Inclusion Formation and Toxicity of the ALS Protein RGNEF and Its Association with the Microtubule Network


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
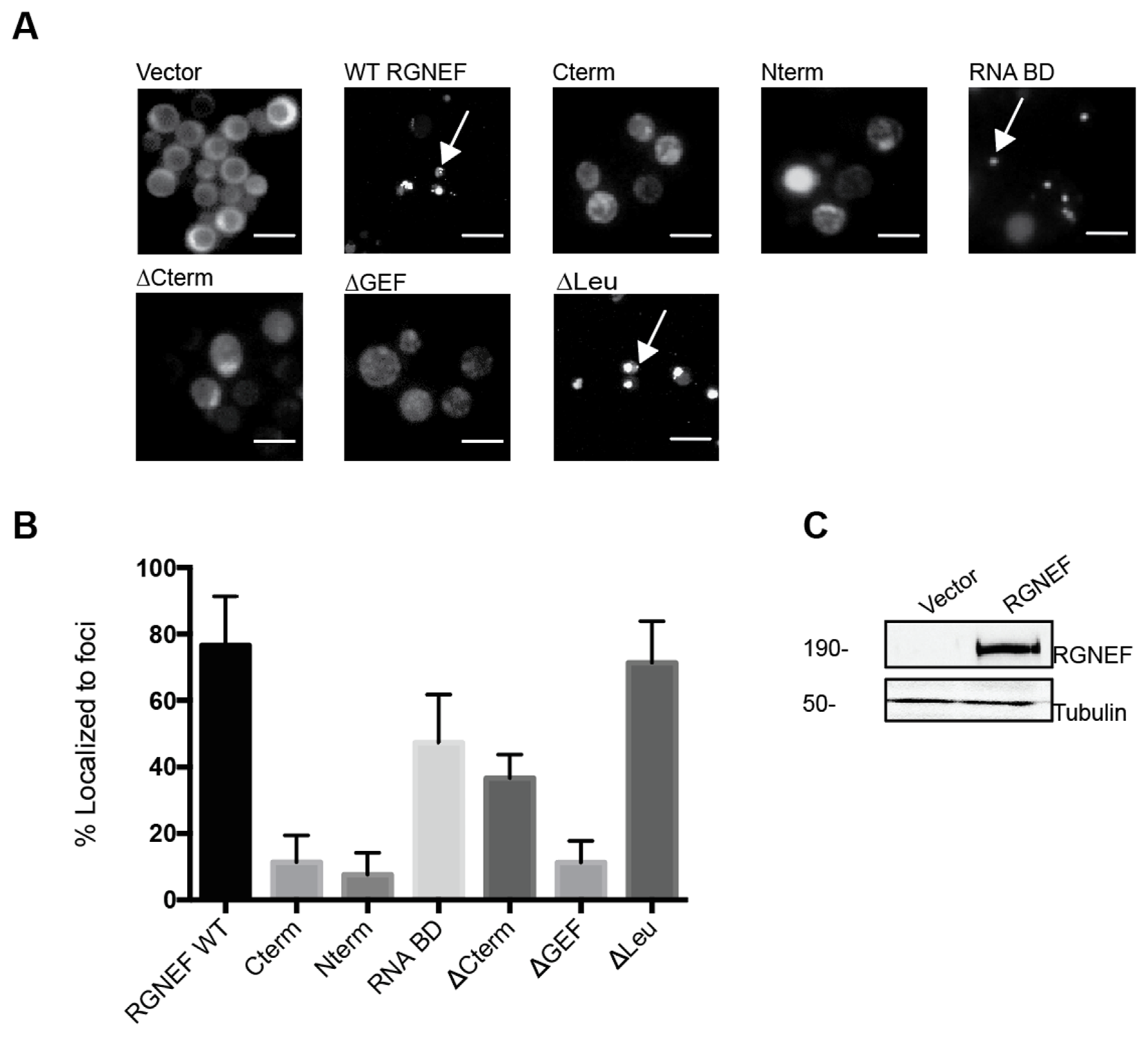
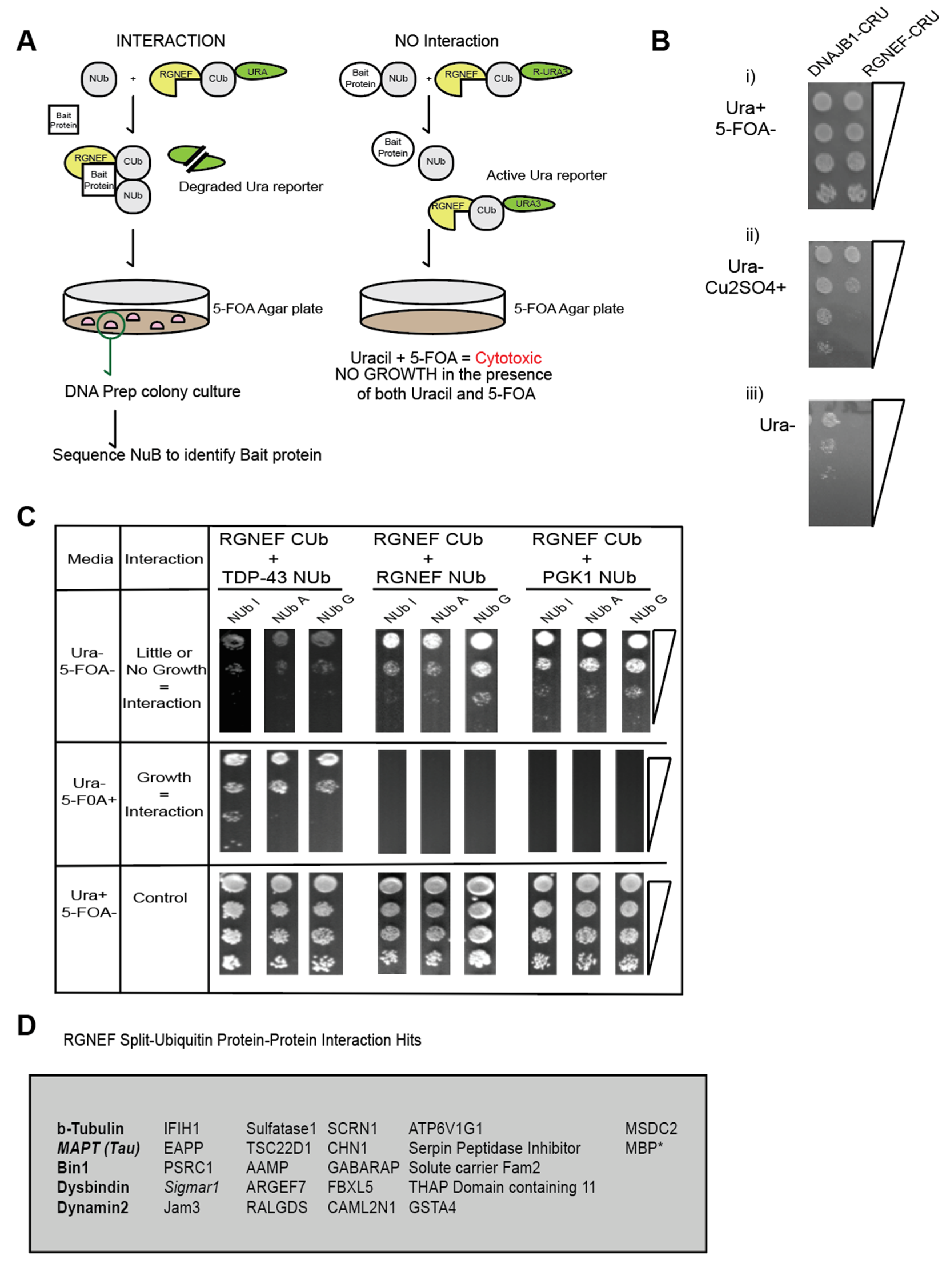
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Yeast Transformations
4.2.2. Spotting Assays
4.2.3. Quantification
4.2.4. Split Ubiquitin Interaction Assay
4.2.5. Microscopy
4.2.6. Mammalian Cell Culture
4.2.7. Immunofluorescence Microscopy
5. Summary Statement
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hardiman, O.; Chalabi, A.; Chio, A.; Corr, M.E.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, H.L. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primers 2017, 3, 17071. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener 2013, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, E.; Rajasekaran, R. A Systematic and Comprehensive Review on Disease-Causing Genes in Amyotrophic Lateral Sclerosis. J. Mol. Neurosci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Fryer, J.D. A matter of balance. eLife 2018, 7, e40034. [Google Scholar] [CrossRef]
- Parakh, S.; Atkin, J.D. Protein folding alterations in amyotrophic lateral sclerosis. Brain Res. 2016, 1648, 633–649. [Google Scholar] [CrossRef]
- Soto, C.; Estrada, L.D. Protein misfolding and neurodegeneration. Arch. Neurol. 2008, 65, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Saberi, S.; Stauffer, J.E.; Schulte, D.J.; Ravits, J. Neuropathology of Amyotrophic Lateral Sclerosis and Its Variants. Neurol. Clin. 2015, 33, 855–876. [Google Scholar] [CrossRef] [Green Version]
- Kryndushkin, D.; Shewmaker, F. Modeling ALS and FTLD proteinopathies in yeast: An efficient approach for studying protein aggregation and toxicity. Prion 2011, 5, 250–257. [Google Scholar] [CrossRef] [Green Version]
- Redler, R.L.; Dokholyan, N.V. The complex molecular biology of amyotrophic lateral sclerosis (ALS). Prog. Mol. Biol. Transl. Sci. 2012, 107, 215–262. [Google Scholar]
- Patel, A.; Lee, H.O.; Jawerth, L.; Drechsel, D.; Hyman, A.A. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, J.R.; Greenamyre, J.T. The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol. Sci. 2011, 124, 225–250. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Mead, R.J.; Shaw, P.J. Oxidative stress in ALS: A mechanism of neurodegeneration and a therapeutic target. Biochim. Biophys. Acta 2006, 1762, 1051–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikis, E.A.; Gidalevitz, T.; Morimoto, R.I. Protein homeostasis in models of aging and age-related conformational disease. Adv. Exp. Med. Biol. 2010, 694, 138–159. [Google Scholar]
- Volkening, K.; Leystra-Lantz, C.; Strong, M.J. Human low molecular weight neurofilament (NFL) mRNA interacts with a predicted p190 RhoGEF homologue (RGNEF) in humans. Amyotroph. Lateral Scler. 2010, 11, 97–103. [Google Scholar] [CrossRef]
- Keller, B.A.; Volkening, K.; Droppelmann, C.A.; Ang, L.C.; Rademakers, R.; Strong, M.J. Co-aggregation of RNA binding proteins in ALS spinal motor neurons: Evidence of a common pathogenic mechanism. Acta Neuropathol. 2012, 124, 733–747. [Google Scholar] [CrossRef]
- Droppelmann, C.; Wang, J.; Campos-Melo, D.; Keller, B.; Volkening, K.; Hegele, R.A.; Strong, M.J. Detection of a novel frameshift mutation and regions with homozygosis within ARHGEF28 gene in familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 444–451. [Google Scholar] [CrossRef]
- Tavolieri, M.V.; Droppelmann, C.A.; Campos-Melo, D.; Volkening, K.; Strong, M.J. A novel overlapping NLS/NES region within the PH domain of Rho Guanine Nucleotide Exchange Factor (RGNEF) regulates its nuclear-cytoplasmic localization. Eur. J. Cell Biol. 2019, 98, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Kleinschmidt, E.G.; Miller, N.L.G.; Ozmadenci, D.; Tancioni, I.; Osterman, C.D.; Barrie, A.M.; Taylor, C.N.; Ye, A.; Jiang, S.; Connolly, D.C.; et al. Rgnef promotes ovarian tumor progression and confers protection from oxidative stress. Oncogene 2019, 38, 6323–6337. [Google Scholar] [CrossRef]
- Cheung, K.; Droppelmann, C.A.; MacLellan, A.; Cameron, A.; Withers, B.; Campos-Melo, D.; Volkening, K.; Strong, M.J. Rho guanine nucleotide exchange factor (RGNEF) is a prosurvival factor under stress conditions. Mol. Cell. Neurosci. 2017, 82, 88–95. [Google Scholar] [CrossRef]
- Wu, J.; Zhai, J.; Lin, H.; Nie, Z.; Ge, W.W.; Garcia-Bermejo, L.; Muschel, R.J.; Schlaepfer, W.W.; Canete-Soler, R. Cytoplasmic retention sites in p190 RhoGEF confer anti-apoptotic activity to an EGFP-tagged protein. Brain Res. Mol. Brain Res. 2003, 117, 27–38. [Google Scholar] [CrossRef]
- Miller, N.L.; Lawson, C.; Chen, X.L.; Lim, S.T.; Schlaepfer, D.D. Rgnef (p190 RhoGEF) knockout inhibits RhoA activity, focal adhesion establishment, and cell motility downstream of integrins. PLoS ONE 2012, 7, e37830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.G.; Nam, J.O.; Miller, N.L.; Tanjoni, I.; Walsh, C.; Shi, L.; Kim, L.; Chen, X.L.; Tomar, A.; Lim, S.T.; et al. RhoGEF (Rgnef) promotes colon carcinoma tumor progression via interaction with focal adhesion kinase. Cancer Res. 2011, 71, 360–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Tang, L.; Chen, L.; Zhang, B.; Deng, P.; Wang, J.; Yang, Y.; Liu, R.; Yang, Y.; Ye, S.; et al. ARHGEF28 gene exon 6/intron 6 junction mutations in Chinese amyotrophic lateral sclerosis cohort. Amyotroph. Lateral Scler. Front. Degener. 2014, 15, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Farhan, S.M.K.; Gendron, T.F.; Petrucelli, L.; Hegele, R.A.; Strong, M.J. ARHGEF28 p.Lys280 Metfs40 Ter in an amyotrophic lateral sclerosis family with a C9 orf72 expansion. Neurol. Genet. 2017, 3, e190. [Google Scholar] [CrossRef] [Green Version]
- Droppelmann, C.A.; Keller, B.A.; Campos-Melo, D.; Volkening, K.; Strong, M.J. Rho guanine nucleotide exchange factor is an NFL mRNA destabilizing factor that forms cytoplasmic inclusions in amyotrophic lateral sclerosis. Neurobiol. Aging 2013, 34, 248–262. [Google Scholar] [CrossRef]
- Schmid, W. The micronucleus test for cytogenetic analysis. In Chemical Mutagens; Springer: Boston, MA, USA, 1976; pp. 31–53. [Google Scholar]
- Kirsch-Volders, M.; Elhajouji, A.; Cundari, E.; Van Hummelen, P. The in vitro micronucleus test: A multi-endpoint assay to detect simultaneously mitotic delay, apoptosis, chromosome breakage, chromosome loss and non-disjunction. Mutat. Res. 1997, 392, 19–30. [Google Scholar] [CrossRef]
- Droppelmann, C.A.; Campos-Melo, D.; Moszczynski, A.J.; Amzil, H.; Strong, M.J. TDP-43 aggregation inside micronuclei reveals a potential mechanism for protein inclusion formation in ALS. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- Johnsson, N.; Varshavsky, A. Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. USA 1994, 91, 10340–10344. [Google Scholar] [CrossRef] [Green Version]
- Lehming, N. Analysis of protein-protein proximities using the split-ubiquitin system. Brief Funct. Genomic. Proteomic 2002, 1, 230–238. [Google Scholar] [CrossRef]
- Muller, J.; Johnsson, N. Split-ubiquitin and the split-protein sensors: Chessman for the endgame. Chembiochem 2008, 9, 2029–2038. [Google Scholar] [CrossRef]
- Van Horck, F.P.; Ahmadian, M.R.; Haeusler, L.C.; Moolenaar, W.H.; Kranenburg, O. Characterization of p190 RhoGEF, a RhoA-specific guanine nucleotide exchange factor that interacts with microtubules. J. Biol. Chem. 2001, 276, 4948–4956. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.K.; Heller, K.K.; Frisèn, L.; Wallack, D.L.; Loayza, D.; Gammie, A.E.; Rose, M.D. The kinesin-related proteins, Kip2 p and Kip3 p, function differently in nuclear migration in yeast. Mol. Biol. Cell 1998, 9, 2051–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuyler, S.C.; Liu, J.Y.; Pellman, D. The molecular function of Ase1 p: Evidence for a MAP-dependent midzone-specific spindle matrix. Microtubule-associated proteins. J. Cell Biol. 2003, 160, 517–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allingham, J.S.; Sproul, L.R.; Rayment, I.; Gilbert, S.P. Vik1 modulates microtubule-Kar3 interactions through a motor domain that lacks an active site. Cell 2007, 128, 1161–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Hart, M.J.; Shinjo, K.; Evans, T.; Bender, A.; Cerione, R.A. Biochemical comparisons of the Saccharomyces cerevisiae Bem2 and Bem3 proteins. Delineation of a limit Cdc42 GTPase-activating protein domain. J. Biol. Chem. 1993, 268, 24629–24634. [Google Scholar]
- Leung, Y.Y.; Yao Hui, L.L.; Kraus, V.B. Colchicine–Update on mechanisms of action and therapeutic uses. Semin. Arthritis Rheum. 2015, 45, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Hoebeke, J.; Van Nijen, G.; De Brabander, M. Interaction of oncodazole (R 17934), a new antitumoral drug, with rat brain tubulin. Biochem. Biophys. Res. Commun. 1976, 69, 319–324. [Google Scholar] [CrossRef]
- Masia-Balague, M.; Izquierdo, I.; Garrido, G.; Cordomí, A.; Pérez-Benito, L.; Miller, N.L.G.; Schlaepfer, D.D.; Gigoux, V.; Aragay, A.M. Gastrin-stimulated Galpha13 Activation of Rgnef Protein (ArhGEF28) in DLD-1 Colon Carcinoma Cells. J. Biol. Chem. 2015, 290, 15197–15209. [Google Scholar] [CrossRef] [Green Version]
- Droppelmann, C.A.; Campos-Melo, D.; Volkening, K.; Strong, M.J. The emerging role of guanine nucleotide exchange factors in ALS and other neurodegenerative diseases. Front Cell Neurosci. 2014, 8, 282. [Google Scholar] [CrossRef] [Green Version]
- Balikova, I.; Devriendt, K.; Fryns, J.P.; Vermeesch, J.R. FOXD1 Duplication Causes Branchial Defects and Interacts with the TFAP2 A Gene Implicated in the Branchio-Oculo-Facial Syndrome in Causing Eye Effects in Zebrafish. Mol. Syndr. 2010, 1, 255–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitler, A.D.; Shorter, J. RNA-binding proteins with prion-like domains in ALS and FTLD-U. Prion 2011, 5, 179–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Gregorio, S.E.; Duennwald, M.L. ALS Yeast Models-Past Success Stories and New Opportunities. Front. Mol. Neurosci. 2018, 11, 394. [Google Scholar] [CrossRef] [PubMed]
- Van Ham, T.J.; Breitling, R.; Swertz, M.A.; Nollen, E.A.A. Neurodegenerative diseases: Lessons from genome-wide screens in small model organisms. EMBO Mol. Med. 2009, 1, 360–370. [Google Scholar] [CrossRef]
- Tenreiro, S.; Outeiro, T.F. Simple is good: Yeast models of neurodegeneration. FEMS Yeast Res. 2010, 10, 970–979. [Google Scholar] [CrossRef]
- Tenreiro, S.; Franssens, V.; Winderickx, J.; Outeiro, T.F. Yeast models of Parkinson’s disease-associated molecular pathologies. Curr. Opin. Genet. Dev. 2017, 44, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Munder, M.C.; Alberti, S.; Outeiro, T.F. Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. J. Neurochem. 2013, 127, 438–452. [Google Scholar] [CrossRef]
- Dreser, A.; Vollrath, J.T.; Sechi, A.; Johann, S.; Roos, A.; Yamoah, A.; Katona, I.; Bohlega, S.; Wiemuth, D.; Tian, Y.; et al. The ALS-linked E102 Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ. 2017, 24, 1655–1671. [Google Scholar] [CrossRef]
- Griffin, J.W.; Watson, D.F. Axonal transport in neurological disease. Ann. Neurol. 1988, 23, 3–13. [Google Scholar] [CrossRef]
- Sleigh, J.N.; Rossor, A.M.; Fellows, A.D.; Tosolini, A.P.; Schiavo, G. Axonal transport and neurological disease. Nat. Rev. Neurol. 2019, 15, 691–703. [Google Scholar] [CrossRef]
- Smith, B.N.; Ticozzi, N.; Fallini, C.; Gkazi, A.S.; Topp, S.; Kenna, K.P.; Scotter, E.L.; Kost, J.; Keagle, P.; Miller, J.W.; et al. Exome-wide rare variant analysis identifies TUBA4 A mutations associated with familial ALS. Neuron 2014, 84, 324–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinhandl, K.; Winkler, M.; Glieder, A.; Camattari, A. Carbon source dependent promoters in yeasts. Microb. Cell Fact. 2014, 13, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, B.J.; Rothstein, R. Elevated recombination rates in transcriptionally active DNA. Cell 1989, 56, 619–630. [Google Scholar] [CrossRef]
- Katzen, F. Gateway((R)) recombinational cloning: A biological operating system. Expert Opin. Drug. Discov. 2007, 2, 571–589. [Google Scholar] [CrossRef]
- Gietz, R.D. Yeast transformation by the LiAc/SS carrier DNA/PEG method. Methods Mol. Biol. 2014, 1163, 33–44. [Google Scholar]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Gregorio, S.E.; Volkening, K.; Strong, M.J.; Duennwald, M.L. Inclusion Formation and Toxicity of the ALS Protein RGNEF and Its Association with the Microtubule Network. Int. J. Mol. Sci. 2020, 21, 5597. https://doi.org/10.3390/ijms21165597
Di Gregorio SE, Volkening K, Strong MJ, Duennwald ML. Inclusion Formation and Toxicity of the ALS Protein RGNEF and Its Association with the Microtubule Network. International Journal of Molecular Sciences. 2020; 21(16):5597. https://doi.org/10.3390/ijms21165597
Chicago/Turabian StyleDi Gregorio, Sonja E., Kathryn Volkening, Michael J. Strong, and Martin L. Duennwald. 2020. "Inclusion Formation and Toxicity of the ALS Protein RGNEF and Its Association with the Microtubule Network" International Journal of Molecular Sciences 21, no. 16: 5597. https://doi.org/10.3390/ijms21165597
APA StyleDi Gregorio, S. E., Volkening, K., Strong, M. J., & Duennwald, M. L. (2020). Inclusion Formation and Toxicity of the ALS Protein RGNEF and Its Association with the Microtubule Network. International Journal of Molecular Sciences, 21(16), 5597. https://doi.org/10.3390/ijms21165597