Pulmonary Artery Thrombosis: A Diagnosis That Strives for Its Independence

 , ,
, , 
Abstract
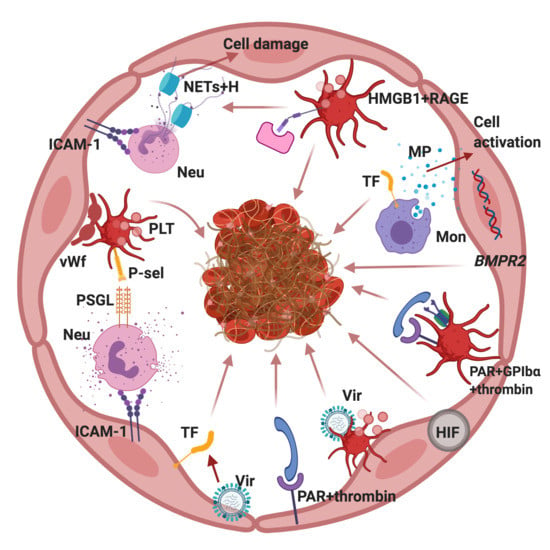
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Clinical Aspects of Pulmonary Thrombosis
3. Potential Mechanisms for Primary Thrombosis in the PA
3.1. Structural Features and PA Endothelium Molecules Potentially Relevant to Thrombosis
3.2. The Role of TNFα in Activation of PA Endothelium
3.3. The Role of Hypoxia in the Activation of PA Endothelium
3.4. The Role of Genetic Factors in the Functioning of PA Endothelium
3.5. Potential Role of Viruses in Predisposition to PA Thrombosis
4. The Role of Platelets in PA Thrombosis
4.1. PAR-Dependent Platelet Activation
4.2. HMGB1-Dependent Platelet Activation
4.3. The Role of Hypoxia in Platelet Activation
4.4. Immune Response and Platelet Activation
4.5. RAGE-Dependent Activation of Endotheliocytes and Platelets and Release of NETs
5. The Role of Microparticles in the Development of PA Thrombosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| VTE | Venous thromboembolism |
| DVT | Deep vein thrombosis |
| PA | Pulmonary artery |
| PE | Pulmonary embolism |
| COPD | chronic obstructive pulmonary disease |
| CT | Computed tomographic imaging |
| SCA | Sickle cell anemia |
| APC | Activated protein C |
| EPCR | Endothelial Protein C Receptor |
| eNOS | Endothelial nitric oxide synthase |
| MP | Microparticles |
| PAR | Protease mediated receptor |
| NO | nitric oxide |
| TNFα | tumor necrosis factor |
| ROS | reactive oxygen species |
| TF | tissue factor |
| ICAM | Inter-Cellular Adhesion Molecule |
| VCAM | Vascular cell adhesion molecule |
| tPA | Tissue plasminogen activator |
| uPA | urokinase-type |
| HIFs | hypoxia inducible factors |
| NF-kB | nuclear factor-KappaB |
| TFPI | Tissue factor pathway inhibitor |
| SMC | smooth muscle cells |
| VEGF | Vascular endothelial growth factor |
| ACEII | angiotensin converting enzyme |
| PSGL-1 | P-selectin glycoprotein ligand |
| vWf | von Willebrand factor |
| HMGB1 | high-mobility group box 1 |
| RAGE | receptor for advanced glycation end products |
| NETs | neutrophil extracellular traps |
| TLR | Toll-like receptor |
| CD40L | CD40 ligand |
| IFITM3 | Interferon-induced transmembrane protein 3 |
| CLR | C-type lectin receptor |
| Mac-1 | Macrophage-1 antigen |
| MCP-1 | Monocyte Chemoattractant Protein 1 |
| BMPR2 | Bone Morphogenetic Protein Receptor Type 2 |
References
- Kumar, D.R.; Hanlin, E.R.; Glurich, I.; Mazza, J.J.; Yale, S.H. Virchow’s contribution to the understanding of thrombosis and cellular biology. Clin. Med. Res. 2010, 8, 168–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevitt, S.; Gallagher, N. Venous thrombosis and pulmonary embolism. A clinico-pathological study in injured and burned patients. Br. J. Surg. 1961, 48, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Velmahos, G.C.; Spaniolas, K.; Tabbara, M.; Abujudeh, H.H.; De Moya, M.; Gervasini, A.; Alam, H.B. Pulmonary embolism and deep venous thrombosis in trauma: Are they related? Arch. Surg. 2009, 144, 928–932. [Google Scholar] [CrossRef] [Green Version]
- Tadlock, M.D.; Chouliaras, K.; Kennedy, M.; Talving, P.; Okoye, O.; Aksoy, H.; Karamanos, E.; Zheng, L.; Grabo, D.J.; Rogers, C.; et al. The origin of fatal pulmonary emboli: A postmortem analysis of 500 deaths from pulmonary embolism in trauma, surgical, and medical patients. Am. J. Surg. 2015, 209, 959–968. [Google Scholar] [CrossRef] [PubMed]
- Van Langevelde, K.; Šrámek, A.; Vincken, P.W.J.; van Rooden, J.K.; Rosendaal, F.R.; Cannegieter, S.C. Finding the origin of pulmonary emboli with a total-body magnetic resonance direct thrombus imaging technique. Haematologica 2012, 98, 309–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, J.; Hunt, B.J.; Moody, A. Magnetic resonance direct thrombus imaging: A novel technique for imaging venous thromboemboli. Thromb. Haemost. 2003, 89, 773–782. [Google Scholar] [PubMed]
- Bertoletti, L.; Quenet, S.; Laporte, S.; Sahuquillo, J.C.; Conget, F.; Pedrajas, J.M.; Martin, M.; Casado, I.; Riera-Mestre, A.; Monreal, M.; et al. Pulmonary embolism and 3-month outcomes in 4036 patients with venous thromboembolism and chronic obstructive pulmonary disease: Data from the RIETE registry. Respir. Res. 2013, 14, 75. [Google Scholar] [CrossRef] [Green Version]
- Bertoletti, L.; Quenet, S.; Mismetti, P.; Hernández, L.; Martín-Villasclaras, J.J.; Tolosa, C.; Valdes, M.; Barron, M.; Todoli, J.A.; Monreal, M. RIETE Investigators. Clinical presentation and outcome of venous thromboembolism in COPD. Eur. Respir. J. 2012, 39, 862–868. [Google Scholar] [CrossRef] [Green Version]
- Børvik, T.; Evensen, L.H.; Morelli, V.M.; Melbye, H.; Braekkan, S.K.; Hansen, J.-B. Impact of respiratory symptoms and oxygen saturation on the risk of incident venous thromboembolism-the Tromsø study. Res. Pract. Thromb. Haemost. 2020, 4, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Gunen, H.; Gulbas, G.; In, E.; Yetkin, O.; Hacievliyagil, S.S. Venous thromboemboli and exacerbations of COPD. Eur. Respir. J. 2010, 35, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Erelel, M.; Çuhadaro, Ǧ.Ç.; Ece, T.; Arseven, O. The frequency of deep venous thrombosis and pulmonary embolus in acute exacerbation of chronic obstructive pulmonary disease. Respir. Med. 2002, 96, 515–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akpinar, E.E.; Hoşgün, D.; Akpinar, S.; Ataç, G.K.; Doğanay, B.; Gülhan, M. Incidence of pulmonary embolism during COPD exacerbation. J. Bras. Pneumol. 2014, 40, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Majoor, C.J.; Kamphuisen, P.W.; Zwinderman, A.H.; Brinke, A.; Amelink, M.; Rijssenbeek-Nouwens, L.; Sterk, P.J.; Buller, H.R.; Bel, E. Risk of deep vein thrombosis and pulmonary embolism in asthma. Eur. Respir. J. 2013, 42, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Dessap, A.M.; Deux, J.F.; Abidi, N.; Lavenu-Bombled, C.; Melica, G.; Renaud, B.; Godeau, B.; Adnot, S.; Brochard, L.; Brun-Buisson, C.; et al. Pulmonary artery thrombosis during acute chest syndrome in sickle cell disease. Am. J. Respir. Crit. Care Med. 2011, 184, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Lundy, J.B.; Oh, J.S.; Chung, K.K.; Ritter, J.L.; Gibb, I.; Nordmann, G.R.; Sonka, B.J.; Tai, N.R.M.; Aden, J.K.; Rasmussen, T.E. Frequency and relevance of acute peritraumatic pulmonary thrombus diagnosed by computed tomographic imaging in combat casualties. J. Trauma Acute Care Surg. 2013, 75, S215–S220. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, J.-M.; Zander, A.L.; Olson, E.J.; Shackford, S.R.; Dunne, C.E.; Badiee, J.; Schechter, M.S.; Sise, M.J. Pulmonary embolism without deep venous thrombosis: De novo or missed deep venous thrombosis? J. Trauma Acute Care Surg. 2014, 76, 1270–1274. [Google Scholar] [CrossRef]
- Cha, S.I.; Choi, K.J.; Shin, K.M.; Lim, J.K.; Yoo, S.S.; Lee, J.; Kim, C.-H.; Park, J.-Y. Clinical characteristics of in-situ pulmonary artery thrombosis in Korea. Blood Coagul. Fibrinolysis 2015, 26, 903–907. [Google Scholar] [CrossRef]
- Ribeiro, D.D.; Lijfering, W.M.; Van Hylckama Vlieg, A.; Rosendaal, F.R.; Cannegieter, S.C. Pneumonia and risk of venous thrombosis: Results from the MEGA study. J. Thromb. Haemost. 2012, 10, 1179–1182. [Google Scholar] [CrossRef]
- Poissy, J.; Goutay, J.; Caplan, M.; Parmentier, E.; Duburcq, T.; Lassalle, F.; Jeanpierre, E.; Rauch, A.; Labreuch, J.; Susen, S. Pulmonary Embolism in COVID-19 Patients: Awareness of an Increased Prevalence. Circulation 2020. [Google Scholar] [CrossRef]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Rotzinger, D.C.; Beigelman-Aubry, C.; von Garnier, C.; Qanadli, S.D. Pulmonary embolism in patients with COVID-19: Time to change the paradigm of computed tomography. Thromb. Res. 2020, 190, 58–59. [Google Scholar] [CrossRef] [PubMed]
- Leonard-Lorant, I.; Delabranche, X.; Severac, F.; Helms, J.; Pauzet, C.; Collange, O.; Schneider, F.; Labani, A.; Bilbaut, P.; Moliere, S.; et al. Acute Pulmonary Embolism in COVID-19 Patients on CT Angiography and Relationship to D-Dimer Levels. Radiology 2020, 201561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, K.H.; Wu, A.K.; Cheng, V.C.; Tang, B.S.; Chan, C.Y.; Yung, C.Y.; Luk, S.H.; Chow, L.; Yuen, K.Y. Pulmonary artery thrombosis in a patient with severe acute respiratory syndrome. Postgrad Med. J. 2005, 81, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, P.Y.; Chui, P.; Ling, A.E.; Franks, T.J.; Tai, D.Y.H.; Kaw, G.J.L.; Wansaicgeong, G.; Chan, K.P.; Oon, L.L.E.; Teo, E.S.; et al. Analysis of deaths during the severe acute respiratory syndrome (SARS) epidemic in Singapore: Challenges in determining a SARS diagnosis. Arch. Pathol. Lab. Med. 2004, 128, 195–204. [Google Scholar] [CrossRef]
- Abgueguen, P.; Delbos, V.; Chennebault, J.M.; Payan, C.; Pichard, E. Vascular Thrombosis and Acute Cytomegalovirus Infection in Immunocompetent Patients: Report of 2 Cases and Literature Review. Clin. Infect. Dis. 2003, 36, e134–e139. [Google Scholar] [CrossRef] [Green Version]
- Delbos, V.; Abgueguen, P.; Chennebault, J.M.; Fanello, S.; Pichard, E. Acute cytomegalovirus infection and venous thrombosis: Role of antiphospholipid antibodies. J. Infect. 2007, 54, e47–e50. [Google Scholar] [CrossRef]
- Van Stralen, K.J.; Doggen, C.J.M.; Bezemer, I.D.; Pomp, E.R.; Lisman, T.; Rosendaal, F.R. Mechanisms of the factor V Leiden paradox. Arter. Thromb. Vasc Biol. 2008, 28, 1872–1877. [Google Scholar] [CrossRef]
- Anand, A.C.; Jha, S.K.; Saha, A.; Sharma, V.; Adya, C.M. Thrombosis as a complication of extended stay at high altitude. Natl. Med. J. India 2001, 14, 197–201. [Google Scholar]
- Pandey, P.; Lohani, B.; Murphy, H. Pulmonary Embolism Masquerading as High Altitude Pulmonary Edema at High Altitude. High Alt. Med. Biol. 2016, 17, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Rao, A.; Attar, N.; Prakasha, R.; Dileep, K.; Sharma, R.; Prakash, P. Hyperhomocysteinemia and pulmonary embolism without deep vein thrombosis in a young patient presenting as pneumonia—A rare case report. J. Health Allied Sci. NU 2012, 2, 36–38. [Google Scholar]
- Duru, S.; Keleşoğlu, A.; ArdIç, S. Clinical update on pulmonary embolism. Arch. Med. Sci. 2012, 10, 557–565. [Google Scholar] [CrossRef]
- Caldera, A.; Mora, J.; Kotler, M.; Eiger, G. Pulmonary embolism in a patient with pernicious anemia and hyperhomocysteinemia. Chest 2002, 122, 1487–1488. [Google Scholar] [CrossRef] [PubMed]
- Millar, F.R.; Summers, C.; Griffiths, M.J.; Toshner, M.R.; Proudfoot, A.G. The pulmonary endothelium in acute respiratory distress syndrome: Insights and therapeutic opportunities. Thorax 2016, 71, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Bae, J.S.; Yang, L.; Manithody, C.; Rezaie, A.R. The ligand occupancy of endothelial protein C receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood 2007, 110, 3909–3916. [Google Scholar] [CrossRef]
- Sun, Y.; Hu, G.; Zhang, X.; Minshall, R.D. Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ. Res. 2009, 105, 676–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, M.W.; Zhang, Y.; Hong, P.K.; Zhou, Z.; Feghali-Bostwick, C.A.; Liu, F. Caveolin-1: A critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J. Exp. Med. 2006, 203, 2895–2906. [Google Scholar] [CrossRef]
- Marudamuthu, A.S.; Bhandary, Y.P.; Fan, L.; Radhakrishnan, V.; MacKenzie, B.A.; Maier, E.; Shetty, S.K.; Nagaraja, M.R.; Gopu, V.; Tiwari, N.; et al. Caveolin-1–derived peptide limits development of pulmonary fibrosis. Sci. Transl. Med. 2019, 11, eaat2848. [Google Scholar] [CrossRef]
- Maeda, K.; Saiki, Y.; Yamaki, S. In Situ Thrombosis of Small Pulmonary Arteries in Pulmonary Hypertension Developing after Chemotherapy for Malignancy. Pulm. Med. 2015, 2015, 230846. [Google Scholar] [CrossRef]
- Hong, K.H.; Lee, Y.J.; Lee, E.; Park, S.O.; Han, C.; Beppu, H.; Li, E.; Raizada, M.K.; Bloch, K.D.; Oh, S.P. Genetic ablation of the Bmpr2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation 2008, 118, 722–730. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.D.S.; Minshall, R.D. Caveolin and Endothelial NO Signaling. Curr. Top. Membr. 2018, 82, 257–279. [Google Scholar] [CrossRef]
- Freedman, J.E.; Loscalzo, J. Nitric oxide and its relationship to thrombotic disorders. J. Thromb. Haemost. 2003, 1, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Fukudome, K.; Tsuneyoshi, N.; Satoh, T.; Tokunaga, O.; Sugawara, K.; Mizokami, H.; Kimoto, M. The endothelial cell protein C receptor (EPCR) functions as a primary receptor for protein C activation on endothelial cells in arteries, veins, and capillaries. Biochem. Biophys. Res. Commun. 1999, 259, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Gresele, P.; Momi, S.; Berrettini, M.; Nenci, G.G.; Schwarz, H.P.; Semeraro, N.; Colucci, M. Activated human protein C prevents thrombin-induced thromboembolism in mice. Evidence that activated protein C reduces intravascular fibrin accumulation through the inhibition of additional thrombin generation. J. Clin. Investig. 1998, 101, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Rezaie, A.R. Protease activated receptor 1 (PAR-1) activation by thrombin is protective in human pulmonary artery endothelial cells if endothelial protein C receptor is occupied by its natural ligand. Thromb. Haemost. 2008, 100, 101109. [Google Scholar] [CrossRef] [Green Version]
- Konecny, F.A. Pulmonary Embolism and vascular injury: What is the role of thrombin? JRMS 2007, 12, 203–216. [Google Scholar]
- Gutmann, C.; Siow, R.; Gwozdz, A.M.; Saha, P.; Smith, A. Reactive oxygen species in venous thrombosis. Int. J. Mol. Sci. 2020, 21, 1918. [Google Scholar] [CrossRef] [Green Version]
- Alberelli, M.A.; De Candia, E. Functional role of protease activated receptors in vascular biology. Vascul. Pharmacol. 2014, 62, 72–81. [Google Scholar] [CrossRef]
- Nieman, M. Protease-activated receptors in hemostasis. Blood 2016, 128, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Page, M.J.; Lourenço, A.L.; David, T.; LeBeau, A.M.; Cattaruzza, F.; Castro, H.C.; VanBrocklin, H.F.; Coughlin, S.R.; Craik, C.S. Non-invasive imaging and cellular tracking of pulmonary emboli by near-infrared fluorescence and positron-emission tomography. Nat. Commun. 2015, 6, 8448. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, E.P.; Yang, Y.; Janssen, W.J.; Gandjeva, A.; Perez, M.J.; Barthel, L. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 2012, 18, 1217–1223. [Google Scholar] [CrossRef] [Green Version]
- Frantzeskaki, F.; Armaganidis, A.; Orfanos, S.E. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respiration 2017, 93, 212–225. [Google Scholar] [CrossRef]
- Kuluöztürk, M.; İn, E.; İlhan, N. Endocan as a marker of disease severity in pulmonary thromboembolism. Clin. Respir. J. 2019, 13, 773–780. [Google Scholar] [CrossRef] [PubMed]
- Dudek, S.M.; Garcia, J.G.N. Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 2001, 91, 1487–1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzales, J.N.; Lucas, R.; Verin, A.D. The Acute Respiratory Distress Syndrome: Mechanisms and Perspective Therapeutic Approaches. Austin J. Vasc. Med. 2015, 2, 1009. [Google Scholar]
- Kumar, N.G.; Clark, A.; Roztocil, E.; Caliste, X.; Gillespie, D.L.; Cullen, J.P. Fibrinolytic activity of endothelial cells from different venous beds. J. Surg. Res. 2015, 194, 297–303. [Google Scholar] [CrossRef]
- Mahemuti, A.; Abudureheman, K.; Aihemaiti, X.; Hu, X.M.; Xia, Y.N.; Tang, B.P.; Upur, H. Association of interleukin-6 and C-reactive protein genetic polymorphisms levels with venous thromboembolism. Chin. Med. J. 2012, 125, 3997–4002. [Google Scholar] [PubMed]
- Branchford, B.R.; Carpenter, S.L. The role of inflammation in venous thromboembolism. Front. Pediatr. 2018, 6, 142. [Google Scholar] [CrossRef] [PubMed]
- Zacho, J.; Tybjærg-Hansen, A.; Nordestgaard, B.G. C-reactive protein and risk of venous thromboembolism in the general population. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1672–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochenek, M.; Rosinus, N.; Lankeit, M.; Hobohm, L.; Bremmer, F.; Schütz, E.; Klok, F.A.; Horke, S.; Wiederoth, C.B.; Munzel, T.; et al. From thrombosis to fibrosis in chronic thromboembolic pulmonary hypertension. Thromb. Haemost. 2017, 117, 769–783. [Google Scholar] [CrossRef]
- Chaurasia, S.N.; Kushwaha, G.; Kulkarni, P.P.; Mallick, R.L.; Latheef, N.A.; Mishra, J.K.; Dash, D. Platelet HIF-2α promotes thrombogenicity through PAI-1 synthesis and extracellular vesicle release. Haematologica 2019, 104, 2482–2492. [Google Scholar] [CrossRef]
- Lim, C.S.; Kiriakidis, S.; Sandison, A.; Paleolog, E.M.; Davies, A.H. Hypoxia-inducible factor pathway and diseases of the vascular wall. Vasc. Surg. 2013, 58, 219–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, N.; Zhao, Y.Y.; Evans, C.E. The stimulation of thrombosis by hypoxia. Thromb. Res. 2019, 181, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Hyman, M.C.; Lawrence, D.A.; Pinsky, D.J. Molecular regulation of the PAI-1 gene by hypoxia: Contributions of Egr-1, HIF-1 α, and C/EBPα. FASEB J. 2007, 21, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Jha, P.K.; Sahu, A.; Prabhakar, A.; Tyagi, T.; Chatterjee, T.; Arvind, P.; Nair, J.; Gupta, N.; Kumari, B.; Nair, V.; et al. Genome-Wide Expression Analysis Suggests Hypoxia-Triggered Hyper-Coagulation Leading to Venous Thrombosis at High Altitude. Thromb. Haemost. 2018, 118, 1279–1295. [Google Scholar] [CrossRef]
- Evans, C.E.; Palazon, A.; Sim, J.; Tyrakis, P.A.; Prodger, A.; Lu, X.; Chan, S.; Bendahl, P.-O.; Belting, M.; Euler, L.V.; et al. Modelling pulmonary microthrombosis coupled to metastasis: Distinct effects of thrombogenesis on tumorigenesis. Biol. Open 2017, 6, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Green, C.E.; Turner, A.M. The role of the endothelium in asthma and chronic obstructive pulmonary disease (COPD). Respir. Res. 2017, 18, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Lee, S.H.; Kim, C.H.; Yang, K.S.; Lee, E.J.; Min, K.H.; Hur, G.Y.; Lee, S.H.; Lee, S.Y.; Kim, J.H.; et al. Increased expression of vascular endothelial growth factor and hypoxia inducible factor-1α in lung tissue of patients with chronic bronchitis. Clin. Biochem. 2014, 47, 552–559. [Google Scholar] [CrossRef]
- Hoshino, M.; Nakamura, Y.; Hamid, Q.A. Gene expression of vascular endothelial growth factor and its receptors and angiogenesis in bronchial asthma. J. Allergy Clin. Immunol. 2001, 107, 1034–1038. [Google Scholar] [CrossRef]
- Yasuo, M.; Mizuno, S.; Kraskauskas, D.; Bogaard, H.J.; Natarajan, R.; Cool, C.D.; Zamora, M.; Voelkel, N.F. Hypoxia inducible factor-1α in human emphysema lung tissue. Eur. Respir. J. 2011, 37, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Amsellem, V.; Gary-Bobo, G.; Marcos, E.; Maitre, B.; Chaar, V.; Validire, P.; Stern, J.-B.; Noureddine, H.; Sapin, E.; Rideau, D.; et al. Telomere dysfunction causes sustained inflammation in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 1358–1366. [Google Scholar] [CrossRef]
- Spira, A.; Beane, J.; Pinto-Plata, V.; Kadar, A.; Liu, G.; Shah, V.; Celli, B.; Brody, J.S. Gene expression profiling of human lung tissue from smokers with severe emphysema. Am. J. Respir. Cell Mol. Biol. 2004, 31, 601–610. [Google Scholar] [CrossRef]
- Banfi, C.; Brioschi, M.; Barbieri, S.S.; Eligini, S.; Barcella, S.; Tremoli, E.; Colli, S.; Mussoni, L. Mitochondrial reactive oxygen species: A common pathway for PAR1- and PAR2-mediated tissue factor induction in human endothelial cells. J. Thromb. Haemost. 2009, 7, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Van Uden, P.; Kenneth, N.S.; Webster, R.; Müller, H.A.; Mudie, S.; Rocha, S. Evolutionary conserved regulation of HIF-1β by nf-κb. PLoS Genet. 2011, 7, e1001285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, H.; Parthasarathi, K.; Quadri, S.; Issekutz, A.C.; Bhattacharya, J. Mechano-oxidative coupling by mitochondria induces proinflammatory responses in lung venular capillaries. J. Clin. Investig. 2003, 111, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visseren, F.L.J.; Bouwman, J.J.M.; Bouter, K.P.; Diepersloot, R.J.A.; de Groot, P.G.; Erkelens, D.W. Procoagulant activity of endothelial cells after infection with respiratory viruses. Thromb. Haemost. 2000, 84, 319–324. [Google Scholar] [PubMed]
- Varga, Z.; Flammer, A.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moh, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Fox, S.E.; Akmatbekov, A.; Harbert, J.L.; Li, G.; Brown, J.Q.; Vander Heide, R.S. Pulmonary and Cardiac Pathology in Covid-19: The First Autopsy Series from New Orleans; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2020. [Google Scholar] [CrossRef]
- Ding, Y.; Wang, H.; Shen, H.; Li, Z.; Geng, J.; Han, H.; Cai, J.; Kang, W.; Weng, D.; Lu, Y.; et al. The clinical pathology of severe acute respiratory syndrome (SARS): A report from China. J. Pathol. 2003, 200, 282–289. [Google Scholar] [CrossRef]
- Li, M.-Y.; Li, L.; Zhang, Y.; Wang, X.-S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef]
- Meng, T.; Cao, H.; Zhang, H.; Kang, Z.; Xu, D.; Gong, H. The Insert Sequence in SARS-CoV-2 Enhances Spike Protein Cleavage by TMPRSS; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2020. [Google Scholar] [CrossRef]
- Guignabert, C.; De Man, F.; Lombès, M. ACE2 as therapy for pulmonary arterial hypertension: The good outweighs the bad. Eur. Respir. J. 2018, 51, 1800848. [Google Scholar] [CrossRef]
- Simes, J.; Becattini, C.; Agnelli, G.; Eikelboom, J.W.; Kirby, A.C.; Mister, R.; Prandoni, P.; Brighton, T.A.; INSPIRE Study Investigators (International Collaboration of Aspirin Trials for Recurrent Venous Thromboembolism). Aspirin for the prevention of recurrent venous thromboembolism: The INSPIRE collaboration. Circulation 2014, 130, 1062–1071. [Google Scholar] [CrossRef]
- Simes, J.; Becattini, C.; Agnelli, G.; Eikelboom, J.W.; Kirby, A.C.; Mister, R.; Prandoni, P.; Brighton, T.A.; INSPIRE Study Investigators (International Collaboration of Aspirin Trials for Recurrent Venous Thromboembolism). Aspirin for preventing the recurrence of venous thromboembolism. N. Engl. J. Med. 2012, 366, 1959–1967. [Google Scholar]
- Hsia, C.W.; Tsai, C.L.; Sheu, J.R.; Lu, W.J.; Hsia, C.H.; Velusamy, M.; Jayakumar, T.; Li, J.Y. Suppression of Human Platelet Activation via Integrin αIIbβ3 Outside-In Independent Signal and Reduction of the Mortality in Pulmonary Thrombosis by Auraptene. Int. J. Mol. Sci. 2019, 20, 5585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branchford, B.R.; Stalker, T.J.; Law, L.; Acevedo, G.; Sather, S.; Brzezinski, C.; Wilson, K.M.; Minson, K.; Lee-Sherick, A.B.; Davizon-Castillo, P.; et al. The small-molecule MERTK inhibitor UNC2025 decreases platelet activation and prevents thrombosis. J. Thromb. Haemost. 2018, 16, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.J.; Hamilton, J.R.; Lease, K.E.; Coughlin, S.R. Protection against thrombosis in mice lacking PAR3. Blood 2002, 100, 3240–3244. [Google Scholar] [CrossRef] [Green Version]
- Lova, P.; Canobbio, I.; Guidetti, G.F.; Balduini, C.; Torti, M. Thrombin induces platelet activation in the absence of functional protease activated receptors 1 and 4 and glycoprotein Ib-IX-V. Cell Signal. 2010, 22, 1681–1687. [Google Scholar] [CrossRef]
- Vretenbrant, K.; Ramström, S.; Bjerke, M.; Lindahl, T.L. Platelet activation via PAR4 is involved in the initiation of thrombin generation and in clot elasticity development. Thromb. Haemost. 2007, 97, 417–424. [Google Scholar]
- De Candia, E. Mechanisms of platelet activation by thrombin: A short history. Thromb. Res. 2012, 129, 250–256. [Google Scholar] [CrossRef]
- Yiming, M.T.; Lederer, D.J.; Sun, L.; Huertas, A.; Issekutz, A.C.; Bhattacharya, S. Platelets enhance endothelial adhesiveness in high tidal volume ventilation. Am. J. Respir. Cell Mol. Biol. 2008, 39, 569–575. [Google Scholar] [CrossRef]
- Emin, M.T.; Sun, L.; Huertas, A.; Das, S.; Bhattacharya, J.; Bhattacharya, S. Platelets induce endothelial tissue factor expression in a mouse model of acid-induced lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L1209–L1220. [Google Scholar] [CrossRef] [Green Version]
- Zarbock, A.; Singbartl, K.; Ley, K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. Clin. Investig. 2006, 116, 3211–3219. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schäffer, T.E.; Bohn, E.; Frick, J.-S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maugeri, N.; Franchini, S.; Campana, L.; Baldini, M.; Ramirez, G.A.; Sabbadini, M.G.; Rovere-Querini, P.; Manfredi, A.A. Circulating platelets as a source of the damage-associated molecular pattern HMGB1 in patients with systemic sclerosis. Autoimmunity 2012, 45, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Philippi, V.; Stockhausen, S.; Busse, J.; Antonelli, A.; Miller, M.; Schubert, I.; Hoseinpour, P.; Chandraratne, S.; von Bruhl, M.-L.; et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 2016, 128, 2435–2449. [Google Scholar] [CrossRef]
- Venereau, E.; Schiraldi, M.; Uguccioni, M.; Bianchi, M.E. HMGB1 and leukocyte migration during trauma and sterile inflammation. Mol. Immunol. 2013, 55, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Savchenko, A.S.; Martinod, K.; Seidman, M.A.; Wong, S.L.; Borissoff, J.I.; Piazza, G.; Libby, P.; Goldhaber, S.Z.; Mitchell, R.N.; Wagner, D.D. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J. Thromb. Haemost. 2014, 12, 860–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, A.-B.E.; Krijnen, P.; Schipper, I.B. Predictive value of cytokines for developing complications after polytrauma. World J. Crit. Care Med. 2016, 5, 187–200. [Google Scholar] [CrossRef]
- Sarkar, J.; Golden, P.J.; Kajiura, L.N.; Murata, L.-A.M.; Uyehara, C.F.T. Vasopressin Decreases Pulmonary–to–Systemic Vascular Resistance Ratio in a Porcine Model of Severe Hemorrhagic Shock. Shock 2015, 43, 475–482. [Google Scholar] [CrossRef]
- Tyagi, T.; Ahmad, S.; Gupta, N.; Sahu, A.; Ahmad, Y.; Nair, V.; Chatterjee, T.; Bajaj, N.; Sengupta, S.; Ganju, L.; et al. Altered expression of platelet proteins and calpain activity mediate hypoxia-induced prothrombotic phenotype. Blood 2014, 123, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Ferroni, P.; Martini, F.; Riondino, S.; La Farina, F.; Magnapera, A.; Ciatti, F.; Guadagni, F. Soluble P-selectin as a marker of in vivo platelet activation. Clin. Chim. Acta 2009, 399, 88–91. [Google Scholar] [CrossRef]
- Prabhakar, A.; Chatterjee, T.; Bajaj, N.; Tyagi, T.; Sahu, A.; Gupta, N.; Kumari, D.; Nair, V.; Kumar, B.; Ashraf, M.Z. Venous thrombosis at altitude presents with distinct biochemical profiles: A comparative study from the Himalayas to the plains. Blood Adv. 2019, 3, 3713–3723. [Google Scholar] [CrossRef]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Bozza, F.A.; Bozza, P.T. Platelets in immune response to virus and immunopathology of viral infections. Front. Med. 2018, 5, 121. [Google Scholar] [CrossRef] [PubMed]
- Lütteke, N.; Raftery, M.J.; Lalwani, P.; Lee, M.H.; Giese, T.; Voigt, S.; Bannert, N.; Schulze, H.; Kruger, D.H.; Schonrich, G. Switch to high-level virus replication and HLA class I upregulation in differentiating megakaryocytic cells after infection with pathogenic hantavirus. Virology 2010, 405, 70–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, R.A.; Schwertz, H.; Hottz, E.D.; Rowley, J.W.; Manne, B.K.; Washington, A.V.; Hunter-Mellado, R.; Tolley, N.D.; Christensen, M.; Eustes, A.S.; et al. Human megakaryocytes possess intrinsic antiviral immunity through regulated induction of IFITM3. Blood 2019, 133, 2013–2026. [Google Scholar] [CrossRef] [PubMed]
- D’Atri, L.P.; Etulain, J.; Rivadeneyra, L.; Lapponi, M.J.; Centurion, M.; Cheng, K.; Yin, H.; Schattner, M. Expression and functionality of Toll-like receptor 3 in the megakaryocytic lineage. J. Thromb. Haemost. 2015, 13, 839–850. [Google Scholar] [CrossRef]
- Anabel, A.S.; Eduardo, P.C.; Pedro Antonio, H.C.; Carlos, S.M.; Juana, N.M.; Honorio, T.A.; Nicolas, V.-S.; Roberto, A.-R.S. Human platelets express Toll-like receptor 3 and respond to poly I:C. Hum. Immunol. 2014, 75, 1244–1251. [Google Scholar] [CrossRef]
- Panigrahi, S.; Ma, Y.; Hong, L.; Gao, D.; West, X.Z.; Salomon, R.G.; Byzova, T.V.; Podrez, E.A. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circ. Res. 2013, 112, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Boilard, E.; Paré, G.; Rousseau, M.; Cloutier, N.; Dubuc, I.; Lévesque, T.; Borgeat, P.; Flamand, L. Influenza virus H1N1 activates platelets through FcγRIIA signaling and thrombin generation. Blood 2014, 123, 2854–2863. [Google Scholar] [CrossRef]
- Guo, L.; Feng, K.; Wang, Y.C.; Mei, J.J.; Ning, R.T.; Zheng, H.W.; Wang, J.J.; Worthen, G.S.; Wang, X.; Song, J.; et al. Critical role of CXCL4 in the lung pathogenesis of influenza (H1N1) respiratory infection. Mucosal Immunol. 2017, 10, 1529–1541. [Google Scholar] [CrossRef]
- Lê, V.B.; Schneider, J.G.; Boergeling, Y.; Berri, F.; Ducatez, M.; Guerin, J.L.; Adrian, I.; Errazuriz-Cerda, E.; Frasquilho, S.; Antunes, L.; et al. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am. J. Respir. Crit. Care Med. 2015, 191, 804–819. [Google Scholar] [CrossRef]
- Ashar, H.K.; Mueller, N.C.; Rudd, J.M.; Snider, T.A.; Achanta, M.; Prasanthi, M.; Ashar, H.K.; Mueller, N.C.; Rudd, J.M.; Snider, T.A.; et al. The Role of Extracellular Histones in Influenza Virus Pathogenesis. Am. J. Pathol. 2018, 188, 135–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangalmurti, N.S.; Friedman, J.L.; Wang, L.C.; Stolz, D.; Muthukumaran, G.; Siegel, D.L.; Schmidt, A.M.; Lee, J.; Albelda, S.M. The receptor for advanced glycation end products mediates lung endothelial activation by RBCs. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L250–L263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frommhold, D.; Kamphues, A.; Hepper, I.; Pruenster, M.; Lukić, I.K.; Socher, I.; Zablotskaya, V.; Buschmann, K.; Lange-Sperandio, B.; Schymeinsky, J.; et al. RAGE and ICAM-1 cooperate in mediating leukocyte recruitment during acute inflammation in vivo. Blood 2010, 116, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating histones are mediators of trauma-associated lung injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [Green Version]
- Wygrecka, M.; Kosanovic, D.; Wujak, L.; Reppe, K.; Henneke, I.; Frey, H.; Didiasova, M.; Kwapiszewska, G.; Marsh, L.M.; Baal, N.; et al. Antihistone properties of C1 esterase inhibitor protect against lung injury. Am. J. Respir. Crit. Care Med. 2017, 196, 186–199. [Google Scholar] [CrossRef]
- McVey, M.; Tabuchi, A.; Kuebler, W.M. Microparticles and acute lung injury. Physiol. Lung Cell Mol. Physiol. 2012, 303, L364–L381. [Google Scholar] [CrossRef] [Green Version]
- Chaar, V.; Romana, M.; Tripette, J.; Broquere, C.; Huisse, M.G.; Hue, O.; Hardy-Dessources, M.D.; Connes, P. Effect of strenuous physical exercise on circulating cell-derived microparticles. Clin. Hemorheol. Microcirc. 2011, 47, 15–25. [Google Scholar] [CrossRef]
- Dignat-George, F.; Boulanger, C.M. The many faces of endothelial microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 27–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abid Hussein, M.N.; Böing, A.N.; Biró, É.; Hoek, F.J.; Vogel, G.M.T.; Meuleman, D.G.; Sturk, A.; Nieuwland, R. Phospholipid composition of in vitro endothelial microparticles and their in vivo thrombogenic properties. Thromb. Res. 2008, 121, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, F.; Roux, V.; Anfosso, F.; Camoin, L.; Sampol, J.; Dignat-George, F. Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood 2002, 99, 3962–3970. [Google Scholar] [CrossRef] [PubMed]
- Del Conde, I.; Shrimpton, C.N.; Thiagarajan, P.; López, J.A. Tissue-factor-bearing microvesicles arise from lipid rafts and fuse with activated platelets to initiate coagulation. Blood 2005, 106, 1604–1611. [Google Scholar] [CrossRef]
- Wang, M.; Fu, Y.; Xu, L.; Xiao, L.; Yue, Y.; Liu, S.; Huang, Q.; Li, Y. Diagnostic value of platelet-derived microparticles in pulmonary thromboembolism: A population-based study. Exp. Ther. Med. 2018, 16, 3099–3106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucciarelli, P.; Martinelli, I.; Artoni, A.; Passamonti, S.M.; Previtali, E.; Merati, G.; Tripodi, A.; Mannucci, P.M. Circulating microparticles and risk of venous thromboembolism. Thromb. Res. 2012, 129, 591–597. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porembskaya, O.; Toropova, Y.; Tomson, V.; Lobastov, K.; Laberko, L.; Kravchuk, V.; Saiganov, S.; Brill, A. Pulmonary Artery Thrombosis: A Diagnosis That Strives for Its Independence. Int. J. Mol. Sci. 2020, 21, 5086. https://doi.org/10.3390/ijms21145086
Porembskaya O, Toropova Y, Tomson V, Lobastov K, Laberko L, Kravchuk V, Saiganov S, Brill A. Pulmonary Artery Thrombosis: A Diagnosis That Strives for Its Independence. International Journal of Molecular Sciences. 2020; 21(14):5086. https://doi.org/10.3390/ijms21145086
Chicago/Turabian StylePorembskaya, Olga, Yana Toropova, Vladimir Tomson, Kirill Lobastov, Leonid Laberko, Viacheslav Kravchuk, Sergey Saiganov, and Alexander Brill. 2020. "Pulmonary Artery Thrombosis: A Diagnosis That Strives for Its Independence" International Journal of Molecular Sciences 21, no. 14: 5086. https://doi.org/10.3390/ijms21145086
APA StylePorembskaya, O., Toropova, Y., Tomson, V., Lobastov, K., Laberko, L., Kravchuk, V., Saiganov, S., & Brill, A. (2020). Pulmonary Artery Thrombosis: A Diagnosis That Strives for Its Independence. International Journal of Molecular Sciences, 21(14), 5086. https://doi.org/10.3390/ijms21145086