelson, D. R., Tuszynski, J. A., Preto, J., & Syed, K. (2020). Comparative Analysis, Structural Insights, and Substrate/Drug Interaction of CYP128A1 in Mycobacterium tuberculosis. International Journal of Molecular Sciences, 21(14), 4816. https://doi.org/10.3390/ijms21144816