Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells

 ,
,  , ,
, ,
Abstract
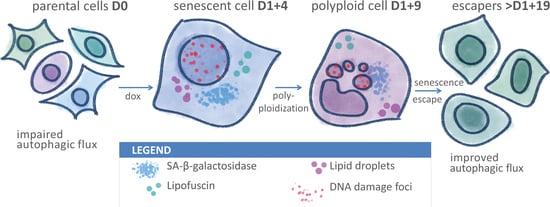
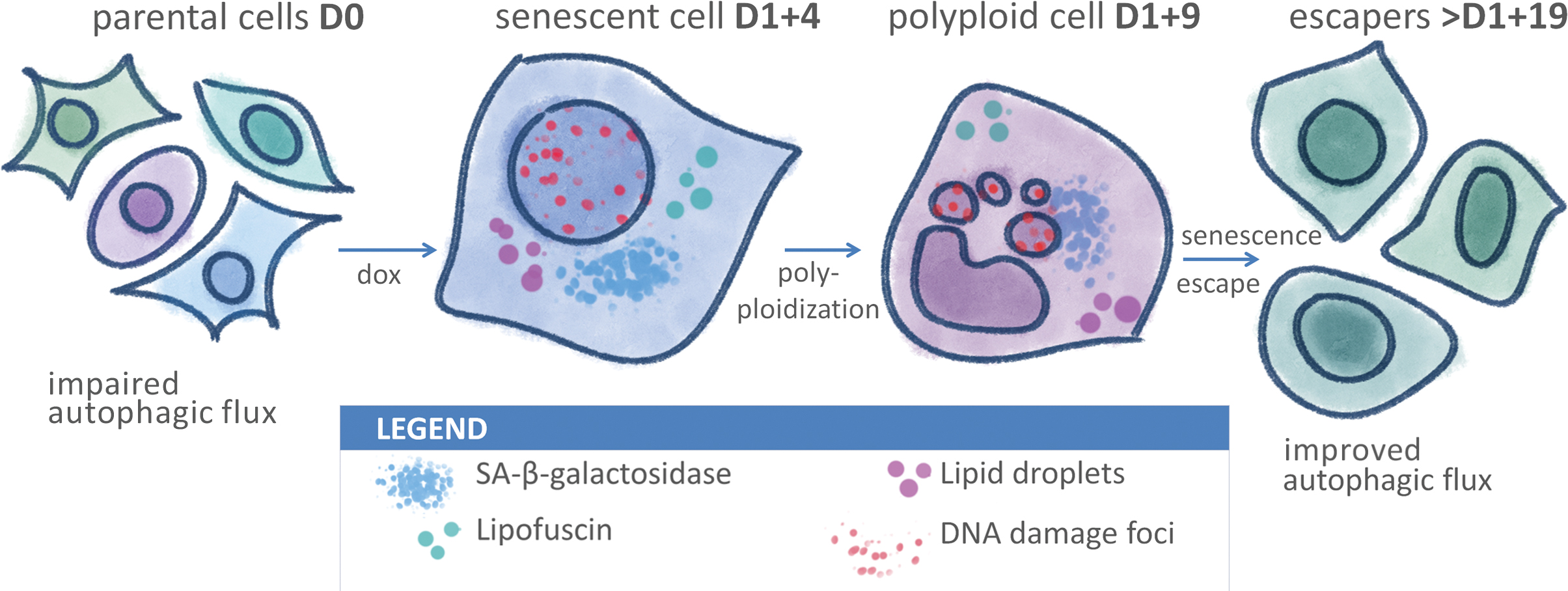
:
1. Introduction
2. Results
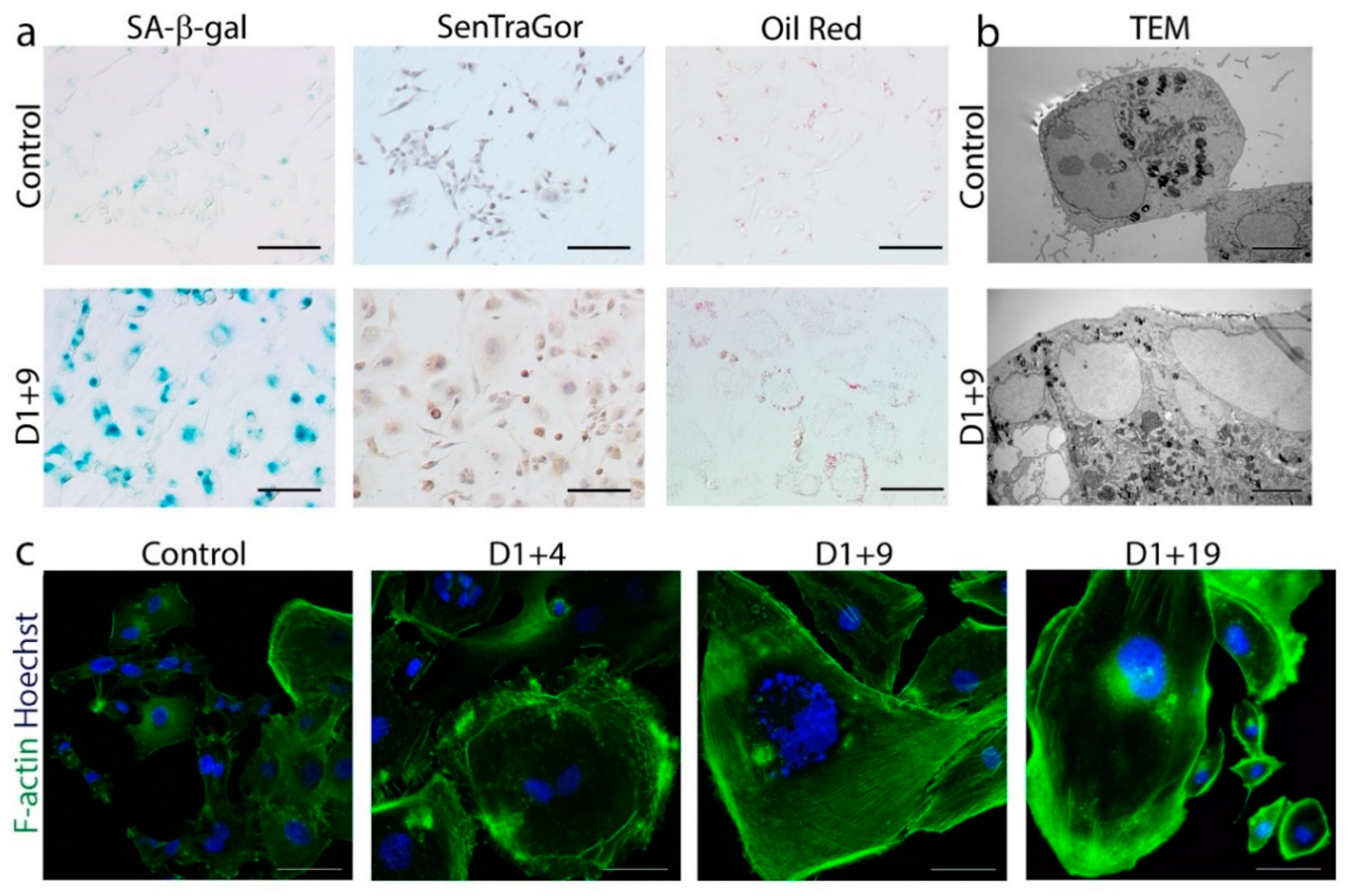
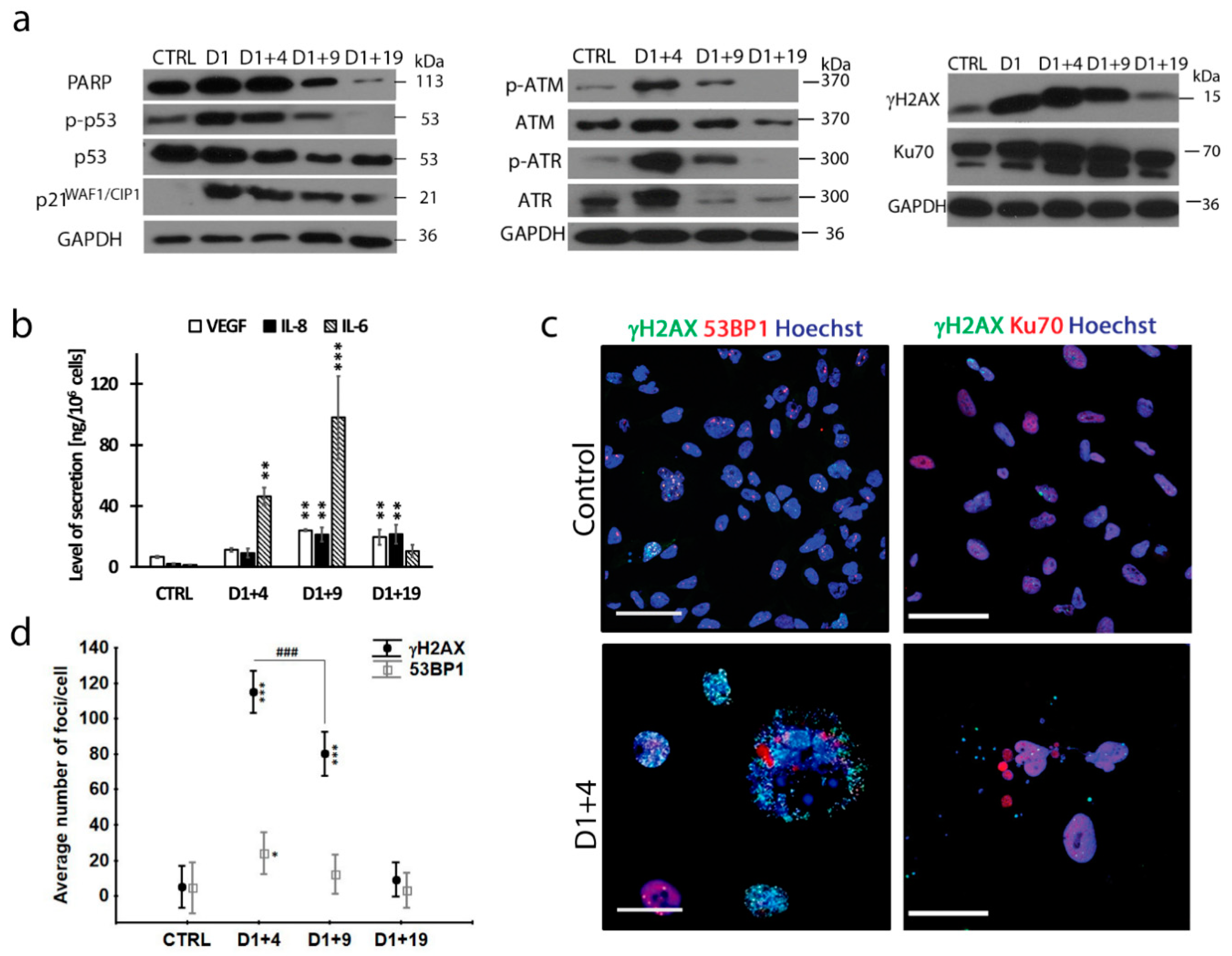
2.1. Doxorubicin-Induced Senescence of MDA-MB-231 Cells
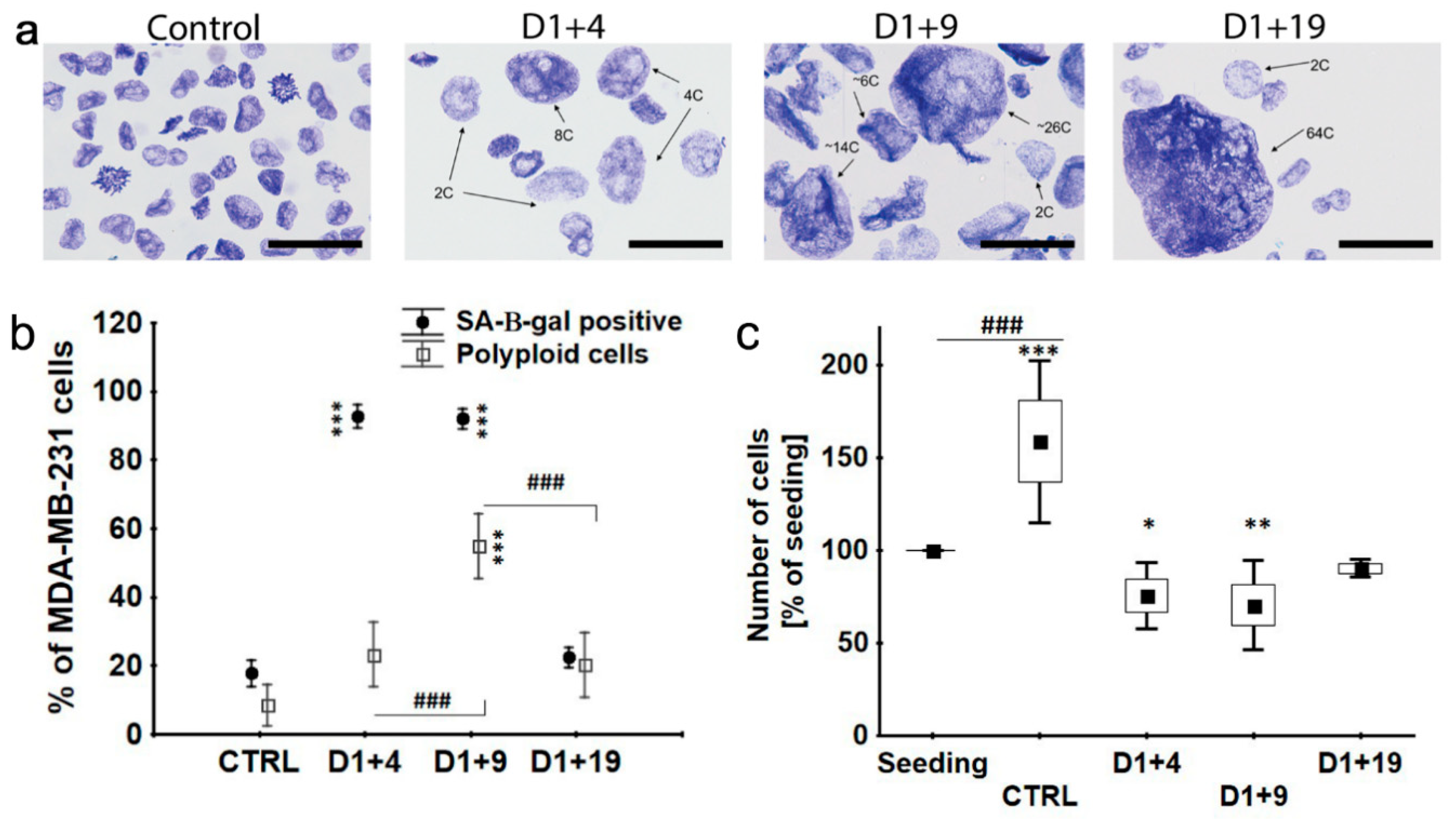
2.2. Transient Polyploidization of Doxorubicin-Treated MDA-MB-231 Cells
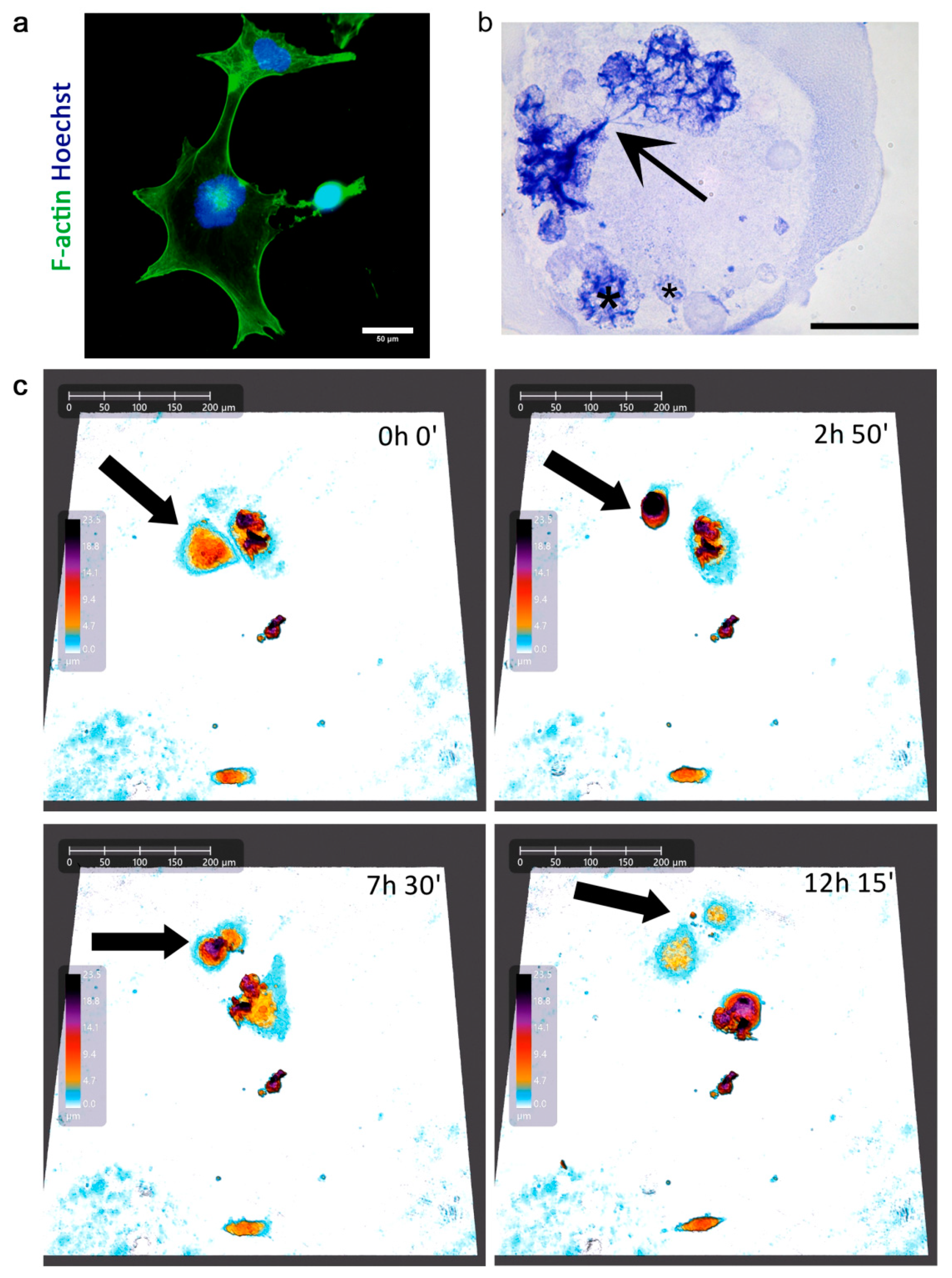
2.3. Atypical Divisions of Polyploid/Senescent Cells
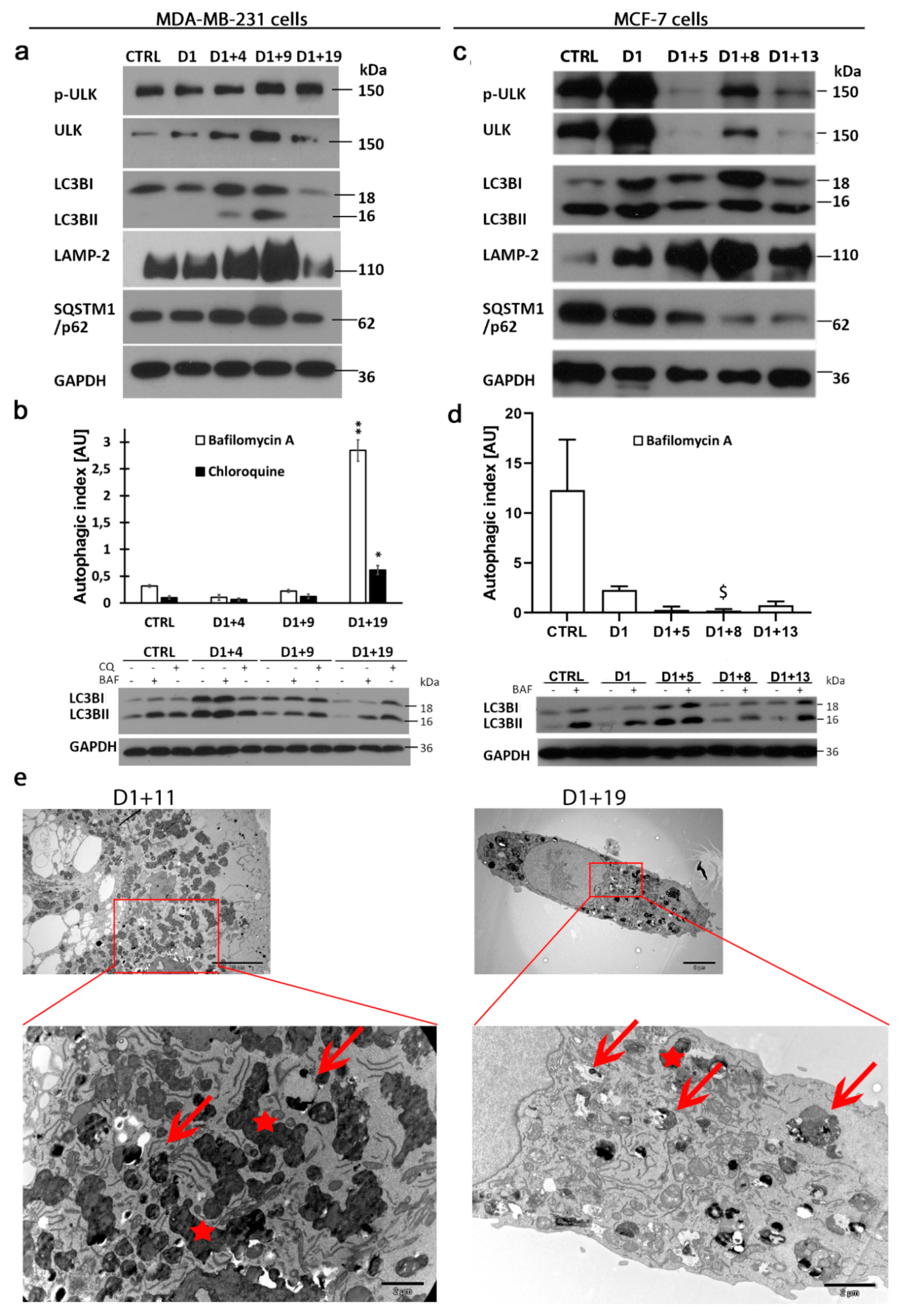
2.4. Resumption of Autophagic Flux in Escape from Dox-Induced Senescence
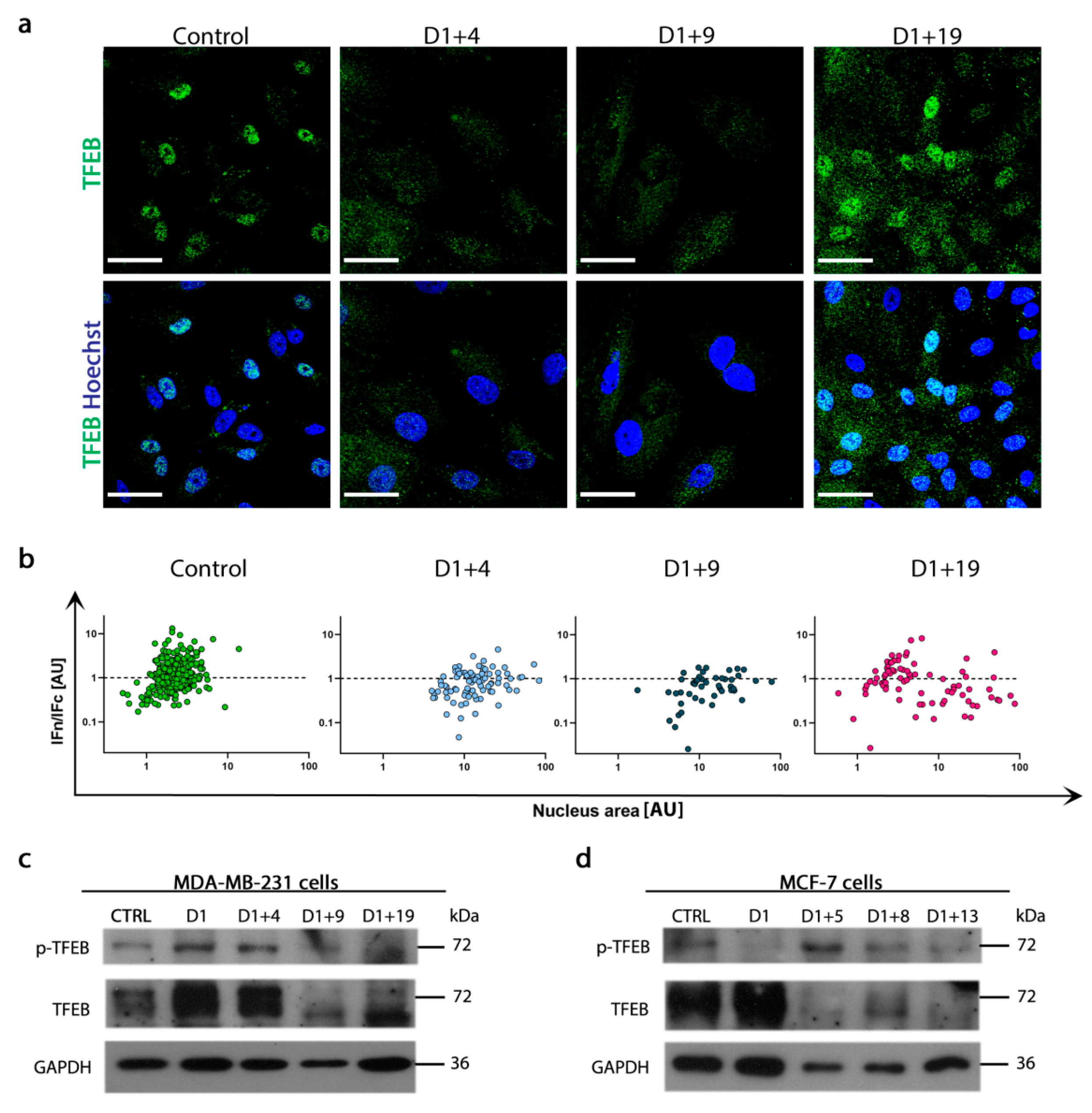
2.5. Resumption of Autophagic Flux Is Accompanied by Translocation of Transcription Factor EB (TFEB) to Nucleus
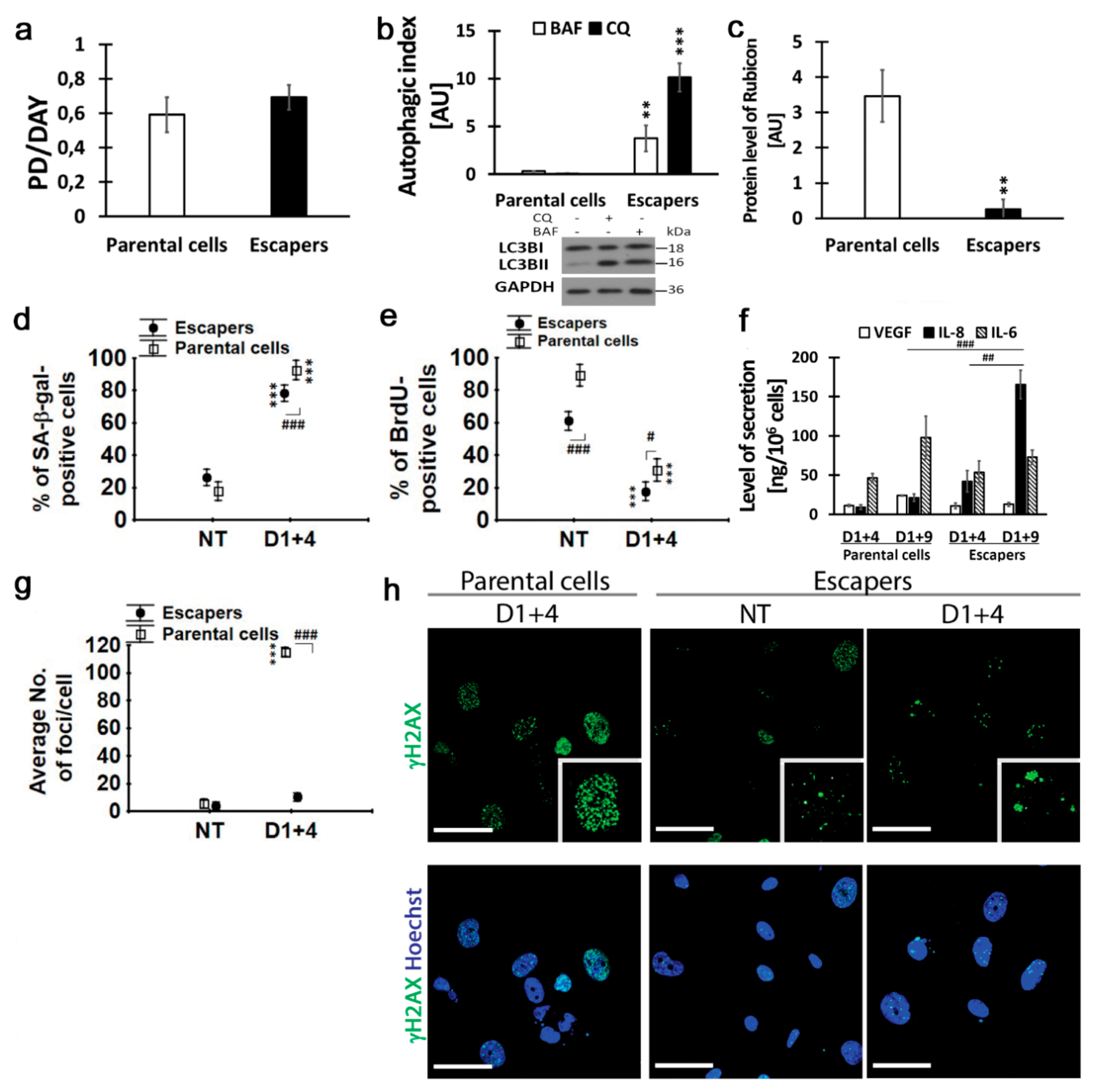
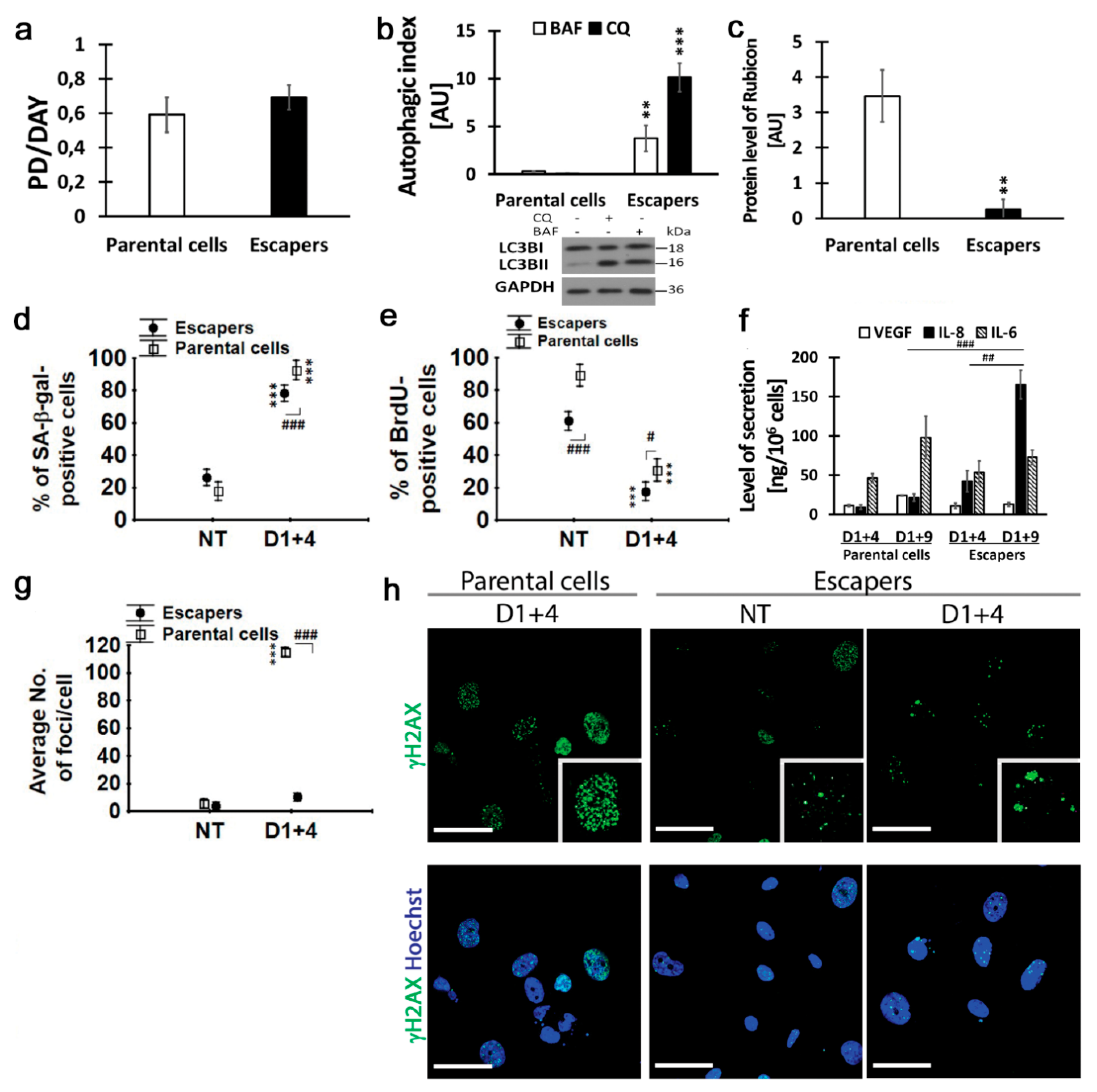
2.6. Escapers from Dox-Induced Senescence Were Characterized by Functional Autophagic Flux and Senescence Capability
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Western Blotting Analysis
4.3. Immunocytochemistry
4.4. Detection of Senescence-Associated β-galactosidase
4.5. Lipofuscin Staining
4.6. Lipid Staining
4.7. Cytokine Measurement
4.8. DNA Content Evaluation by Toluidine Blue Staining
4.9. Bromodeoxyuridine Incorporation Assay
4.10. Cell Size and DNA Index Estimation by Flow Cytometry Analysis
4.11. TEM Sample Preparation
4.12. Population Doubling (PD)
4.13. Double Staining
4.14. Autophagic Index
4.15. Live Imaging
4.16. Image Acquisition
4.17. Quantitative Analysis
4.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AI | Autophagy Index |
| DDR | DNA Damage Response |
| DOX | Doxorubicin |
| DSBs | Double-Strand Breaks |
| HR | Homologous Recombination |
| NHEJ | Non-Homologous End Joining |
| SA-β-gal | Senescence-Associated β-galactosidase |
| SASP | Senescence-Associated Secretory Phenotype |
| TIP | Therapy-Induced Polyploidy |
| TIS | Therapy -Induced Senescence |
References
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. JNCI J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-senescence therapy for cancer treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Chitikova, Z.V.; Gordeev, S.A.; Bykova, T.V.; Zubova, S.G.; Pospelov, V.A.; Pospelova, T.V. Sustained activation of DNA damage response in irradiated apoptosis-resistant cells induces reversible senescence associated with mTOR downregulation and expression of stem cell markers. Cell Cycle 2014, 13, 1424–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Cragg, M.S. Three steps to the immortality of cancer cells: Senescence, polyploidy and self-renewal. Cancer Cell Int. 2013, 13, 92. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Mansilla, S.; Bataller, M.; Portugal, J. A nuclear budding mechanism in transiently arrested cells generates drug-sensitive and drug-resistant cells. Biochem. Pharm. 2009, 78, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Mosieniak, G.; Sikora, E. Polyploidy: The link between senescence and cancer. Curr. Pharm. Des. 2010, 16, 734–740. [Google Scholar] [CrossRef] [Green Version]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef]
- Sabisz, M.; Skladanowski, A. Cancer stem cells and escape from drug-induced premature senescence in human lung tumor cells: Implications for drug resistance and in vitro drug screening models. Cell Cycle 2009, 8, 3208–3217. [Google Scholar] [CrossRef] [Green Version]
- Sliwinska, M.A.; Mosieniak, G.; Wolanin, K.; Babik, A.; Piwocka, K.; Magalska, A.; Szczepanowska, J.; Fronk, J.; Sikora, E. Induction of senescence with doxorubicin leads to increased genomic instability of HCT116 cells. Mech. Ageing Dev. 2009, 130, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wu, P.C.; Dong, D.Z.; Ivanova, I.; Chu, E.; Zeliadt, S.; Vesselle, H.; Wu, D.Y. Polyploidy road to therapy-induced cellular senescence and escape. Int. J. Cancer 2013, 132, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Czarnecka, J.; Kominek, A.; Barszcz, K.; Bernas, T.; Piwocka, K.; Kaminska, B. Some chemotherapeutics-treated colon cancer cells display a specific phenotype being a combination of stem-like and senescent cell features. Cancer Biol. Ther. 2018, 19, 63–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakradeo, S.; Elmore, L.W.; Gewirtz, D.A. Is senescence reversible? Curr. Drug Targets 2016, 17, 460–466. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Nakamura, S.; Oba, M.; Suzuki, M.; Takahashi, A.; Yamamuro, T.; Fujiwara, M.; Ikenaka, K.; Minami, S.; Tabata, N.; Yamamoto, K.; et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun. 2019, 10, 847. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Passos, J.F. Mitochondria: Are they causal players in cellular senescence? Biochim. Biophys. Acta 2015, 1847, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Bielak-Zmijewska, A.; Mosieniak, G.; Sikora, E. Is DNA damage indispensable for stress-induced senescence? Mech. Ageing Dev. 2018, 170, 13–21. [Google Scholar] [CrossRef]
- Gewirtz, D.A. Autophagy and senescence in cancer therapy. J. Cell. Physiol. 2014, 229, 6–9. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Salmina, K.; Cragg, M.S. Accelerated Senescence of Cancer Stem Cells: A Failure to Thrive or a Route to Survival. In Senescence Physiology or Pathology; Dorszewska, J., Kozubsk, W., Eds.; IntechOpen: London, UK, 2017; pp. 45–64. Available online: https://www.intechopen.com/books/senescence-physiology-or-pathology/accelerated-senescence-of-cancer-stem-cells-a-failure-to-thrive-or-a-route-to-survival- (accessed on 30 August 2017).
- Erenpreisa, J.; Huna, A.; Salmina, K.; Jackson, T.R.; Cragg, M.S. Macroautophagy-aided elimination of chromatin: Sorting of waste, sorting of fate? Autophagy 2012, 8, 1877–1881. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome formation in mammalian cells. Cell Struct. Funct. 2002, 27, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagelkerke, A.; Bussink, J.; Geurts-Moespot, A.; Sweep, F.C.; Span, P.N. Therapeutic targeting of autophagy in cancer. Part II: Pharmacological modulation of treatment-induced autophagy. Semin. Cancer Biol. 2015, 31, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. Autophagy and senescence: A partnership in search of definition. Autophagy 2013, 9, 808–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, J. Animal Models for Studying Prevention and Treatment of Breast Cancer. In Animal Models for the Study of Human Disease; American Press: Cambridge, MA, USA, 2013; pp. 997–1018. [Google Scholar] [CrossRef]
- Leroy, B.; Girard, L.; Hollestelle, A.; Minna, J.D.; Gazdar, A.F.; Soussi, T. Analysis of TP53 mutation status in human cancer cell lines: A reassessment. Hum. Mutat. 2014, 35, 756–765. [Google Scholar] [CrossRef]
- Soule, H.D.; Vazguez, J.; Long, A.; Albert, S.; Brennan, M. A human cell line from a pleural effusion derived from a breast carcinoma. J. Natl. Cancer Inst. 1973, 51, 1409–1416. [Google Scholar] [CrossRef]
- Staniak, K. The role of lisosomes and autophagy in cancer cell response to tacrin-melatonin heterodimer. PhD Def. 2020, in press. [Google Scholar]
- Salmina, K.; Bojko, A.; Inashkina, I.; Staniak, K.; Dudkowska, M.; Podlesniy, P.; Rumnieks, F.; Vainshelbaum, N.M.; Pjanova, D.; Sikora, E.; et al. “Mitotic Slippage” and Extranuclear DNA in Cancer Chemoresistance: A Focus on Telomeres. Int. J. Mol. Sci. 2020, 21, 2779. [Google Scholar] [CrossRef] [Green Version]
- Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501. [Google Scholar] [CrossRef] [Green Version]
- Mosieniak, G.; Sliwinska, M.A.; Alster, O.; Strzeszewska, A.; Sunderland, P.; Piechota, M.; Was, H.; Sikora, E. Polyploidy Formation in Doxorubicin-Treated Cancer Cells Can Favor Escape from Senescence. Neoplasia 2015, 17, 882–893. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Georgakopoulou, E.A.; Tsimaratou, K.; Evangelou, K.; Fernandez Marcos, P.J.; Zoumpourlis, V.; Trougakos, I.P.; Kletsas, D.; Bartek, J.; Serrano, M.; Gorgoulis, V.G. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging 2013, 5, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.; Tchkonia, T.; Krüger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. 2019, 29, 1061–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Hartlerode, A.J.; Scully, R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 2009, 423, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Indiviglio, S.M.; Bertuch, A.A. Ku‘s essential role in keeping telomeres intact. Proc. Natl. Acad. Sci. USA 2009, 106, 12217–12218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmina, K.; Huna, A.; Kalejs, M.; Pjanova, D.; Scherthan, H.; Cragg, M.S.; Erenpreisa, J. The Cancer Aneuploidy Paradox: In the Light of Evolution. Genes 2019, 10, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Liu, J. The “life code”: A theory that unifies the human life cycle and the origin of human tumors. Semin. Cancer Biol. 2020, 60, 380–397. [Google Scholar] [CrossRef]
- Amend, S.R.; Torga, G.; Lin, K.C.; Kostecka, L.G.; de Marzo, A.; Austin, R.H.; Pienta, K.J. Polyploid giant cancer cells: Unrecognized actuators of tumorigenesis, metastasis, and resistance. Prostate 2019, 79, 1489–1497. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Murray, D. Roles of Polyploid/Multinucleated Giant Cancer Cells in Metastasis and Disease Relapse Following Anticancer Treatment. Cancers 2018, 10, 118. [Google Scholar] [CrossRef] [Green Version]
- Sikora, E.; Mosieniak, G.; Sliwinska, M.A. Morphological and Functional Characteristic of Senescent Cancer Cells. Curr. Drug Targets 2016, 17, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Erenpreisa, J.A.; Cragg, M.S.; Fringes, B.; Sharakhov, I.; Illidge, T.M. Release of mitotic descendants by giant cells from irradiated Burkitt’s lymphoma cell line. Cell Biol. Int. 2000, 24, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Illidge, T.M.; Cragg, M.S.; Fringes, B.; Olive, P.; Erenpreisa, J.A. Polyploid giant cells provide a survival mechanism for p53 mutant cells after DNA damage. Cell Biol. Int. 2000, 24, 621–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Jackson, T.R.; Vazquez-Martin, A.; Cragg, M.S. The “virgin birth“, polyploidy, and the origin of cancer. Oncoscience 2015, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartek, J.; Lukas, J.; Bartkova, J. DNA damage response as an anti-cancer barrier: Damage threshold and the concept of ‘conditional haploinsufficiency’. Cell Cycle 2007, 6, 2344–2347. [Google Scholar] [CrossRef] [Green Version]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [Green Version]
- Junk, D.J.; Vrba, L.; Watts, G.S.; Oshiro, M.M.; Martinez, J.D.; Futscher, B.W. Different mutant/wild-type p53 combinations cause a spectrum of increased invasive potential in nonmalignant immortalized human mammary epithelial cells. Neoplasia 2008, 10, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Strzeszewska, A.; Alster, O.; Mosieniak, G.; Ciolko, A.; Sikora, E. Insight into the role of PIKK family members and NF-small ka, CyrillicB in DNAdamage-induced senescence and senescence-associated secretory phenotype of colon cancer cells. Cell Death Dis. 2018, 9, 44. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.R.; Menck, C.F.M.; Leandro, G.S. Autophagy Roles in the Modulation of DNA Repair Pathways. Int. J. Mol. Sci. 2017, 18, 2351. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res. 2013, 750, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodarzi, A.A.; Jeggo, P.; Lobrich, M. The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax. DNA Repair 2010, 9, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.Q.; Zhong, Z.H.; Henson, J.D.; Reddel, R.R. Identification of candidate alternative lengthening of telomeres genes by methionine restriction and RNA interference. Oncogene 2007, 26, 4635–4647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, G.J.; Moulding, H.A.; Caldecott, K.W.; Rulten, S.L. One ring to bring them all—The role of Ku in mammalian non-homologous end joining. DNA Repair 2014, 17, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Jackson, J.G.; Pereira-Smith, O.M. Primary and compensatory roles for RB family members at cell cycle gene promoters that are deacetylated and downregulated in doxorubicin-induced senescence of breast cancer cells. Mol. Cell. Biol. 2006, 26, 2501–2510. [Google Scholar] [CrossRef] [Green Version]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Dabritz, J.H.M.; Zhao, Z.; Yu, Y.; Dorr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- Weihua, Z.; Lin, Q.; Ramoth, A.J.; Fan, D.; Fidler, I.J. Formation of solid tumors by a single multinucleated cancer cell. Cancer 2011, 117, 4092–4099. [Google Scholar] [CrossRef] [Green Version]
- Tyutyunyk-Massey, L.; Gewirtz, D.A. Roles of autophagy in breast cancer treatment: Target, bystander or benefactor. In Seminars in Cancer Biology; American Press: Cambridge, MA, USA, 2019. [Google Scholar] [CrossRef]
- Jakhar, R.; Luijten, M.N.H.; Wong, A.X.F.; Cheng, B.; Guo, K.; Neo, S.P.; Au, B.; Kulkarni, M.; Lim, K.J.; Maimaiti, J.; et al. Autophagy Governs Protumorigenic Effects of Mitotic Slippage-induced Senescence. Mol. Cancer Res. 2018, 16, 1625–1640. [Google Scholar] [CrossRef] [Green Version]
- Was, H.; Barszcz, K.; Czarnecka, J.; Kowalczyk, A.; Bernas, T.; Uzarowska, E.; Koza, P.; Klejman, A.; Piwocka, K.; Kaminska, B.; et al. Bafilomycin A1 triggers proliferative potential of senescent cancer cells in vitro and in NOD/SCID mice. Oncotarget 2017, 8, 9303–9322. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gammoh, N.; Fraser, J.; Puente, C.; Syred, H.M.; Kang, H.; Ozawa, T.; Lam, D.; Acosta, J.C.; Finch, A.J.; Holland, E.; et al. Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 2016, 12, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Goehe, R.W.; Di, X.; Sharma, K.; Bristol, M.L.; Henderson, S.C.; Valerie, K.; Rodier, F.; Davalos, A.R.; Gewirtz, D.A. The autophagy-senescence connection in chemotherapy: Must tumor cells (self) eat before they sleep? J. Pharmacol. Exp. Ther. 2012, 343, 763–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosieniak, G.; Sliwinska, M.; Piwocka, K.; Sikora, E. Curcumin abolishes apoptosis resistance of calcitriol-differentiated HL-60 cells. FEBS Lett. 2006, 580, 4653–4660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erenpreisa, J.; Salmina, K.; Huna, A.; Kosmacek, E.A.; Cragg, M.S.; Ianzini, F.; Anisimov, A.P. Polyploid tumour cells elicit paradiploid progeny through depolyploidizing divisions and regulated autophagic degradation. Cell Biol. Int. 2011, 35, 687–695. [Google Scholar] [CrossRef]
- Piechota, M.; Sunderland, P.; Wysocka, A.; Nalberczak, M.; Sliwinska, M.A.; Radwanska, K.; Sikora, E. Is senescence-associated beta-galactosidase a marker of neuronal senescence? Oncotarget 2016, 7, 81099–81109. [Google Scholar] [CrossRef] [Green Version]
- Sunderland, P.; Augustyniak, J.; Lenart, J.; Buzanska, L.; Carlessi, L.; Delia, D.; Sikora, E. ATM-deficient neural precursors develop senescence phenotype with disturbances in autophagy. Mech. Ageing Dev. 2020, 190, 111296. [Google Scholar] [CrossRef]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Munoz-Espin, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef]
- Erenpreisa, J.; Freivalds, T. Anisotropic staining of apurinic acid with toluidine blue. Histochemistry 1979, 60, 321–325. [Google Scholar] [CrossRef]
- Borczyk, M.; Sliwinska, M.A.; Caly, A.; Bernas, T.; Radwanska, K. Neuronal plasticity affects correlation between the size of dendritic spine and its postsynaptic density. Sci. Rep. 2019, 9, 1693. [Google Scholar] [CrossRef]
- Hanson, H.H.; Reilly, J.E.; Lee, R.; Janssen, W.G.; Phillips, G.R. Streamlined embedding of cell monolayers on gridded glass-bottom imaging dishes for correlative light and electron microscopy. Microsc. Microanal. 2010, 16, 747–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bojko, A.; Reichert, K.; Adamczyk, A.; Ligeza, J.; Ligeza, J.; Klein, A. The effect of tyrphostins AG494 and AG1478 on the autocrine growth regulation of A549 and DU145 cells. Folia Histochem. Cytobiol. 2012, 50, 186–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Concentration | Product No. and Manufacturer | |
|---|---|---|---|
| PARP1 | Mouse monoclonal | 1:500 | #556494, BD Bioscences, Franklin Lakes, NJ, USA |
| p53 (DO-1) | Mouse monoclonal | 1:500 | sc-126, Santa Cruz, Dallas, TX, USA |
| p-p53 (S15) | Rabbit polyclonal | 1:500 | #9284, Cell Signaling Technology, Danvers, MA, USA |
| p21 WAF1/CIP1 | Mouse monoclonal | 1:500 | P1484, Sigma-Aldrich, St. Louis, MO, USA |
| ATM (Y170) | Rabbit monoclonal | 1:500 | ab32420, Abcam, Cambridge, UK |
| p-ATM (S1981) | Mouse monoclonal | 1:500 | ab36810, Abcam, Cambridge, UK |
| ATR | Rabbit polyclonal | 1:500 | #2790, Cell Signaling Technology, Danvers, MA, USA |
| p-ATR (S428) | Rabbit polyclonal | 1:500 | #2853, Cell Signaling Technology, Danvers, MA, USA |
| Ku70 (E-5) | Mouse monoclonal | 1:500 | sc-17789, Santa Cruz, Dallas, TX, USA |
| ὙH2AX (S139) | Mouse monoclonal | 1:1000 | ab26350, Abcam, Cambridge, UK |
| ὙH2AX (S139) | Rabbit monoclonal | 1:500 | #9718, Cell Signaling Technology, Danvers, MA, USA |
| 53BP1 | Rabbit polyclonal | 1:500 | NB100, Novus Biologicals, Centennial, CO, USA |
| Ki67 | Rabbit polyclonal | 1:500 | ab15580, Abcam, Cambridge, UK |
| SQSTM1/p62 | Mouse monoclonal | 1:1000 | #610832, BD Bioscences, Franklin Lakes, NJ, USA |
| SQSTM1/p62 | Guinea pig polyclonal | 1:500 | GP62-C, Progen, Heidelberg, Germany |
| LC3B | Rabbit polyclonal | 1:500 | L7543, Sigma-Aldrich, St. Louis, MO, USA |
| ULK1 (D8H5) | Rabbit monoclonal | 1:250 | #8054, Cell Signaling Technology, Danvers, MA, USA |
| p-ULK1 (S757) | Rabbit monoclonal | 1:250 | #14202, Cell Signaling Technology, Danvers, MA, USA |
| m-TOR (7C10) | Rabbit monoclonal | 1:500 | #2983, Cell Signaling Technology, Danvers, MA, USA |
| p-m-TOR (S2448) | Rabbit monoclonal | 1:500 | #5536, Cell Signaling Technology, Danvers, MA, USA |
| p70S6K | Rabbit monoclonal | 1:500 | #2708, Cell Signaling Technology, Danvers, MA, USA |
| p-p70S6K (T389) | Rabbit monoclonal | 1:500 | #9234, Cell Signaling Technology, Danvers, MA, USA |
| p-S6 (S235/236) | Rabbit monoclonal | 1:500 | #4858, Cell Signaling Technology, Danvers, MA, USA |
| 4EBP1 | Rabbit polyclonal | 1:500 | #9452, Cell Signaling Technology, Danvers, MA, USA |
| p-4EBP1 (T37/46) | Rabbit monoclonal | 1:500 | #2855, Cell Signaling Technology, Danvers, MA, USA |
| p-AMPK(T172) | Rabbit monoclonal | 1:500 | #2531, Cell Signaling Technology, Danvers, MA, USA |
| AMPK | Rabbit monoclonal | 1:250 | #2532, Cell Signaling Technology, Danvers, MA, USA |
| p-Akt(S473) | Rabbit monoclonal | 1:2000 | #4060, Cell Signaling Technology, Danvers, MA, USA |
| Akt | Rabbit monoclonal | 1:1000 | #4691, Cell Signaling Technology, Danvers, MA, USA |
| LAMP-2 | Mouse monoclonal | 1:500 | #14-1078-82, eBioscience, Thermo Fisher Scientific, Waltham, MA, USA |
| TFEB | Rabbit polyclonal | 1:250 | #4240, Cell Signaling Technology, Danvers, MA, USA |
| p-TFEB (S142) | Rabbitpolyclonal | 1:500 | ABE-1971, Merck Millipore, Burlington, MA, USA |
| Rubicon | Rabbit monoclonal | 1:500 | #8465, Cell Signaling Technology, Danvers, MA, USA |
| GAPDH | Mouse monoclonal | 1:50,000 | MAB374, Merck Millipore, Burlington, MA, USA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bojko, A.; Staniak, K.; Czarnecka-Herok, J.; Sunderland, P.; Dudkowska, M.; Śliwińska, M.A.; Salmina, K.; Sikora, E. Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 6084. https://doi.org/10.3390/ijms21176084
Bojko A, Staniak K, Czarnecka-Herok J, Sunderland P, Dudkowska M, Śliwińska MA, Salmina K, Sikora E. Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells. International Journal of Molecular Sciences. 2020; 21(17):6084. https://doi.org/10.3390/ijms21176084
Chicago/Turabian StyleBojko, Agnieszka, Karolina Staniak, Joanna Czarnecka-Herok, Piotr Sunderland, Magdalena Dudkowska, Małgorzata Alicja Śliwińska, Kristine Salmina, and Ewa Sikora. 2020. "Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells" International Journal of Molecular Sciences 21, no. 17: 6084. https://doi.org/10.3390/ijms21176084
APA StyleBojko, A., Staniak, K., Czarnecka-Herok, J., Sunderland, P., Dudkowska, M., Śliwińska, M. A., Salmina, K., & Sikora, E. (2020). Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells. International Journal of Molecular Sciences, 21(17), 6084. https://doi.org/10.3390/ijms21176084