Copper Homeostasis in Mammals, with Emphasis on Secretion and Excretion. A Review


Abstract
:
1. Introduction
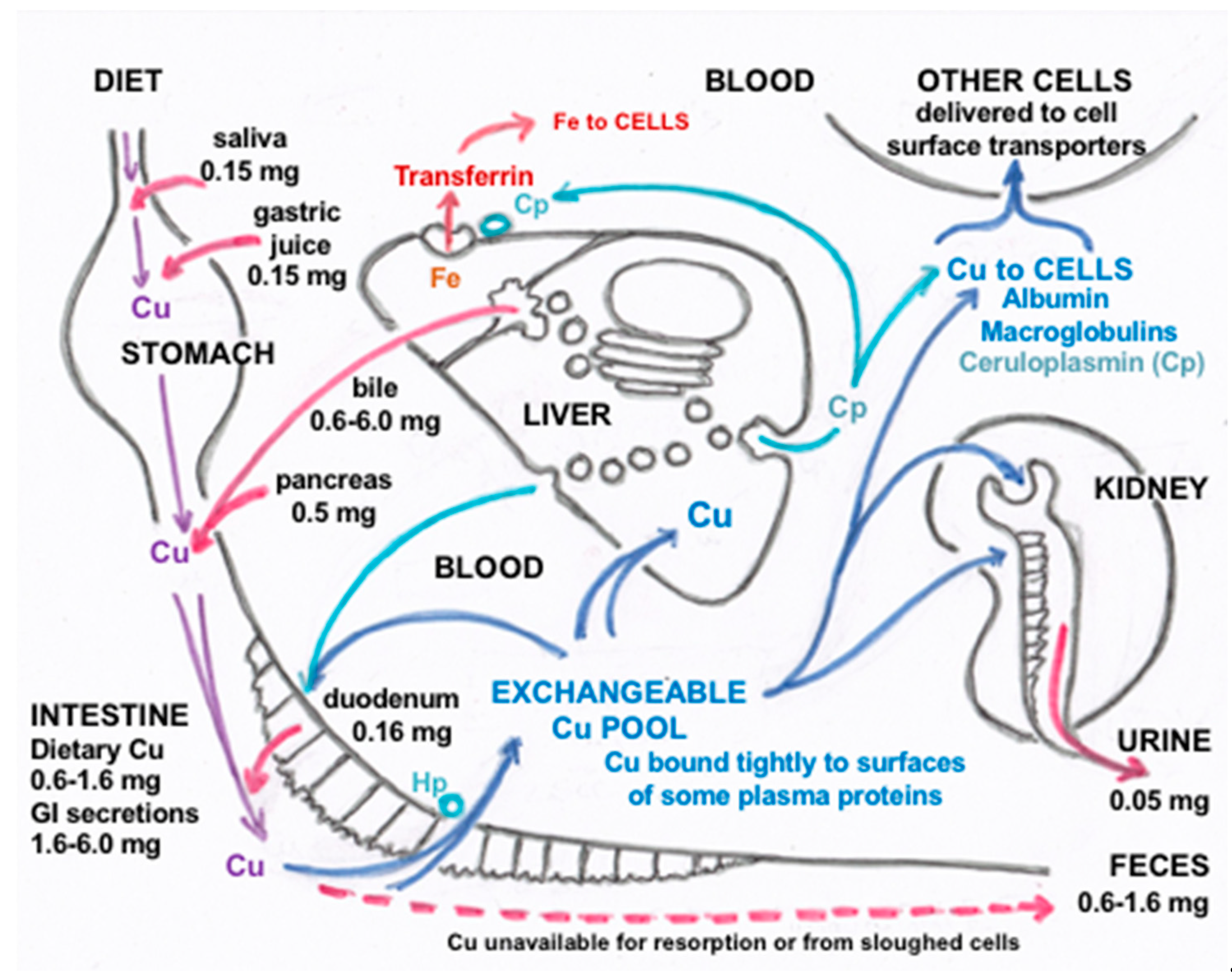
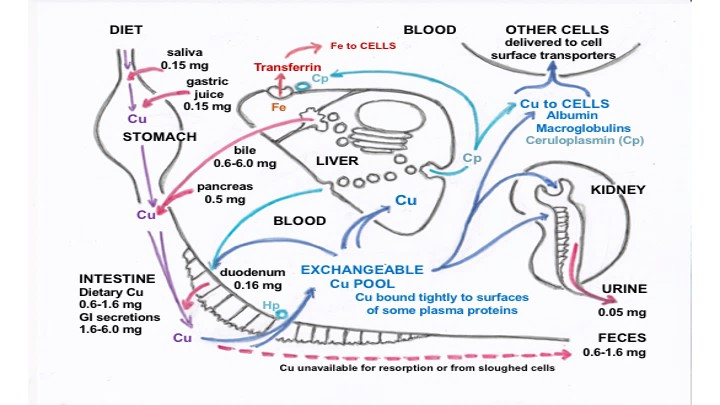
2. Secretion and Recycling of Cu in Gastrointestinal Fluids
3. Biliary Cu Excretion
4. Forms of Cu in Bile and Their Origins
- (a)
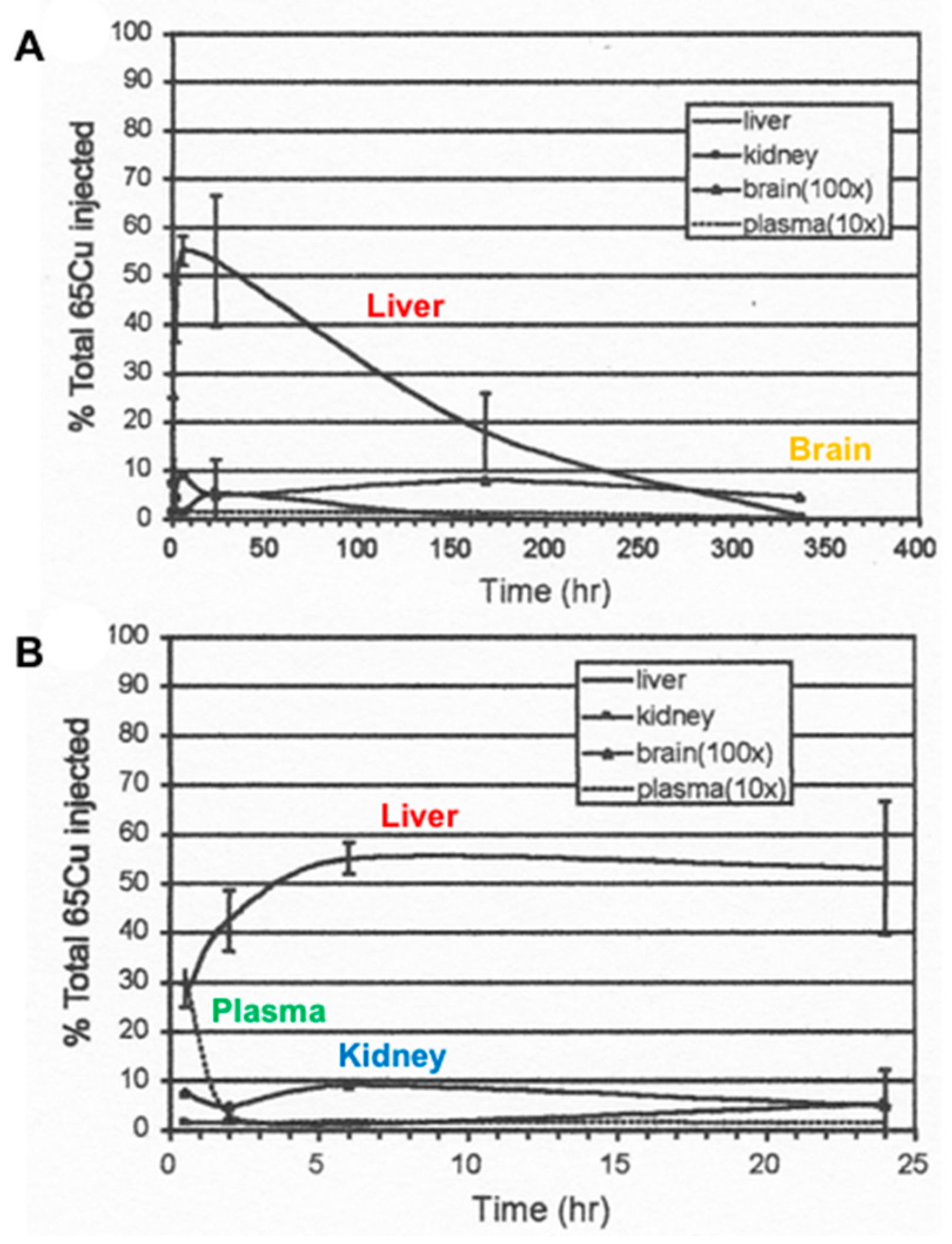
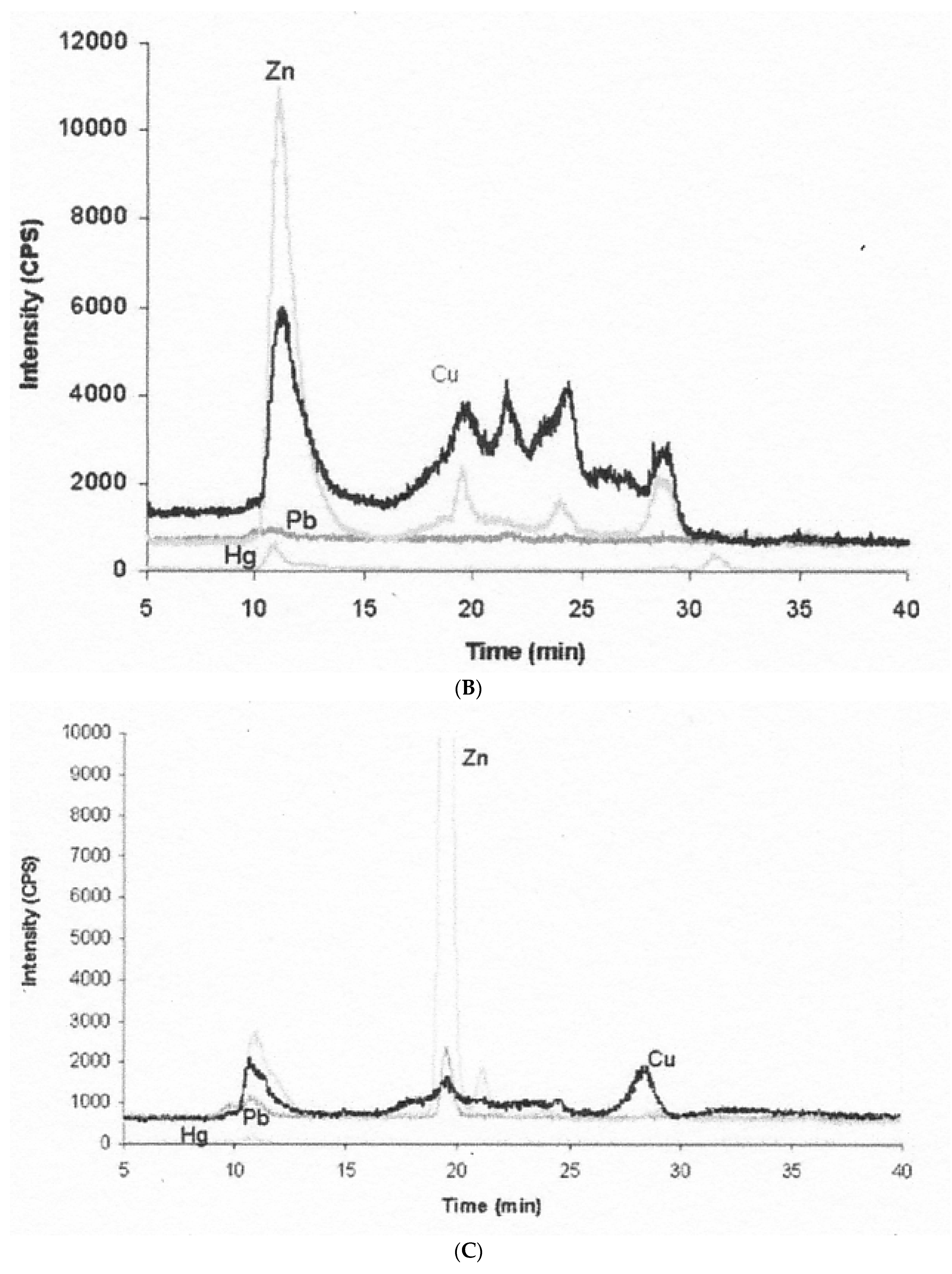
- The nature of the components varies in terms of how easily the attached Cu can be dialyzed away and how easily it is re-absorbed by the GI tract, as shown for example in rats injected with 100 µg of 64Cu, measuring biliary 64Cu radioactivity over time, dialyzing the bile (into 0.9% NaCl), and administering portions into new rats by intrapyloric intubation, to measure rates of intestinal uptake [1]. Table 2 shows that the bile collected early on (zero to 4h after 64Cu administration) had half of its Cu loosely bound (and released by dialysis); but dialyzability decreased substantially over time and was accompanied by greater susceptibility to TCA precipitation (a characteristic of proteins). This suggests that a portion of the Cu in bile becomes more tightly bound and is associated with proteins. Reabsorption of the dialyzed and undialysed biliary Cu, however, was similar but quite low (10–15%), reinforcing the concept that biliary Cu is more difficult to re-absorb than that in other GI fluids. In another study with human GI fluids (Table 2), gastric and salivary Cu was easily dialyzed away (85–98%) vs. that of gall bladder bile (12%), using 0.9% NaCl/10 mM Tris HCl buffer, pH 8. Additionally, the absorbability of salivary and gastric fluid Cu (administered intraduodenally to rats) was twice that of gall bladder bile, and the same as for Cu2+-acetate or histidine.
- (b)
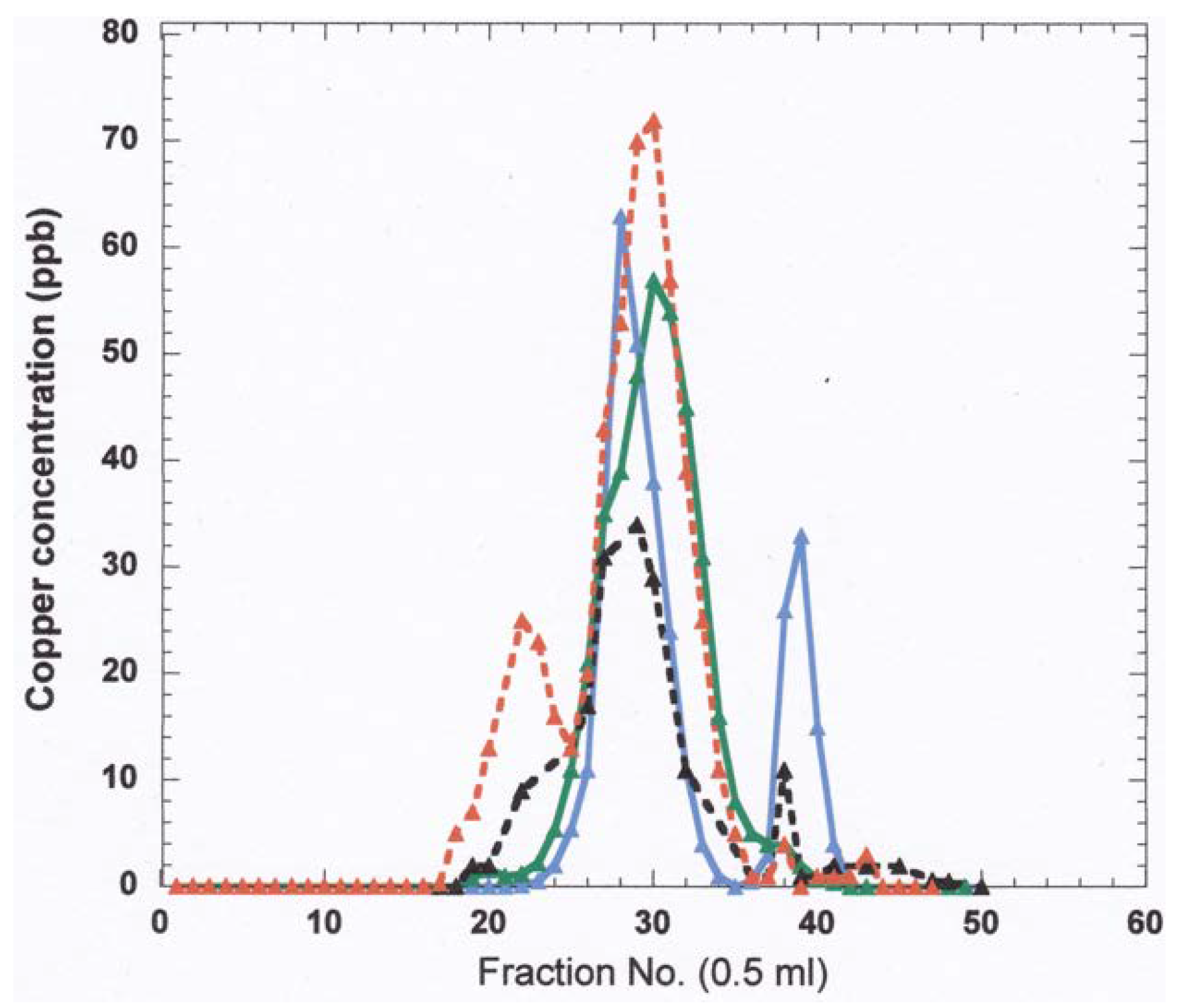
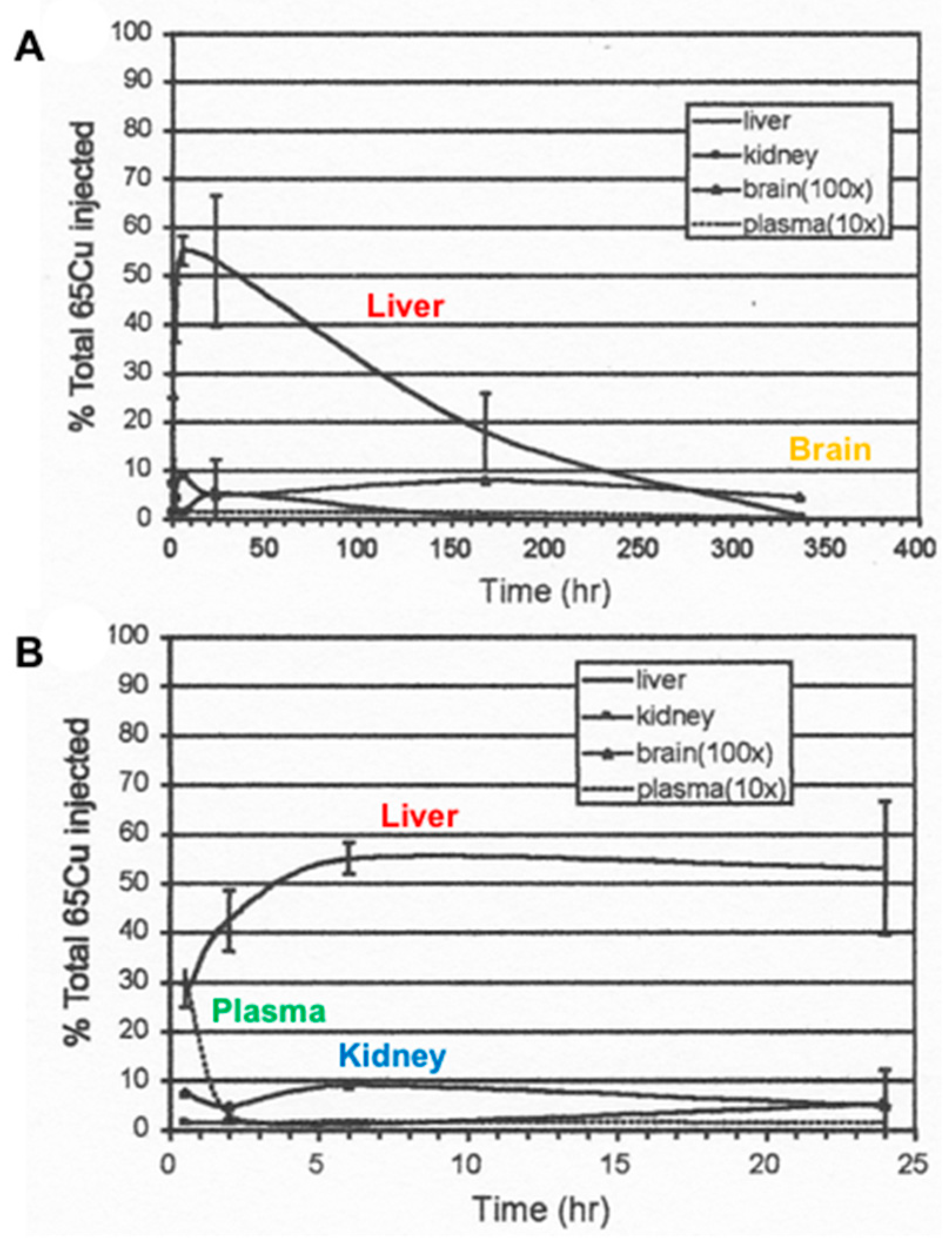
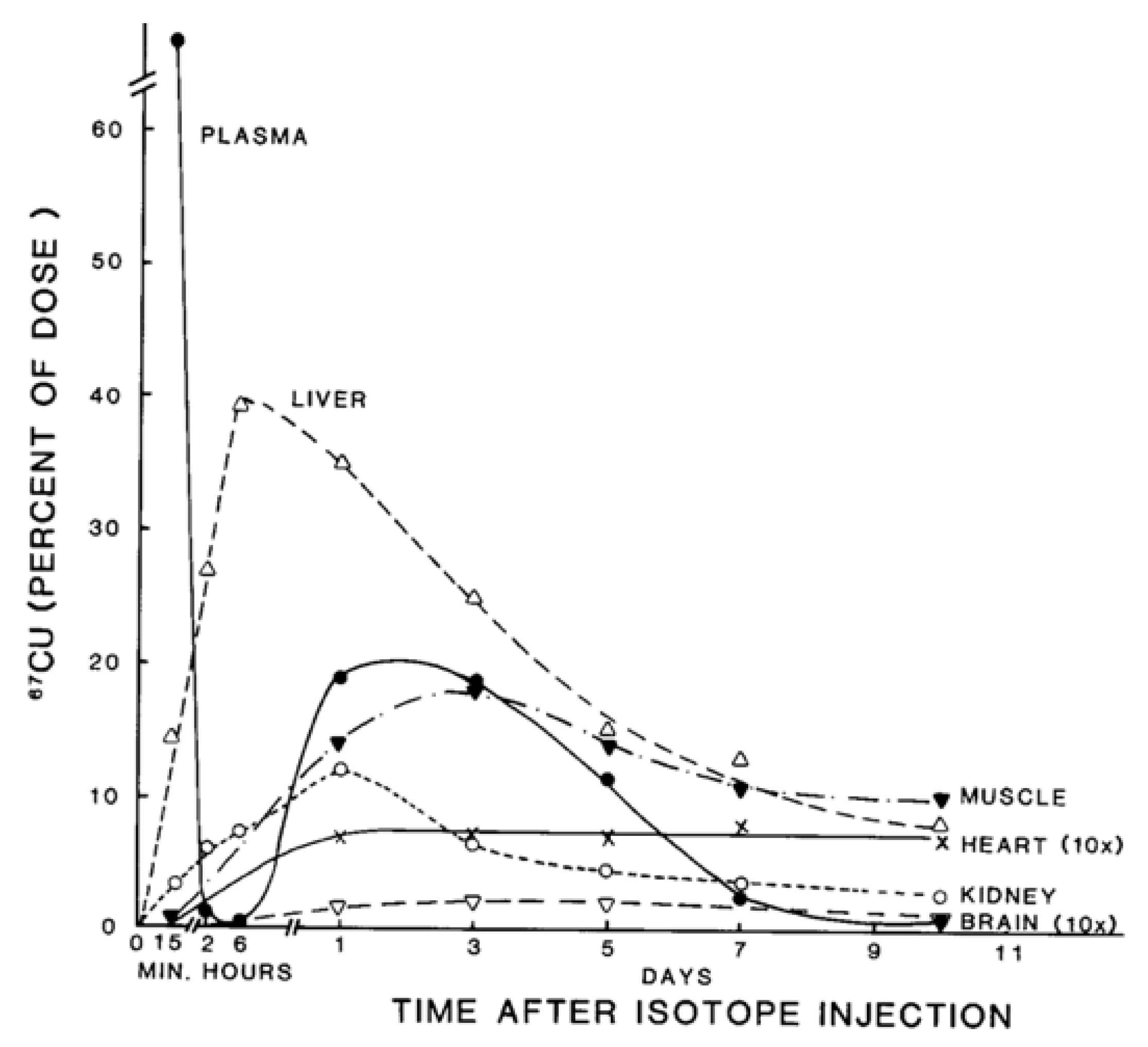
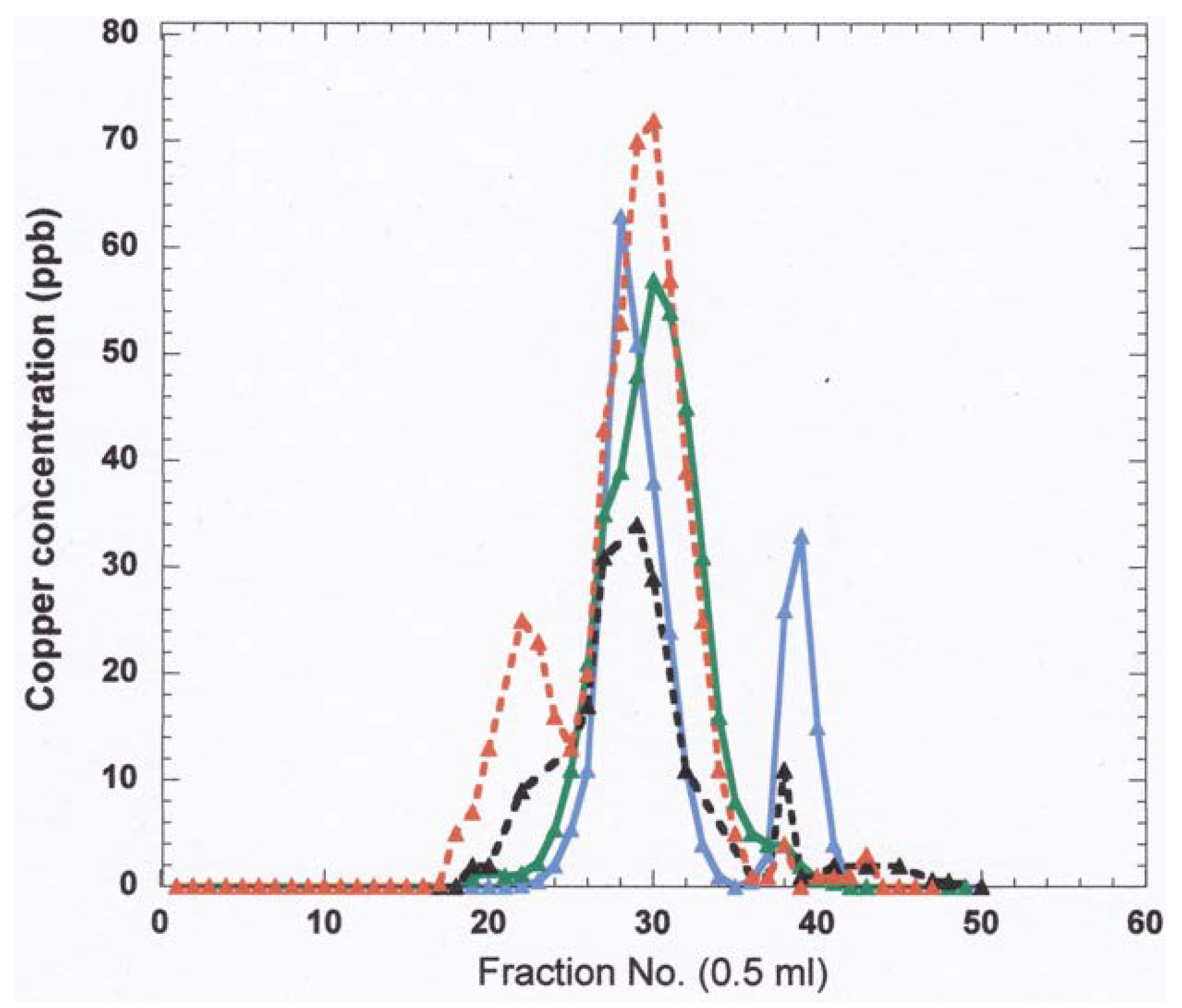
- The nature and size of the biliary Cu components was determined largely by size exclusion chromatography of human and rat bile that had been labeled in vitro or in vivo with 64Cu2+. It showed two major peaks in Sephadex G50 and G75, one in the void volume and the other at about 5 kDa. Bile acids (detergents like glycocholate, taurodeoxycholate and lecithin) disaggregated the larger component into the smaller one. Traces of an 8 kDa component [43] and another below 5 kDa [44] were also reported. Where tested, elution of these Cu complexes was not abolished or changed by treating with trypsin or other digestive proteases. This suggests they are not polypeptides, and would explain how they survive the gut for excretion in the feces. Alternatively, the proteins/peptides might for some other reason be resistant to degradation by digestive enzymes. The Cu attached to these components was bound very tightly, as it was not removed by strong chelating agents [1]. Other work suggested that one or more of these components may be fragments of ceruloplasmin that are hard to digest by proteases. In one case [45], but not another [46], some of the Cu of bile eluting in the void volume reacted with the antibody against ceruloplasmin. The former is consistent with physiological studies in rats by another group [47], where ceruloplasmin that had been radiolabeled in vitro with 67Cu or 64Cu was given i.v., and some of the radiolabeled ceruloplasmin-Cu ended up not only in the liver but in the bile. One caveat related to these studies, however, is that it is very difficult to insert Cu into ceruloplasmin in vitro, and it was not ascertained that the radiolabeled Cu added and used in these experiments was in its native state.)
- (c)
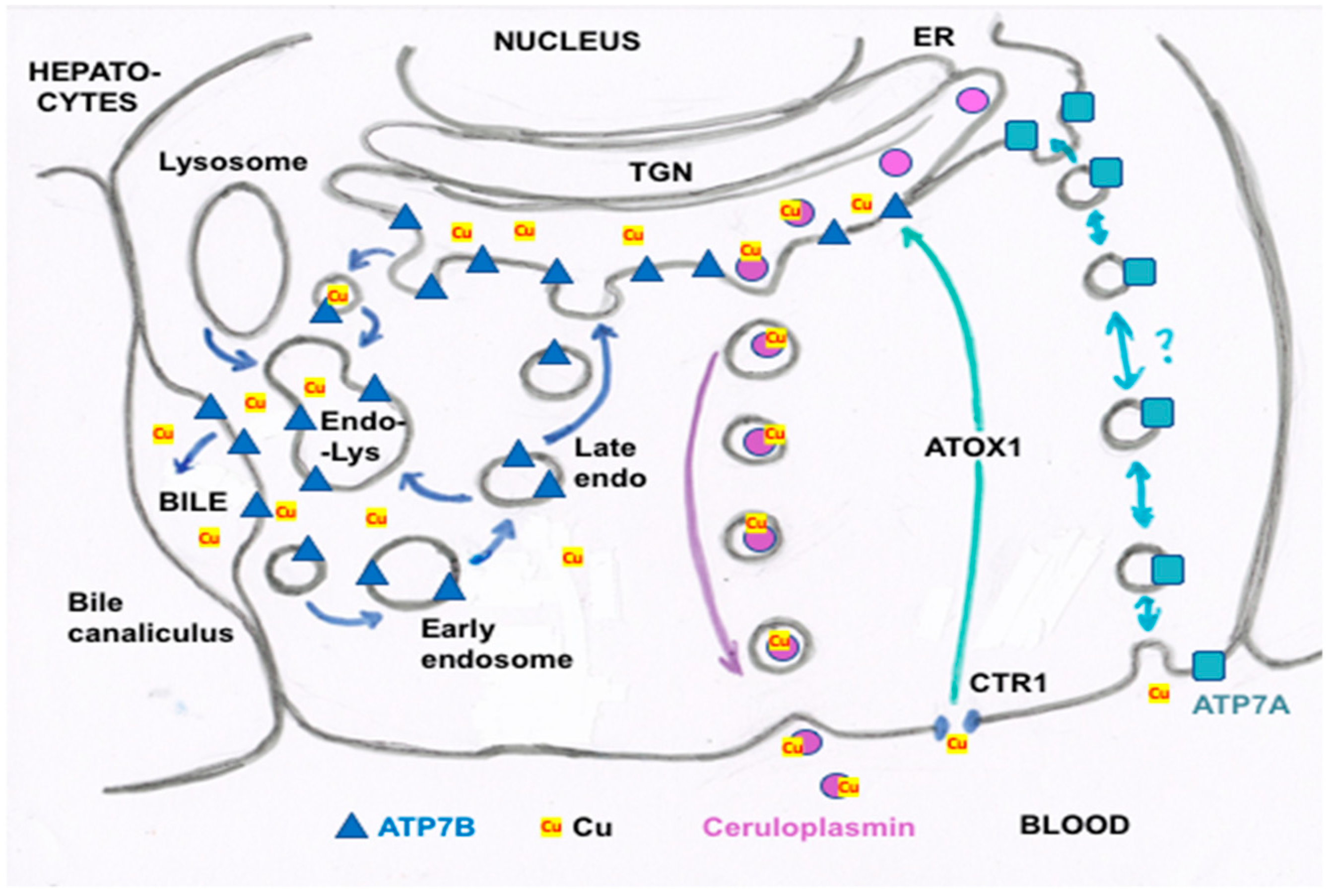
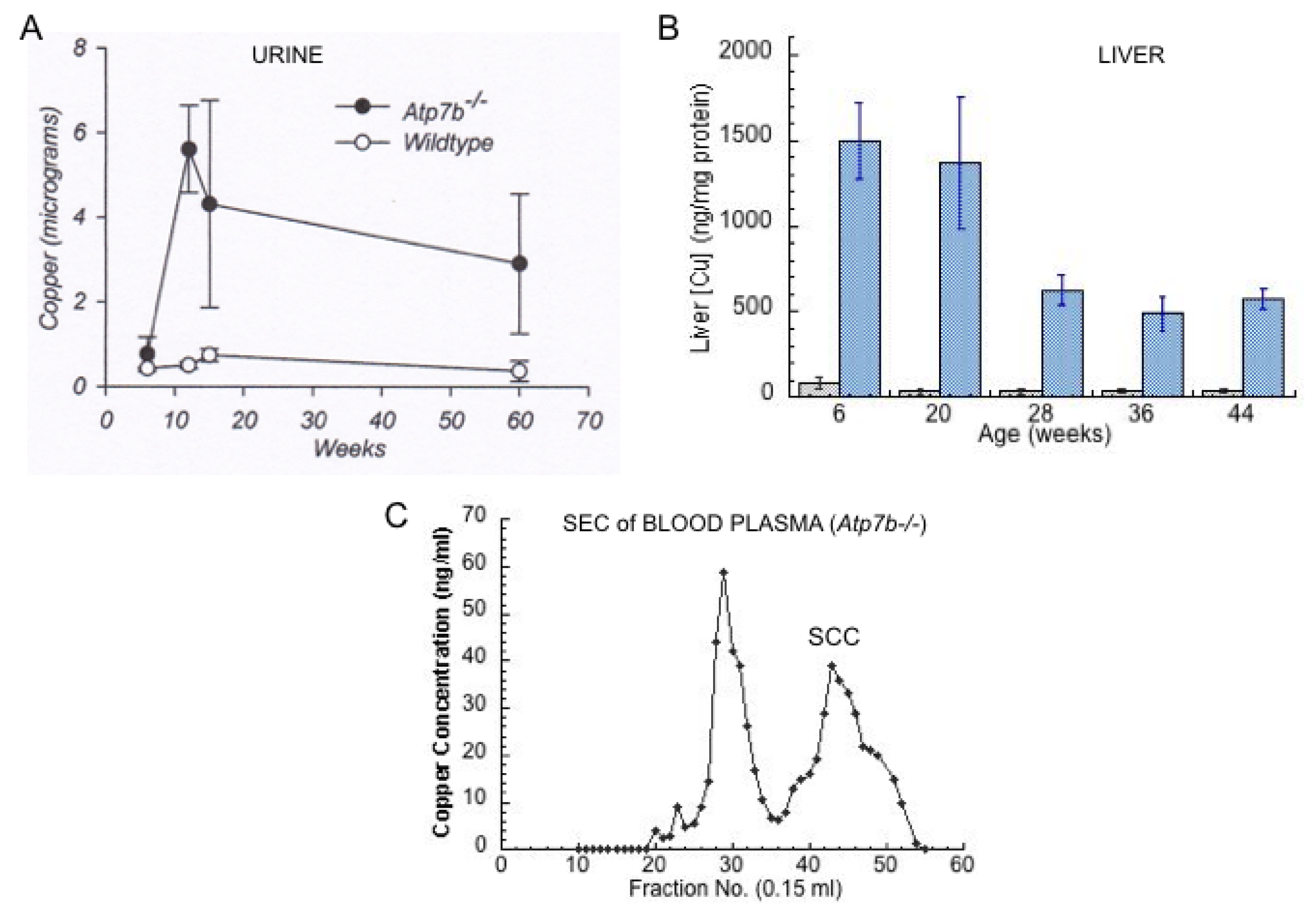
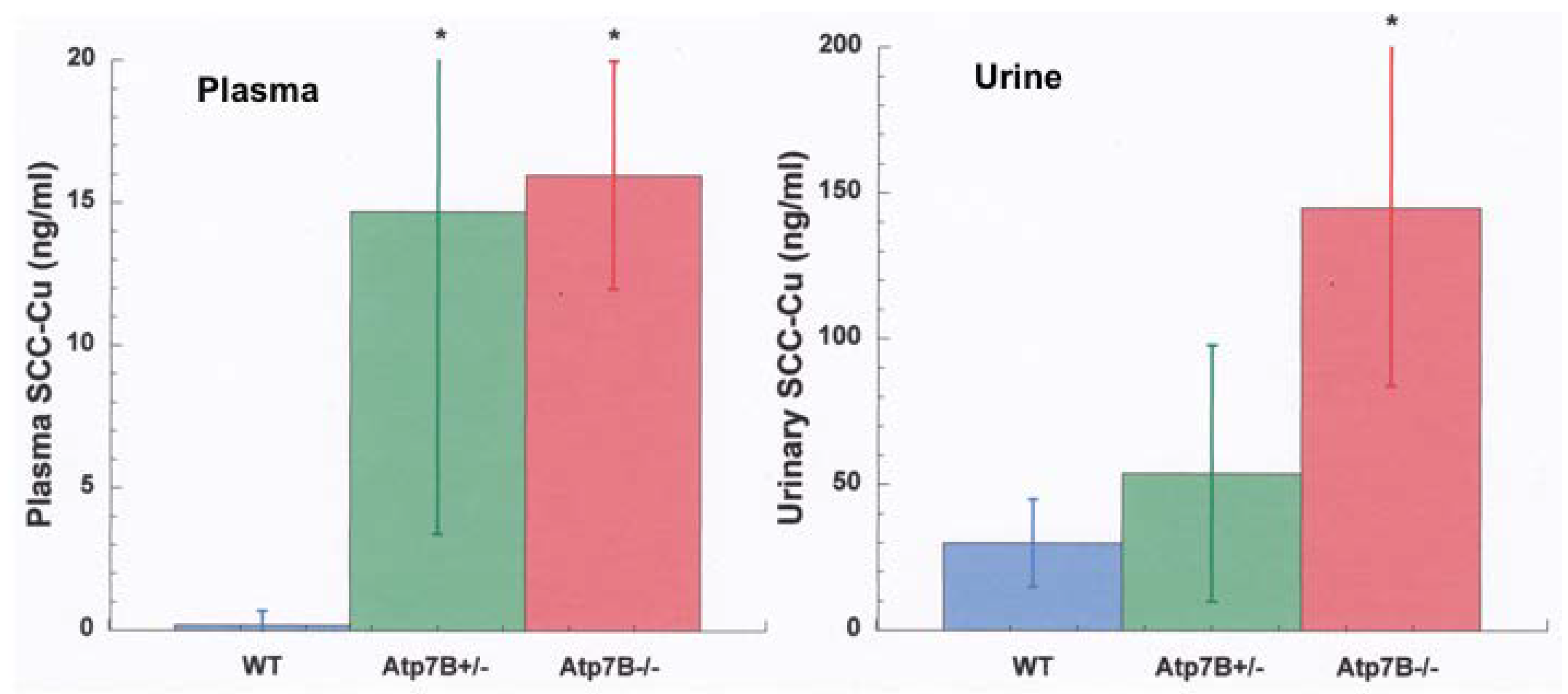
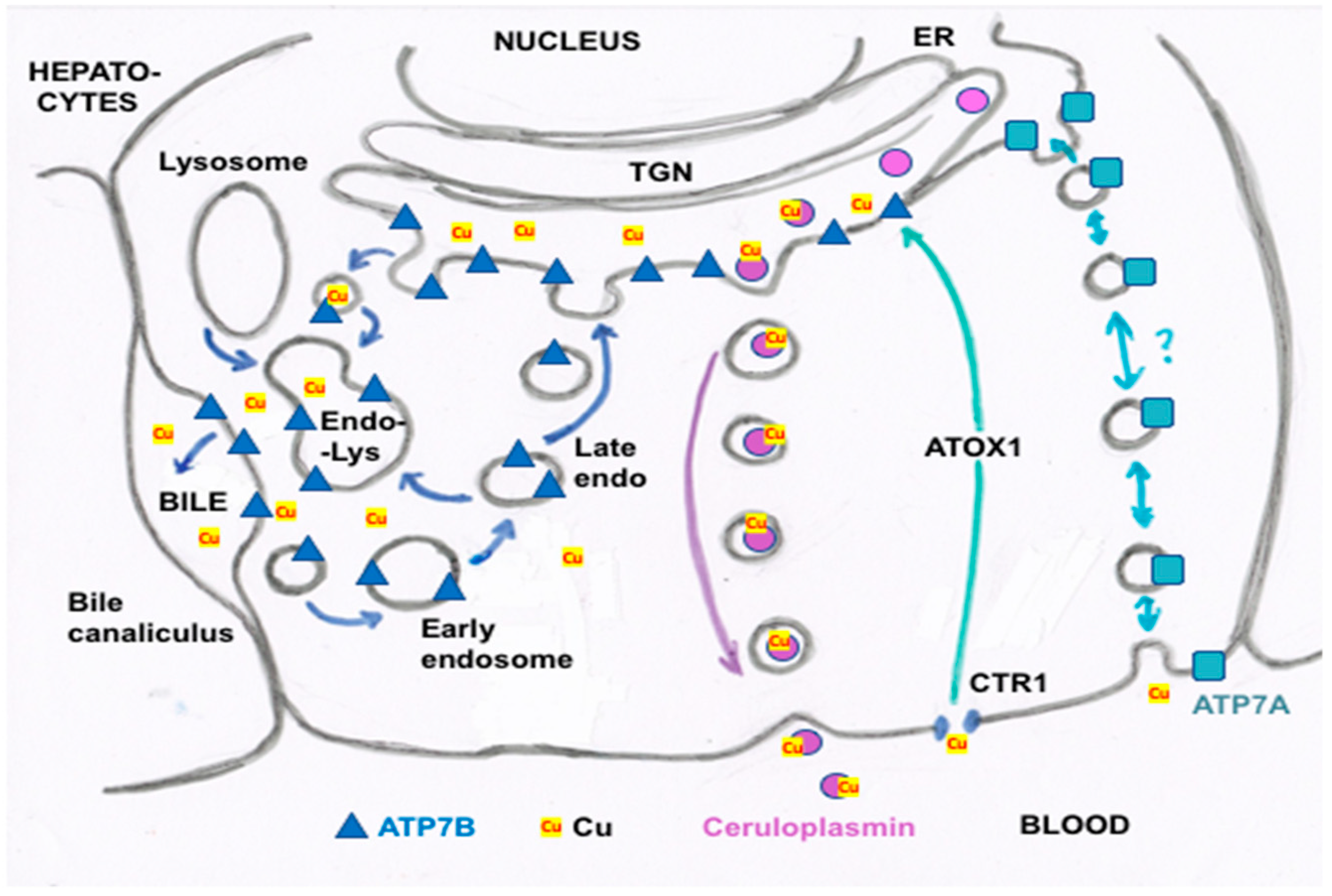
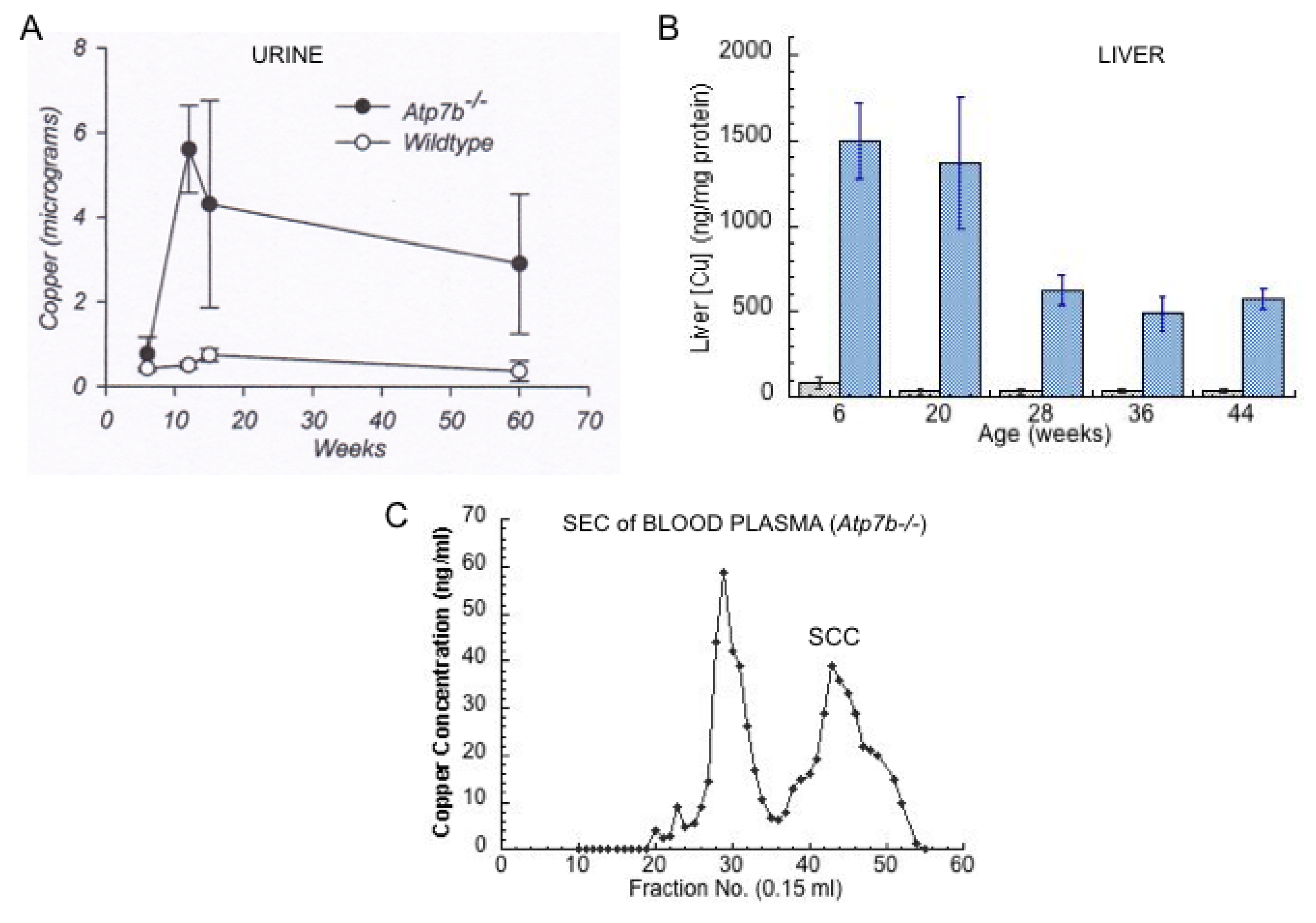
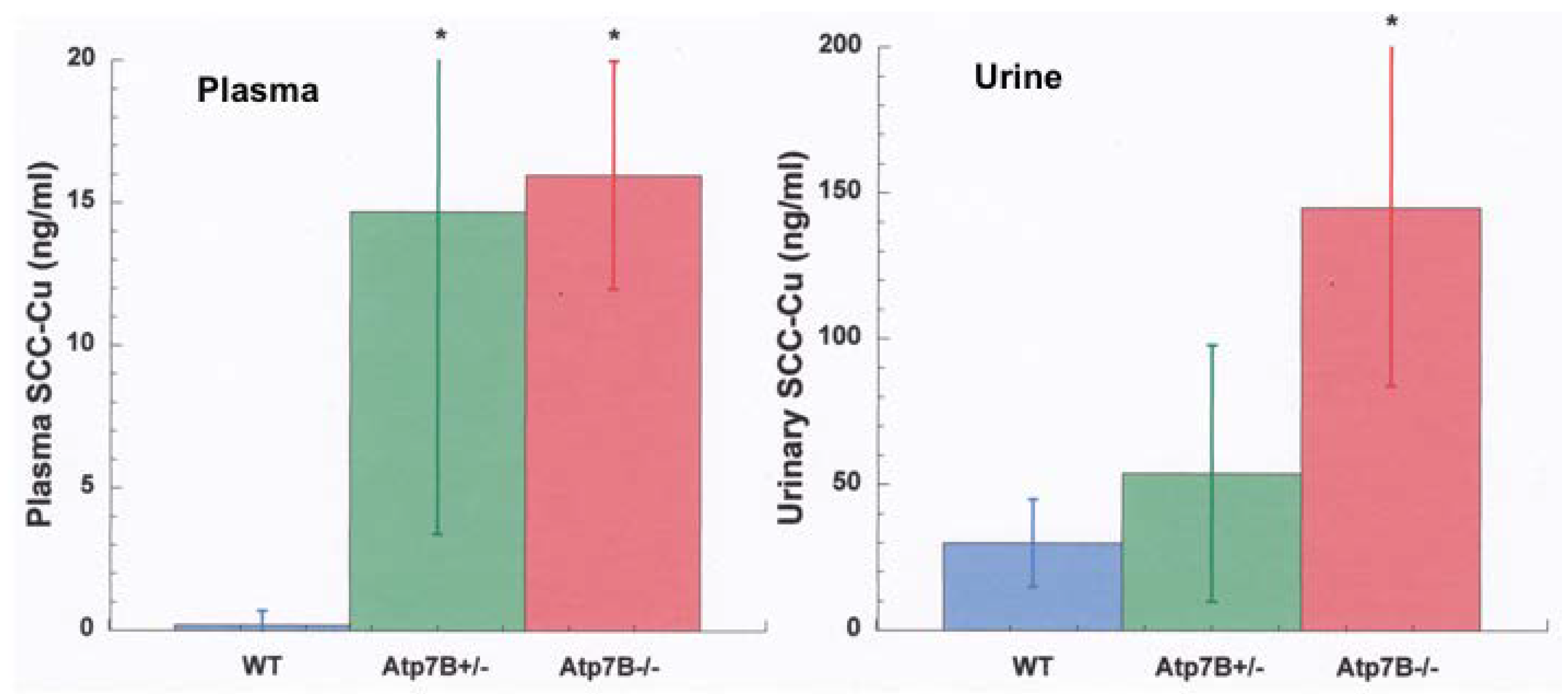
- Other kinds of studies also support the idea that ceruloplasmin or its fragments can enter bile. In other rat studies, more of the radioactive Cu associated with ceruloplasmin entered the bile when administered as asialo-ceruloplasmin [48]. Sialic acid is known to be removed from several plasma proteins by hepatic endothelial cells. This has also been demonstrated for ceruloplasmin [48,49]. Desialylation occurred during transcytosis of the hepatic endothelial cells that form a porous layer between the blood and the hepatocytes sheaves, and the asialo-form then entered the hepatocytes via the galactose receptor [48,49]. In line with this, the binding of ceruloplasmin to specific “receptors” on the surface of hepatic endothelial cells but not hepatocytes was demonstrated histologically and through binding studies by Tavassoli et al. [50]. If ceruloplasmin enters hepatocytes via the galactose receptor, it will be transferred into the lysosomal compartment, which is also a source of bile components (Figure 4). These various studies (which have not been followed up) suggested that a portion or fragment of asialo-ceruloplasmin could be the main form of biliary Cu not re-absorbed by the intestine that ends up in the feces.
5. Forms of Cu in Other GI Fluids and Their Origins
6. Forms of Cu in Other Fluids and Their Origins
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Linder, M.C. Biochemistry of Copper; Plenum: New York, NY, USA, 1991. [Google Scholar]
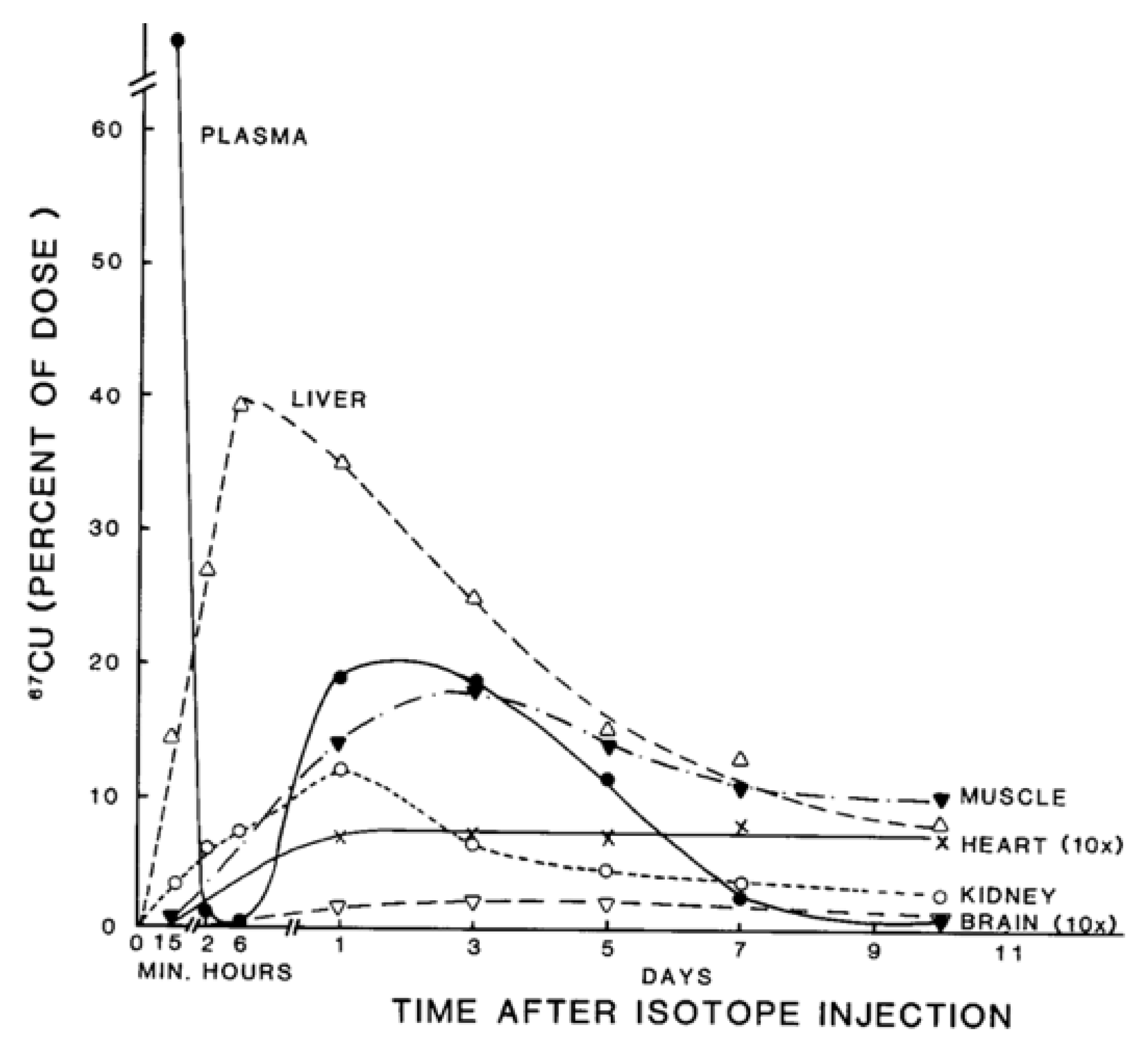
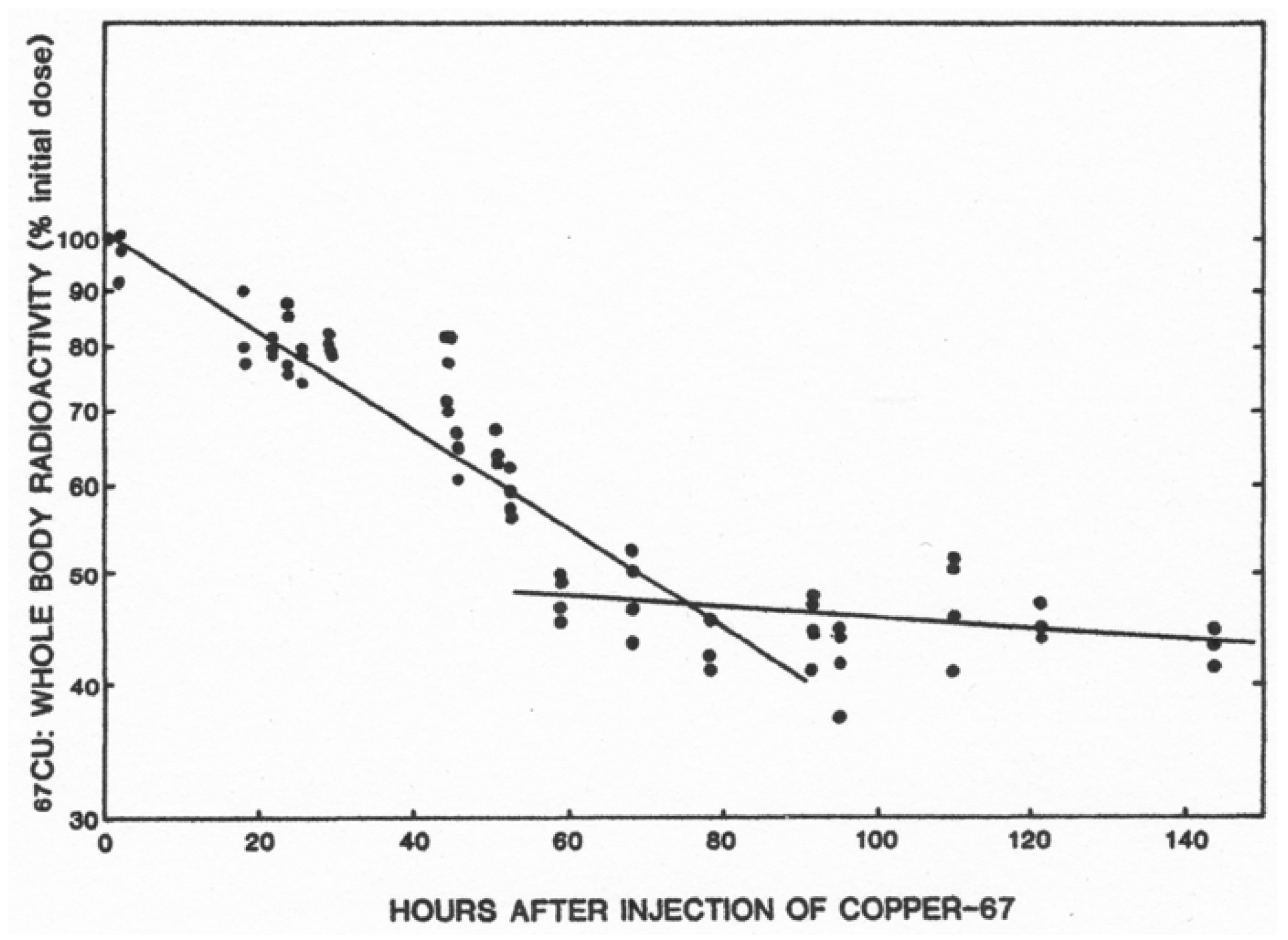
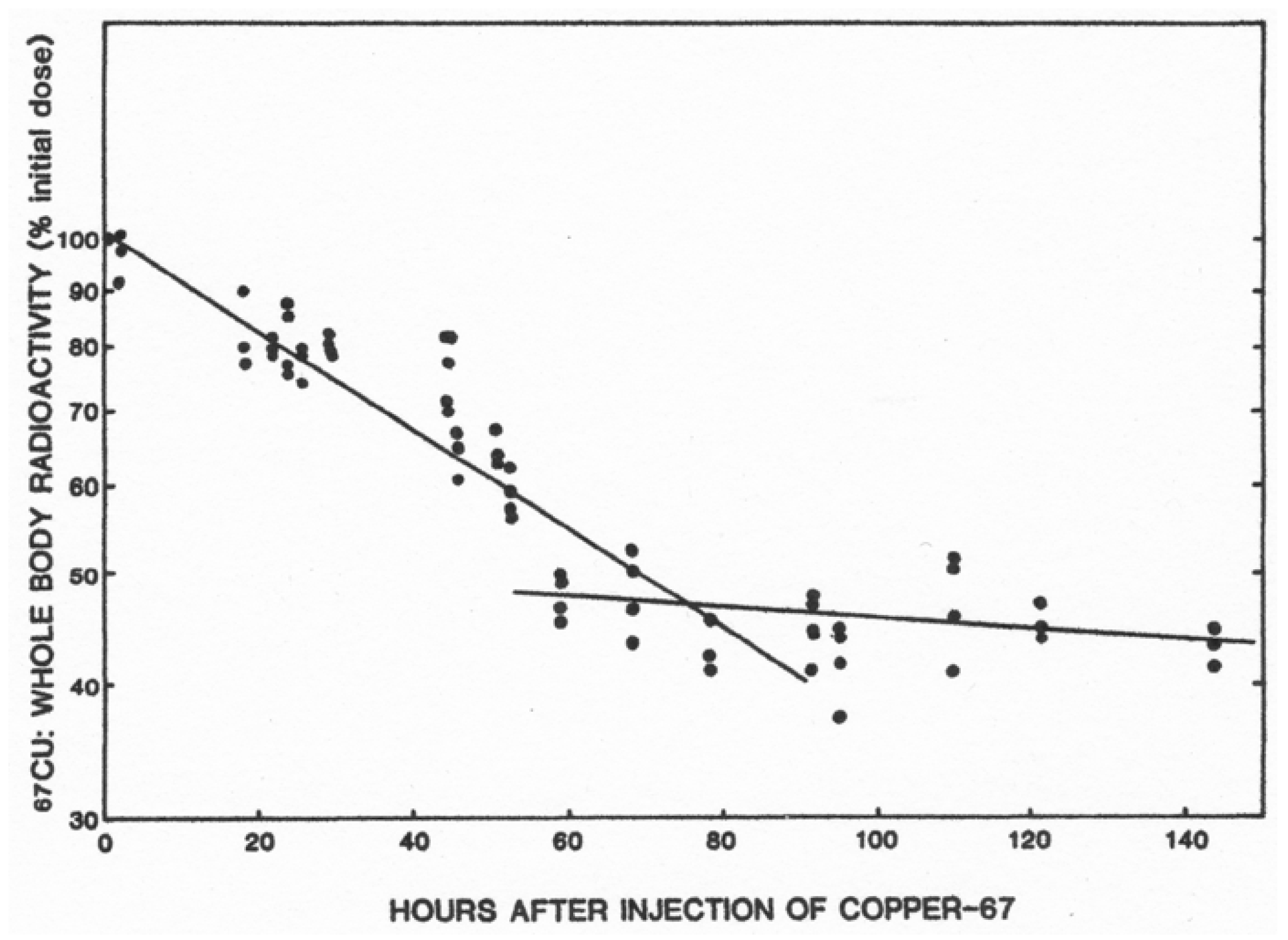
- Linder, M.C.; Roboz, M. Turnover and excretion of copper in rats as measured with 67Cu. Am. J. Physiol. Endocrinol. Metab. 1986, 251, E551–E555. [Google Scholar] [CrossRef] [PubMed]
- Czlonkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.L.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Wilson disease. Nat. Rev. Dis. Primers 2018, 4, 21. [Google Scholar] [CrossRef]
- Guindi, M. Wilson disease. Semin. Diagn. Pathol. 2019, 36, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Van de Sluis, B.; Rothuizen, J.; Pearson, P.L.; Wijmenga, C. Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum. Mol. Genet. 2002, 11, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Fedoseienko, A.; Bartuzi, P.; van der Sluis, B. Functional understanding of the versatile protein copper metabolism MURR1 domain (COMMD1) in copper homeostasis. Ann. N.Y. Acad. Sci. 2014, 1314, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Haywood, S.; Boursnell, M.; Loughran, M.J.; Trafford, J.; Isherwood, D.; Liu, X.; Olohan, L.; Carter, S.D. Copper toxicosis in non-COMMD1 Bedlington terriers is associated with metal transport gene ABCA12. J. Trace Elem. Biol. Med. 2016, 35, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.C.; Hazegh-Azam, M. Copper biochemistry and molecular biology. Am. J. Clin. Nutr. 1996, 63, 797S–811S. [Google Scholar] [CrossRef]
- Linder, M.C. Nutritional biochemistry of copper, with emphasis on the perinatal period. In Biochemical Aspects of Human Nutrition; Avigliano, L.A., Rossi, L., Eds.; Transworld Research Network: Trivandrum, Kerala, India, 2010; pp. 143–179. ISBN 978-81-7895-478-3. [Google Scholar]
- Turnlund, J.R.; Keyes, W.R.; Anderson, H.L.; Acord, L.L. Copper absorption in young men at three levels of dietary copper. Am. J. Clin. Nutr. 1989, 49, 870–878. [Google Scholar] [CrossRef]
- Hausmann, D.H.; Porstmann, T.; Weber, I.; Hausmann, S.; Dummler, W.; Liebe, S.; Emmrich, J. Cu/Zn-SOD in human pancreatic tissue and pancreatic juice. Int. J. Pancreatol. 1997, 22, 207–213. [Google Scholar] [CrossRef]
- Evering, W.E.N.D.; Haywood, S.; Bremner, I.; Wood, A.M.; Trafford, J. The protective role of metallothionein in copper overload: II. Transport and excretion of immunoreactive MT-1 in blood, bile and urine of copper-loaded rats. Chem. Biol. Interact. 1991, 78, 297–305. [Google Scholar] [CrossRef]
- Cumings, J.N. Heavy Metals and the Brain; Blackwells Scientific Publications: Oxford, UK, 1959. [Google Scholar]
- Weiss, K.C.; Linder, M.C. Copper transport in rats involving a new plasma protein. Am. J. Physiol. Endocrinol. 1985, 249, E77–E88. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, A.; Alonzo, E.; Sauble, E.; Chu, Y.L.; Nguyen, D.; Linder, M.C.; Sato, D.S.; Mason, A.Z. Copper binding components of blood plasma and organs and their response to influx of large doses of 65Cu, in the mouse. Biometals 2008, 21, 525–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Ray, K. Wilson’s disease: An update. Nat. Clin. Pract. Neurol. 2006, 2, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Shilsky, M.L.; Irani, A.N.; Gorla, G.R.; Volenberg, I.; Gupta, S. Biliary copper excretion capacity in intact animals: Correlation between ATP7B function hepatic mass, and biliary copper excretion. J. Biochem. Mol. Toxicol. 2000, 14, 210–214. [Google Scholar] [CrossRef]
- Harada, M.; Sakisaka, S.; Yoshitake, M.; Shakadoh, S.; Gondoh, K.; Sata, M.; Tanikawa, K. Biliary copper excretion in acutely and chronically copper loaded rats. Hepatology 1993, 17, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Lech, T.; Sedlick, J.K. Copper concentration in body tissues and fluids in normal subjects of southern Poland. Biol. Trace Elem. Res. 2007, 118, 10–15. [Google Scholar] [CrossRef]
- Lutsenko, S.; Barnes, N.L.; Bartee, M.Y.; Dmitriev, O.Y. Function and regulation of human copper-transporting ATPases. Physiol. Rev. 2007, 87, 1011–1046. [Google Scholar] [CrossRef]
- Linz, R.; Barnes, N.L.; Zimnicka, A.M.; Kaplan, J.H.; Eipper, B.; Lutsenko, S. Intracellular targeting of copper-transporting ATPase ATP7A in a normal and Atpb-/- kidney. Am. J. Physiol. Renal Physiol. 2008, 294, F53–F61. [Google Scholar] [CrossRef] [Green Version]
- Pierson, H.; Muchenditsi, A.; Kim, B.-E.; Ralle, M.; Zachos, N.; Huster, D.; Lutsenko, S. The function of ATPase copper transporter ATP7B in intestine. Gastroenterology 2018, 154, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Middleton, R.B.; Linder, M.C. Synthesis and turnover of ceruloplasmin in rats treated with 17β-estradiol. Arch. Biochem. Biophys. 1983, 302, 362–368. [Google Scholar] [CrossRef]
- Polishchuk, E.V.; Concilli, M.; Iacobacci, S.; Chesi, G.; Pastore, N.; Piccolo, P.; Paladino, S.; Baldantoni, D.; van Ijzendoorn, S.C.; Chan, J.; et al. Wilson disease protein ATP7B utilizes lysosomal exocytosis to maintain copper homeostasis. Dev. Cell 2014, 29, 686–700. [Google Scholar] [CrossRef] [Green Version]
- Polishchuk, R.; Lutsenko, S. Golgi in copper homeostasis: A view from the membrane trafficking field. Histochem. Cell Biol. 2013, 140, 285–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polishchuk, R.S.; Polishchuk, E.V. From and to the Golgi—Defining the Wilson disease protein road map. FEBS Lett. 2019, 593, 2341–2350. [Google Scholar] [CrossRef] [Green Version]
- Stewart, D.J.; Short, K.K.; Maniaci, B.N.; Burkhead, J.L. COMMD1 and PtdIns (4,5) P2 interaction maintain ATP7B copper transporter trafficking fidelity in HepG2 cells. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef]
- Muller, P.; van Bakel, H.; van de Sluis, B.; Holstege, F.; Wijmenga, C.; Klomp, L.W. Gene expression profiling of liver cells after copper overload in vivo and in vitro reveals new copper-regulated genes. J. Biol. Inorg. Chem. 2007, 12, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Van den Berghe, P.V.E.; Klomp, L.W.J. Posttranslational regulation of copper transporters. J. Biol. Inorg. Chem. 2010, 15, 37–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, W.J.; Kim, E.K.; Park, S.H.; Hahn, S.H.; Yoo, O.Y. Cloning and characterization of the promoter region of the Wilson disease gene. Biochem. Biophys. Res. Commun. 1999, 259, 206–211. [Google Scholar] [CrossRef]
- Bauerly, K.A.; Kelleher, S.L.; Lonnerdal, B. Effects of copper supplementation on copper absorption, tissue distribution, and copper transporter expression in an infant rat model. Am. J. Physiol Gastrointest. Liver. Physiol. 2005, 288, G1007–G1014. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.L.; Ashwell, M.S.; Fry, R.S.; Lloyd, K.E.; Flowers, W.L.; Spears, J.W. Effect of dietary copper amount and source on copper metabolism and oxidative stress of weanling pigs in short-term feeding. J. Animal. Sci. 2015, 93, 2948–2955. [Google Scholar] [CrossRef]
- Minghetti, M.; Leaver, M.J.; George, S.G. Multiple Cu-ATPase genes are differentially expressed and transcriptionally regulated by Cu exposure in sea bream, Sparus aurata. Aquat. Toxicol. 2010, 97, 23–33. [Google Scholar] [CrossRef]
- Stalke, A.; Pfister, E.D.; Baumann, U.; Illig, T.; Reischl, E.; Sandbothe, M.; Vajen, B.; Huge, N.; Schlegelberger, B.; von Neuhoff, N.; et al. MTF1 binds to metal-responsive element e within the ATP7B promoter and is a strong candidate in regulating the ATP7B expression. Ann. Human Genet. 2020, 84, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, E.S.; Abril, S.I.; Zanette, J.; Bianchini, A. Salinity-dependent copper accumulation in the guppy Poecilia vivipara is associated with CTR1 and ATP7B transcriptional regulation. Aquat. Toxicol. 2014, 152, 300–307. [Google Scholar] [CrossRef]
- Lenartowicz, M.; Moos, T.; Ogorek, M.; Jensen, T.G.; Moller, L.B. Metal-dependent regulation of ATP7A and ATP7B in fibroblast cultures. Front. Mol. Neurosci. 2016, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Ravia, J.J.; Stephen, R.M.; Ghishan, F.K.; Collins, J.F. Menkes copper ATPase (Atp7b) is a novel metal-responsive gene in rat duodenum, and immunoreactive protein is present on brush-border and basolateral membrane domains. J. Biol. Chem. 2005, 280, 36221–36227. [Google Scholar] [CrossRef] [Green Version]
- Chun, H.; Catterton, T.; Kim, H.; Lee, J.; Kim, B.-E. Organ specific regulation of ATP7A abundance is coordinated with systemic copper homeostasis. Sci. Rep. 2017, 7, 12001. [Google Scholar] [CrossRef] [Green Version]
- De Bie, P.; van de Sluis, B.; Burstein, E.; van der Berghe, P.; Muller, P.; Berger, R.; Gitlin, J.D.; Wijmenga, C.; Klomp, L.W.J. Distinct Wilson-disease mutations in ATP7B are associated with enhanced binding to COMMD1 and reduced stability of ATP7B. Gastroenterology 2007, 133, 1316–1326. [Google Scholar] [CrossRef] [Green Version]
- Materia, S.; Cater, M.A.; Klomp, L.W.J.; Mercer, J.F.B.; La Fontaine, S. Clusterin (apolipoprotein J), a molecular chaperone that facilitates degradation of the copper ATPases ATP7A and ATP7B. J. Biol. Chem. 2011, 286, 10073–10083. [Google Scholar] [CrossRef] [Green Version]
- Materia, S.; Cater, M.A.; Klomp, L.W.J.; Mercer, J.F.B.; La Fontaine, S. Clusterin and CommD1 independently regulate degradation of the mammalian copper ATPases ATP7A and ATP7B. J. Biol. Chem. 2012, 287, 2485–2499. [Google Scholar] [CrossRef] [Green Version]
- Hatori, Y.; Lutsenko, S. the role of copper chaperone Atox1 in coupling redox homeostasis to intracellular copper distribution. Antioxidants 2016, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Frommer, D.J. Biliary copper excretion in man and the rat. Digestion 1977, 15, 390–396. [Google Scholar] [CrossRef]
- Martin, M.T.; Jacobs, F.A.; Brushmiller, J.G. Low molecular weight copper-binding ligands in human bile. Proc. Soc. Exp. Biol. Med. 1986, 181, 249–255. [Google Scholar] [CrossRef]
- Iyengar, V.; Brewer, G.J.; Dick, R.D.; Owyang, C. Studies of cholecystokinin-stimulated biliary secretions reveal a high molecular weight copper-binding substance in normal subjects that is absent in patients with Wilson’s disease. J. Lab. Clin. Med. 1988, 111, 267–274. [Google Scholar] [PubMed]
- Samuels, A.R.; Freedman, J.H.; Bhargave, M.M. Purification and characterization of a novel abundant protein in rat bile that binds azo dye metabolites and copper. Biochim. Biophys. Acta 1983, 759, 23–31. [Google Scholar] [CrossRef]
- Kressner, M.S.; Stockert, R.J.; Morell, A.G.; Sternlieb, I. Origins of biliary copper. Hepatology 1984, 4, 867–870. [Google Scholar] [CrossRef]
- Van den Hammer, C.J.A.; Morell, A.G.; Scheinberg, I.H. Physical and chemical studies on ceruloplasmin. IX. The role of galactosyl receptors in clearance of ceruloplasmin from the circulation. J. Biol. Chem. 1970, 245, 4397–4402. [Google Scholar]
- Morell, A.G.; Irvine, R.A.; Sternlieb, I.; Scheinberg, I.H.; Ashwell, G. Physical and chemical studies on ceruloplasmin. V. Metabolic studies on sialic acid-free ceruloplasmin in vivo. J. Biol. Chem. 1968, 243, 155–159. [Google Scholar]
- Tavassoli, M. Liver endothelium binds, transports, and desialates ceruloplasmin which is then recognized by galactosyl receptors of hepatocytes. Trans. Assoc. Am. Phys. 1985, 98, 370–377. [Google Scholar]
- Hauser-Davis, R.A.; Goncalves, R.A.; Ziolli, R.L.; de Campos, R.C. A novel report of metallothionein in fish bile: SDS-PAGE analysis, spectrophotometry quantification and metal speciation characterization by liquid chromatography coupled to ICP-MS. Aquat. Toxicol. 2012, 116, 54–60. [Google Scholar] [CrossRef]
- Land, S.N.; Rocha, R.C.C.; Bordon, I.C.; Saint Pierre, T.; Ziolli, R.L.; Hauser-Davis, R.A. Biliary and hepatic metallothionein, metals and trace elements in environmentally exposed neutropical cichlids Geophagus brasiliensis. J. Trace Elem. Med. Biol. 2018, 50, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos, D.; Mar, D.; Ishida, M.; Vargas, R.; Gaite, M.; Montgomery, A.; Linder, M.C. Direct uptake of copper from blood plasma ceruloplasmin by mammalian cells. PLoS ONE 2016, 11, e0149516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piperno, A.; Alessio, M. Aceruloplasminemia: Waiting for an efficient therapy. Front. Neurosci. 2018, 12, 903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenawi, M.; Rouger, E.; Island, M.L.; Leroyer, P.; Robin, F.; Remy, S.; Tesson, L.; Anegon, I.; May, K.; Derbre, F.; et al. Ceruloplasmin deficiency does not induce macrophagic iron overload: Lessons from a new rat model of hereditary aceruloplasminemia. FASEB J. 2019, fj201901106R. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, L.A.; Durley, A.P.; Prohaska, J.R.; Harris, Z.L. Copper transport and metabolism are normal in aceruloplasminemic mice. J. Biol. Chem. 2001, 276, 36857–36861. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.-L.; Sauble, E.N.; Cabrera, A.; Roth, A.; Ackland, M.L.; Mercer, J.F.B.; Linder, M.C. Lack of ceruloplasmin expression alters aspects of copper transport to the fetus and newborn, as determined in mice. Biometals 2012, 25, 373–382. [Google Scholar] [CrossRef]
- Ke, B.X.; Llanos, R.M.; Wright, M.; Deal, Y.; Mercer, J.F. Alteration of copper physiology in mice overexpressing the human Menkes protein ATP7A. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R1460–R1467. [Google Scholar] [CrossRef] [Green Version]
- Wadwa, J.; Chu, Y.-H.; Ngyyen, N.; Henson, T.; Figueroa, A.; Llanos, A.; Ackland, M.L.; Mercer, J.F.B.; Fullriede, H.; Brennan, G.; et al. Effects of ATP7A overexpression on copper transport and metabolism in lactation and gestation, as measured in transgenic mice. Physiol. Rep. 2014, 2, e00195. [Google Scholar] [CrossRef]
- Yatsunyk, L.A.; Rosenzweig, A.C. Cu(I) binding and transfer by the N terminus of the Wilson disease protein. J. Biol. Chem. 2007, 282, 8622–8631. [Google Scholar] [CrossRef] [Green Version]
- Masuoka, J.; Hegenauer, J.; Van Dyke, G.R.; Saltman, P. Intrinsic stoichiometric equilibrium constants for the binding of zinc (II) and copper (II) to the high affinity site of serum albumin. J. Biol. Chem. 1993, 268, 21533–21537. [Google Scholar]
- Liu, N.M.; Lo, L.S.L.; Askary, H.; Jones, L.T.; Goforth, J.; Kidane, T.Z.; Vivas, E.; Sebastian, S.; Efron, A.; Tsai, M.T.; et al. Transcuprein is a macroglobulin regulated by copper availability. J. Nutr. Biochem. 2007, 18, 597–608. [Google Scholar] [CrossRef] [Green Version]
- Samimi, G.; Safael, R.; Katano, K.; Holzer, A.K.; Tomioka, M.; Goodman, M.; Howell, S.B. Increased expression of the copper efflux transporter ATP7A mediates resistance to cisplatin, carboplatin, oxaloplatin in ovarian cancer cells. Clin. Cancer Res. 2004, 10, 4661–4669. [Google Scholar] [CrossRef] [Green Version]
- Mariniello, M.; Petruzzelli, R.; Wanderlingh, L.G.; La Montagna, R.; Carissimo, A.; Pane, F.; Amoresano, A.; Ilyechova, E.Y.; Galagudza, M.M.; Catalano, F.; et al. Synthetic lethality screening identifies FDA-approved drugs that overcome ATP7B-mediated tolerance of tumor cells to cisplatin. Cancers 2020, 12, 608. [Google Scholar] [CrossRef] [Green Version]
- Ishida, S.; Lee, J.; Thiele, D.J.; Herskowitz, I. Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 14298–14302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.Y.; Choi, C.H.; Do, I.G.; Song, S.Y.; Lee, W.; Park, H.S.; Song, T.J.; Kim, M.K.; Kim, T.J.; Lee, J.W.; et al. Prognostic value of the copper transporters, CTR1 and CTR2, in patients with ovarian carcinoma receiving platinum-based chemotherapy. Gynecol. Oncol. 2011, 122, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.C. Ceruloplasmin and other copper binding components of blood plasma and their functions: An update. Metallomics 2016, 8, 887–905. [Google Scholar] [CrossRef] [PubMed]
- Kidane, T.Z.; Farhad, R.; Cabrera, A.; Roth, A.; Ackland, M.L.; Mercer, J.F.B.; Linder, M.C. Uptake of copper from plasma proteins in cells where expression of CTR1 has been modulated. Biometals 2012, 25, 697–709. [Google Scholar] [CrossRef]
- Moriya, M.; Ho, Y.-H.; Grana, A.; Nguyen, L.; Alvarez, A.; Jamil, R.; Ackland, M.L.; Michalczyk, A.; Hamer, P.; Ramos, D.; et al. Copper is taken up efficiently from albumin and a2-macroglobulin by cultured human cells by more than one mechanism. Am. J. Physiol. Cell Physiol. 2008, 295, C708–C721. [Google Scholar] [CrossRef] [Green Version]
- George, T.; Hoonmurthy, J.B.; Shiashankara, A.R.; Suresh, S.; Baliga, M.S. Correlation of blood and salivary levels of zinc, iron and copper in head and neck cancer patients: An investigative study. Avicenna J. Med. Biochem. 2017, 5, 35–39. [Google Scholar] [CrossRef] [Green Version]
- Faraz, M.; Manohar, V.; D’Souza, N. Estimation of copper in saliva and areca nut products and its correlation with histological grades of oral submucous fibrosis. J. Oral Pathol. Med. 2015, 44, 208–213. [Google Scholar] [CrossRef]
- Hong, J.H.; Duncan, S.E.; Dietrich, A.M.; O’Keefe, S.F.; Eigel, W.N. Interaction of copper and human salivary proteins. J. Agric. Food Chem. 2009, 57, 6967–6975. [Google Scholar] [CrossRef]
- Robinson, R.; Kauffman, D.L.; Waye, M.M.Y.; Blum, M.; Bennick, A.; Keller, P.J. Primary structure and possible origin of the non-glycosylated basic proline-rich protein of human submandibular/sublingual saliva. Biochem. J. 1989, 263, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Conklin, S.E.; Bridgman, E.C.; Su, Q.; Riggs-Gelasco, P.; Haas, K.L.; Franz, K.J. Specific histidine residues confer histatin peptides with copper-dependent activity against Candida albicans. Biochemistry 2017, 56, 4244–4255. [Google Scholar] [CrossRef] [PubMed]
- Blotnick, E.; Sol, A.; Bachrach, G.; Muhlrad, A. Interaction of histatin-3 and histatin-5 with actin. BMC Biochem. 2017, 18, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Salih, E.; Oppenheim, F.G.; Helmerhorst, E.J. Kinetics of histatin proteolysis in whole saliva and the effect on bioactive domains with metal-binding, antifungal, and wound-healing properties. FASEB J. 2009, 23, 2691–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moffa, E.B.; Machado, M.A.A.M.; Mussi, M.C.M.; Xiao, Y.; Garrido, S.S.; Giampaolo, E.T.; Siqueira, W.L. In vitro identification of histatin 5 salivary complexes. PLoS ONE 2015, 10, e0142517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, C.A., Jr. Physiological Aspects of Copper: Copper in Organs and Systems; Noyes Publications: Park Ridge, IL, USA, 1982. [Google Scholar]
- Powell, J.J.; Greenfield, S.M.; Thompson, R.P.H. Concentrations of metals in gastric juice in health and peptic ulcer disease. Gut 1992, 33, 1617–1620. [Google Scholar] [CrossRef] [Green Version]
- Miyata, S.; Toyoshima, M.; Takashima, M.; Itokawa, Y. Metal contents in duodenal aspirates of normal subjects during pancreozymin-secretin test. Gastroenterol. Jpn. 1982, 17, 207–213. [Google Scholar] [CrossRef]
- Carrigan, P.E.; Hentz, J.G.; Gordon, G.; Morgan, J.L.; Raimondo, M.; Anbar, A.D.; Miller, L.J. Distinctive heavy metal composition of pancreatic juice in patients with pancreatic carcinoma. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2656–2663. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Yoshida, A.; Koizumi, M. Distinctive metal compositioin of human pancreatic juice in patients with pancreatic carcinoma. Arch. Environ. Health 1987, 42, 356–360. [Google Scholar] [CrossRef]
- Heacox, H.N.; Gillman, P.L.; Zwart, S.R.; Smith, S.M. Excretion of zinc and copper increases in men during 3 weeks of bed rest, with and without artificial gravity. J. Nutr. 2017, 147, 1113–1120. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, N.; Matsushiro, T. Biliary and urinary excretion of metals in humans. Arch. Environ. Health 1986, 41, 324–330. [Google Scholar] [CrossRef]
- Ranjkesh, F.; Jaliseh, H.K.; Abutorabi, S. Monitoring the copper content of serum and urine in pregnancies complicated by preeclampsia. Biol. Trace Elem. Res. 2011, 144, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Grimes, A.; Hearn, C.J.; Lockhart, P.; Newgreen, D.F.; Mercer, J.F.B. Molecular basis of the brindled mouse mutant (Mobr): A murine model of Menkes disease. Hum. Mol. Genet. 1997, 6, 1037–1042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, S.D.P.; Cox, D.W. Expression in mouse kidney f membrane copper transporters Atp7a and Atp7b. Nephron 2002, 92, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Barnes, N.; Bartee, M.Y.; Braiterman, L.; Gupta, A.; Ustiyan, V.; Zuzel, V.; Kaplan, J.H.; Hubbard, A.L.; Lutsenko, S. Cell-specific trafficking suggests a new role for renal ATP7B in the intracellular copper storage. Traffic 2009, 10, 767–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Toda, K.; Koike, S.; Yoshikawa, H. Cadmium, copper and zinc in the urine of welders using cadmium-containing silver solder. Ind. Health 1981, 19, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Yoshikawa, H. Cadmium, copper, and zinc excretion and their binding to metallothionein in urine of cadmium-exposed rats. J. Toxicol. Environ. Health 1981, 8, 479–487. [Google Scholar] [CrossRef]
- Squitti, R.; Ventriglia, M.; Barbati, G.; Cassetta, E.; Ferreri, F.; Dal Forno, G.; Ramires, S.; Zappasodi, F.; Rossini, P.M. “Free” copper in serum of Alzheimer’s disease patients correlates with markers of liver function. J. Neural. Transm. 2007, 114, 1589–1594. [Google Scholar] [CrossRef]
- Walshe, J.M. The pattern of urinary copper excretion and its response to treatment in patients with Wilson’s disease. Q. J. Med. 2011, 104, 775–778. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.W.; Peng, F.; Molloy, S.A.; Pendyala, V.S.; Muchenditsi, A.; Muzik, O.; Lee, J.; Kaplan, J.; Lutsenko, S. Urinary copper elevation in a mouse model of Wilson’s disease is a regulated process to specifically decrease the hepatic copper load. PLoS ONE 2012, 7, e38327. [Google Scholar] [CrossRef]
- Huster, D.; Finegold, M.J.; Morgan, C.T.; Burkhard, J.L.; Nixon, R.; Vanderwerf, S.M.; Gilliam, C.T.; Lutsenko, S. Consequences of copper accumulation in the livers of Atp7b-/- (Wilson Disease gene) knockout mice. Am. J. Path. 2006, 168, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Squitti, R.; Siotto, M.; Cassetta, E.; El Idrissi, I.G.; Colabufo, N.A. Measurement of serum non-ceruloplasmin copper by direct fluorescent method specific to Cu (II). Clin. Chem. Lab. Med. 2017, 55, 1360–1367. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fluid | [Cu] mg/L | (uM) | Volume (L/day) | Total Cu (mg) |
|---|---|---|---|---|
| Saliva | 0.1 | 1.6 | 1–2 | 0.15 |
| Gastric juice | 0.08 | 1.2 | 2.5 | 0.18 |
| Pancreatic juice | 0.3 | 4 | 1.5 | 0.5 |
| Bile | 4 ± 2 | 19–63 | 0.5–0.75 | 0.6–6 |
| Duodenal fluid | 0.016 | (1?) | 0.016 | |
| Urine | (0.02–0.1) ** | (0.03–0.10) | 1.5 | 0.04–0.05 *** |
| Fluid | Dialysable Cu (Percent 64Cu) | Intestinal Absorption (Percent) | Body retention (Percent 64Cu) |
|---|---|---|---|
| In vitro 64Cu labeled a | |||
| Saliva | 98 ± 2 (6) | 13 ± 3 (6) | |
| Gastric juice | 97 ± 2 (6) | 12 ± 10 (6) | |
| Bile (gall bladder) | 12 ± 12 (5) | 6 ± 1 (6) | |
| In vivo 64Cu-labeled b | |||
| Time after intubation | |||
| Bile 0–4 h | 50 ± 7 (9) | 16 ± 3 (8) | |
| 4–8 h | 28 ± 6 (9) | 13 ± 4 (8) | |
| 8–24 h | 16 ± 3 (9) | 10 ± 3 (4) |
| Sham-Operated Controls | Bile Duct Ligation | |
|---|---|---|
| Slope [Means ± SD (N)] * 0–63 h 0–86 h | −552 ± 28 (23) −623 ± 56 (28) | −258 ± 97 (15) ** −272 ± 67 (17) *** |
| Half-life (h) 0–63 h 0–86 h | 54 h 49 h | 117 h 110 h |
| Liver [Means ± SD (N)] Cu (ug/g) 67Cu specific activity (cpm/ug) | 4.6 ± 0.5 (6) 64 ± 11 (4) | 3.8 ± 1.1 (6) 105 ± 42 (5) |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linder, M.C. Copper Homeostasis in Mammals, with Emphasis on Secretion and Excretion. A Review. Int. J. Mol. Sci. 2020, 21, 4932. https://doi.org/10.3390/ijms21144932
Linder MC. Copper Homeostasis in Mammals, with Emphasis on Secretion and Excretion. A Review. International Journal of Molecular Sciences. 2020; 21(14):4932. https://doi.org/10.3390/ijms21144932
Chicago/Turabian StyleLinder, Maria C. 2020. "Copper Homeostasis in Mammals, with Emphasis on Secretion and Excretion. A Review" International Journal of Molecular Sciences 21, no. 14: 4932. https://doi.org/10.3390/ijms21144932