N-Terminal Regions of Prion Protein: Functions and Roles in Prion Diseases

Abstract
1. Introduction
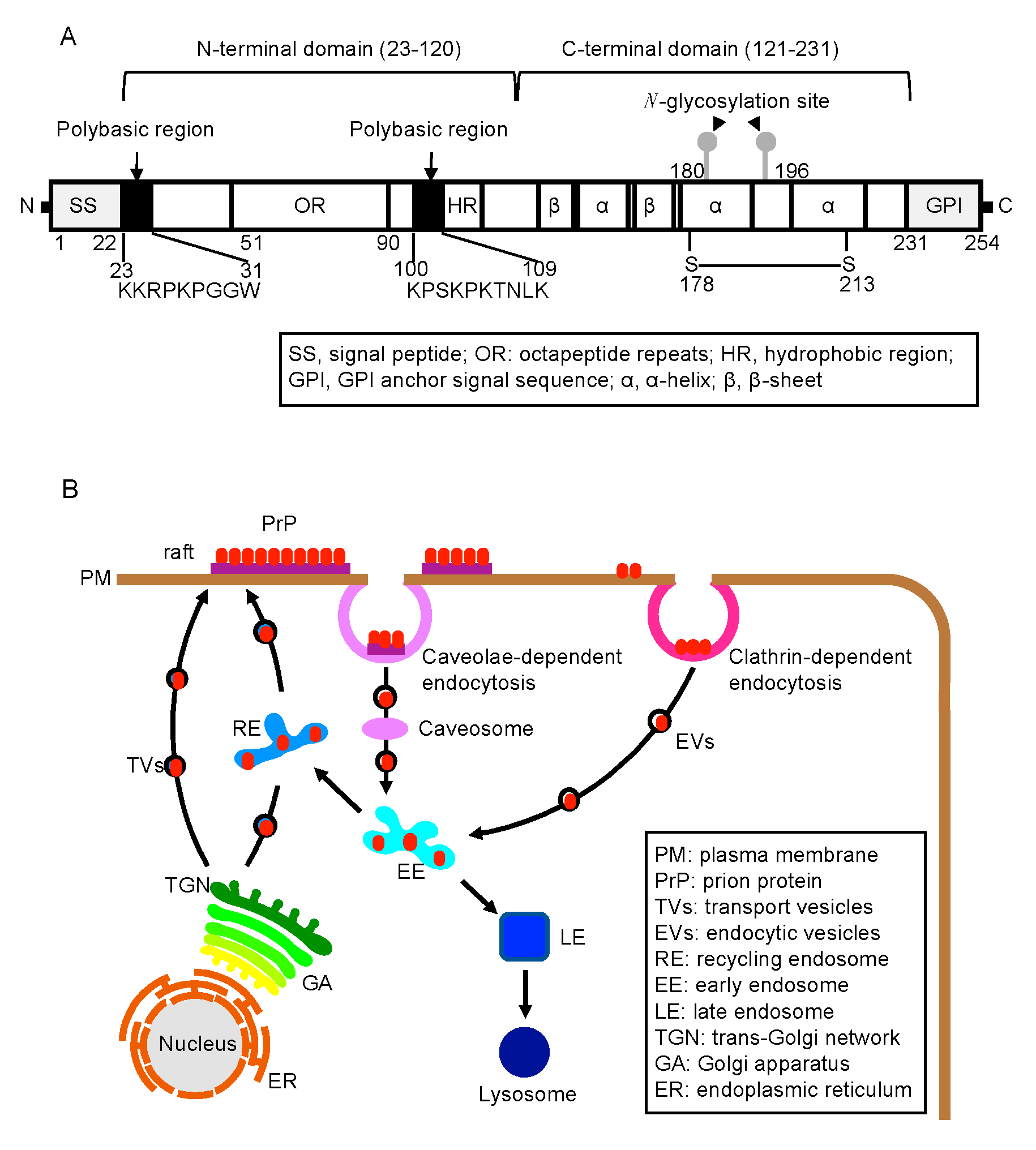
2. The N-Terminal Domain in the Function of PrPC
2.1. Biosynthesis of PrPC
2.2. Various Abnormal Phenotypes Are Spontaneously Observed in Prnp0/0 Mice
2.3. The OR Region in the Cell-Protective Role of PrPC
2.4. The Polybasic Region in the Function of PrPC
3. The N-Terminal Domain of PrPC in Prion Disease
3.1. The Polybasic Region in Prion Disease
3.2. The OR Region in Prion Disease
3.3. The Post-OR Region in Prion Diseases
4. The N-Terminal Domain in Conversion of PrPC to PrPSc
5. The N-Terminal Domain and Neurotoxic PrP Molecules
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PrP | Prion protein |
| OR | Octapeptide repeat |
| WT | Wild-type |
| CJD | Creutzfeldt-Jakob disease |
References
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. The prion diseases. Brain Pathol. 1998, 8, 499–513. [Google Scholar] [CrossRef] [PubMed]
- Scheckel, C.; Aguzzi, A. Prions, prionoids and protein misfolding disorders. Nat. Rev. Genet. 2018, 19, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Giles, K.; Olson, S.H.; Prusiner, S.B. Developing Therapeutics for PrP Prion Diseases. Cold Spring Harb. Perspect. Med. 2017, 7, a023747. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Early evidence that a protease-resistant protein is an active component of the infectious prion. Cell 2004, 116, S109. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef]
- Igel-Egalon, A.; Bohl, J.; Moudjou, M.; Herzog, L.; Reine, F.; Rezaei, H.; Beringue, V. Heterogeneity and Architecture of Pathological Prion Protein Assemblies: Time to Revisit the Molecular Basis of the Prion Replication Process? Viruses 2019, 11, 429. [Google Scholar] [CrossRef]
- Bueler, H.; Aguzzi, A.; Sailer, A.; Greiner, R.A.; Autenried, P.; Aguet, M.; Weissmann, C. Mice devoid of PrP are resistant to scrapie. Cell 1993, 73, 1339–1347. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Groth, D.; Serban, A.; Koehler, R.; Foster, D.; Torchia, M.; Burton, D.; Yang, S.L.; DeArmond, S.J. Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc. Natl. Acad. Sci. USA 1993, 90, 10608–10612. [Google Scholar] [CrossRef]
- Manson, J.C.; Clarke, A.R.; McBride, P.A.; McConnell, I.; Hope, J. PrP gene dosage determines the timing but not the final intensity or distribution of lesions in scrapie pathology. Neurodegeneration 1994, 3, 331–340. [Google Scholar]
- Sakaguchi, S.; Katamine, S.; Shigematsu, K.; Nakatani, A.; Moriuchi, R.; Nishida, N.; Kurokawa, K.; Nakaoke, R.; Sato, H.; Jishage, K.; et al. Accumulation of proteinase K-resistant prion protein (PrP) is restricted by the expression level of normal PrP in mice inoculated with a mouse-adapted strain of the Creutzfeldt-Jakob disease agent. J. Virol. 1995, 69, 7586–7592. [Google Scholar] [CrossRef] [PubMed]
- Schatzl, H.M.; Da Costa, M.; Taylor, L.; Cohen, F.E.; Prusiner, S.B. Prion protein gene variation among primates. J. Mol. Biol. 1995, 245, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Oesch, B.; Westaway, D.; Walchli, M.; McKinley, M.P.; Kent, S.B.; Aebersold, R.; Barry, R.A.; Tempst, P.; Teplow, D.B.; Hood, L.E.; et al. A cellular gene encodes scrapie PrP 27–30 protein. Cell. 1985, 40, 735–746. [Google Scholar] [CrossRef]
- Riek, R.; Hornemann, S.; Wider, G.; Glockshuber, R.; Wuthrich, K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23–231). FEBS Lett. 1997, 413, 282–288. [Google Scholar] [CrossRef]
- Donne, D.G.; Viles, J.H.; Groth, D.; Mehlhorn, I.; James, T.L.; Cohen, F.E.; Prusiner, S.B.; Wright, P.E.; Dyson, H.J. Structure of the recombinant full-length hamster prion protein PrP(29–231): The N terminus is highly flexible. Proc. Natl. Acad. Sci. USA 1997, 94, 13452–13457. [Google Scholar] [CrossRef] [PubMed]
- Calzolai, L.; Lysek, D.A.; Perez, D.R.; Guntert, P.; Wuthrich, K. Prion protein NMR structures of chickens, turtles, and frogs. Proc. Natl. Acad. Sci. USA 2005, 102, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef]
- Hackl, S.; Becker, C.F.W. Prion protein-Semisynthetic prion protein (PrP) variants with posttranslational modifications. J. Pept. Sci. 2019, 25, e3216. [Google Scholar] [CrossRef]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
- Stahl, N.; Baldwin, M.A.; Hecker, R.; Pan, K.M.; Burlingame, A.L.; Prusiner, S.B. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemical 1992, 31, 5043–5053. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, T.A. Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 2007, 450, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luis, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling - From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef] [PubMed]
- Needham, P.G.; Guerriero, C.J.; Brodsky, J.L. Chaperoning Endoplasmic Reticulum-Associated Degradation (ERAD) and Protein Conformational Diseases. Cold Spring Harb. Perspect. Biol. 2019, 11, a033928. [Google Scholar] [CrossRef] [PubMed]
- Puig, B.; Altmeppen, H.C.; Linsenmeier, L.; Chakroun, K.; Wegwitz, F.; Piontek, U.K.; Tatzelt, J.; Bate, C.; Magnus, T.; Glatzel, M. GPI-anchor signal sequence influences PrPC sorting, shedding and signalling, and impacts on different pathomechanistic aspects of prion disease in mice. PLoS Pathog. 2019, 15, e1007520. [Google Scholar] [CrossRef]
- Wulf, M.A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Biol. 2017, 15, 34. [Google Scholar] [CrossRef]
- Peters, P.J.; Mironov, A., Jr.; Peretz, D.; van Donselaar, E.; Leclerc, E.; Erpel, S.; DeArmond, S.J.; Burton, D.R.; Williamson, R.A.; Vey, M.; et al. Trafficking of prion proteins through a caveolae-mediated endosomal pathway. J. Cell Biol. 2003, 162, 703–717. [Google Scholar] [CrossRef]
- Campana, V.; Sarnataro, D.; Zurzolo, C. The highways and byways of prion protein trafficking. Trends Cell Biol. 2005, 15, 102–111. [Google Scholar] [CrossRef]
- Vilette, D.; Courte, J.; Peyrin, J.M.; Coudert, L.; Schaeffer, L.; Andreoletti, O.; Leblanc, P. Cellular mechanisms responsible for cell-to-cell spreading of prions. Cell Mol. Life Sci. 2018, 75, 2557–2574. [Google Scholar] [CrossRef]
- Taylor, D.R.; Watt, N.T.; Perera, W.S.; Hooper, N.M. Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J. Cell Sci. 2005, 118, 5141–5153. [Google Scholar] [CrossRef]
- Uchiyama, K.; Tomita, M.; Yano, M.; Chida, J.; Hara, H.; Das, N.R.; Nykjaer, A.; Sakaguchi, S. Prions amplify through degradation of the VPS10P sorting receptor sortilin. PLoS Pathog. 2017, 13, e1006470. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.R.; Hooper, N.M. The low-density lipoprotein receptor-related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem. J. 2007, 402, 17–23. [Google Scholar] [CrossRef]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.C.; Clarke, A.R.; Hooper, M.L.; Aitchison, L.; McConnell, I.; Hope, J. 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 1994, 8, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Katamine, S.; Shigematsu, K.; Nakatani, A.; Sakamoto, N.; Hasegawa, S.; Nakaoke, R.; Atarashi, R.; Kataoka, Y.; Miyamoto, T. Prion protein is necessary for latent learning and long-term memory retention. Cell Mol. Neurobiol. 1997, 17, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Whittington, M.A.; Sidle, K.C.; Smith, C.J.; Palmer, M.S.; Clarke, A.R.; Jefferys, J.G. Prion protein is necessary for normal synaptic function. Nature 1994, 370, 295–297. [Google Scholar] [CrossRef]
- Tobler, I.; Gaus, S.E.; Deboer, T.; Achermann, P.; Fischer, M.; Rulicke, T.; Moser, M.; Oesch, B.; McBride, P.A.; Manson, J.C. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 1996, 380, 639–642. [Google Scholar] [CrossRef]
- Nishida, N.; Tremblay, P.; Sugimoto, T.; Shigematsu, K.; Shirabe, S.; Petromilli, C.; Erpel, S.P.; Nakaoke, R.; Atarashi, R.; Houtani, T.; et al. A mouse prion protein transgene rescues mice deficient for the prion protein gene from purkinje cell degeneration and demyelination. Lab. Investig. 1999, 79, 689–697. [Google Scholar]
- Kim, C.K.; Sakudo, A.; Taniuchi, Y.; Shigematsu, K.; Kang, C.B.; Saeki, K.; Matsumoto, Y.; Sakaguchi, S.; Itohara, S.; Onodera, T. Late-onset olfactory deficits and mitral cell loss in mice lacking prion protein with ectopic expression of Doppel. Int. J. Mol. Med. 2007, 20, 169–176. [Google Scholar] [CrossRef][Green Version]
- Le Pichon, C.E.; Valley, M.T.; Polymenidou, M.; Chesler, A.T.; Sagdullaev, B.T.; Aguzzi, A.; Firestein, S. Olfactory behavior and physiology are disrupted in prion protein knockout mice. Nat. Neurosci. 2009, 12, 60–69. [Google Scholar] [CrossRef]
- Lledo, P.M.; Tremblay, P.; DeArmond, S.J.; Prusiner, S.B.; Nicoll, R.A. Mice deficient for prion protein exhibit normal neuronal excitability and synaptic transmission in the hippocampus. Proc. Natl. Acad. Sci. USA 1996, 93, 2403–2407. [Google Scholar] [CrossRef] [PubMed]
- Weise, J.; Crome, O.; Sandau, R.; Schulz-Schaeffer, W.; Bahr, M.; Zerr, I. Upregulation of cellular prion protein (PrPC) after focal cerebral ischemia and influence of lesion severity. Neurosci. Lett. 2004, 372, 146–150. [Google Scholar] [CrossRef] [PubMed]
- McLennan, N.F.; Brennan, P.M.; McNeill, A.; Davies, I.; Fotheringham, A.; Rennison, K.A.; Ritchie, D.; Brannan, F.; Head, M.W.; Ironside, J.W.; et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 2004, 165, 227–235. [Google Scholar] [CrossRef]
- Sakurai-Yamashita, Y.; Sakaguchi, S.; Yoshikawa, D.; Okimura, N.; Masuda, Y.; Katamine, S.; Niwa, M. Female-specific neuroprotection against transient brain ischemia observed in mice devoid of prion protein is abolished by ectopic expression of prion protein-like protein. Neuroscience 2005, 136, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Cowden, D.; Zhang, F.; Yuan, J.; Siedlak, S.; Abouelsaad, M.; Zeng, L.; Zhou, X.; O’Toole, J.; Das, A.S.; et al. Prion Protein Protects against Renal Ischemia/Reperfusion Injury. PLoS ONE 2015, 10, e0136923. [Google Scholar]
- Zanetti, F.; Carpi, A.; Menabo, R.; Giorgio, M.; Schulz, R.; Valen, G.; Baysa, A.; Massimino, M.L.; Sorgato, M.C.; Bertoli, A.; et al. The cellular prion protein counteracts cardiac oxidative stress. Cardiovasc. Res. 2014, 104, 93–102. [Google Scholar] [CrossRef]
- Chida, J.; Hara, H.; Yano, M.; Uchiyama, K.; Das, N.R.; Takahashi, E.; Miyata, H.; Tomioka, Y.; Ito, T.; Kido, H.; et al. Prion protein protects mice from lethal infection with influenza A viruses. PLoS Pathog. 2018, 14, e1007049. [Google Scholar] [CrossRef] [PubMed]
- Mitteregger, G.; Vosko, M.; Krebs, B.; Xiang, W.; Kohlmannsperger, V.; Nolting, S.; Hamann, G.F.; Kretzschmar, H.A. The role of the octarepeat region in neuroprotective function of the cellular prion protein. Brain Pathol. 2007, 17, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Wong, B.S.; Hafiz, F.; Clive, C.; Haswell, S.J.; Jones, I.M. Normal prion protein has an activity like that of superoxide dismutase. Biochem. J. 1999, 344, 1–5. [Google Scholar] [CrossRef]
- Jones, S.; Batchelor, M.; Bhelt, D.; Clarke, A.R.; Collinge, J.; Jackson, G.S. Recombinant prion protein does not possess SOD-1 activity. Biochem. J. 2005, 392, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Heppner, F.L.; Aguzzi, A. No superoxide dismutase activity of cellular prion protein In vivo. J. Biol. Chem. 2003, 384, 1279–1285. [Google Scholar] [CrossRef]
- Bounhar, Y.; Zhang, Y.; Goodyer, C.G.; LeBlanc, A. Prion protein protects human neurons against Bax-mediated apoptosis. J. Biol. Chem. 2001, 276, 39145–39149. [Google Scholar] [CrossRef]
- Oh, J.M.; Shin, H.Y.; Park, S.J.; Kim, B.H.; Choi, J.K.; Choi, E.K.; Carp, R.I.; Kim, Y.S. The involvement of cellular prion protein in the autophagy pathway in neuronal cells. Mol. Cell Neurosci. 2008, 39, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.L.; Drew, S.C.; Boland, M.P.; Masters, C.L.; Barnham, K.J.; Lawson, V.A.; Collins, S.J. Dominant roles of the polybasic proline motif and copper in the PrP23–89-mediated stress protection response. J. Cell Sci. 2009, 122, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Haigh, C.L.; Tumpach, C.; Drew, S.C.; Collins, S.J. The Prion Protein N1 and N2 Cleavage Fragments Bind to Phosphatidylserine and Phosphatidic Acid; Relevance to Stress-Protection Responses. PLoS ONE 2015, 10, e0134680. [Google Scholar] [CrossRef]
- Osiecka, K.M.; Nieznanska, H.; Skowronek, K.J.; Karolczak, J.; Schneider, G.; Nieznanski, K. Prion protein region 23–32 interacts with tubulin and inhibits microtubule assembly. Proteins 2009, 77, 279–296. [Google Scholar] [CrossRef]
- Pan, T.; Wong, B.S.; Liu, T.; Li, R.; Petersen, R.B.; Sy, M.S. Cell-surface prion protein interacts with glycosaminoglycans. Biochem. J. 2002, 368, 81–90. [Google Scholar] [CrossRef]
- Warner, R.G.; Hundt, C.; Weiss, S.; Turnbull, J.E. Identification of the heparan sulfate binding sites in the cellular prion protein. J. Biol. Chem. 2002, 277, 18421–18430. [Google Scholar] [CrossRef]
- Taubner, L.M.; Bienkiewicz, E.A.; Copie, V.; Caughey, B. Structure of the flexible amino-terminal domain of prion protein bound to a sulfated glycan. J. Mol. Biol. 2010, 395, 475–490. [Google Scholar] [CrossRef]
- Parkin, E.T.; Watt, N.T.; Hussain, I.; Eckman, E.A.; Eckman, C.B.; Manson, J.C.; Baybutt, H.N.; Turner, A.J.; Hooper, N.M. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer’s amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2007, 104, 11062–11067. [Google Scholar] [CrossRef] [PubMed]
- Bravard, A.; Auvre, F.; Fantini, D.; Bernardino-Sgherri, J.; Sissoeff, L.; Daynac, M.; Xu, Z.; Etienne, O.; Dehen, C.; Comoy, E.; et al. The prion protein is critical for DNA repair and cell survival after genotoxic stress. Nucleic Acids Res. 2015, 43, 904–916. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, J.A.; Unterberger, U.; Saa, P.; Massignan, T.; Fluharty, B.R.; Bowman, F.P.; Miller, M.B.; Supattapone, S.; Biasini, E.; Harris, D.A. The N-terminal, polybasic region of PrPC dictates the efficiency of prion propagation by binding to PrPSc. J. Neurosci. 2012, 32, 8817–8830. [Google Scholar] [CrossRef] [PubMed]
- Das, N.R.; Miyata, H.; Hara, H.; Uchiyama, K.; Chida, J.; Yano, M.; Watanabe, H.; Kondoh, G.; Sakaguchi, S. Effects of prion protein devoid of the N-terminal residues 25–50 on prion pathogenesis in mice. Arch. Virol. 2017, 162, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Khalife, M.; Reine, F.; Paquet-Fifield, S.; Castille, J.; Herzog, L.; Vilotte, M.; Moudjou, M.; Moazami-Goudarzi, K.; Makhzami, S.; Passet, B.; et al. Mutated but Not Deleted Ovine PrPC N-Terminal Polybasic Region Strongly Interferes with Prion Propagation in Transgenic Mice. J. Virol. 2016, 90, 1638–1646. [Google Scholar] [CrossRef]
- Das, N.R.; Miyata, H.; Hara, H.; Chida, J.; Uchiyama, K.; Masujin, K.; Watanabe, H.; Kondoh, G.; Sakaguchi, S. The N-Terminal Polybasic Region of Prion Protein Is Crucial in Prion Pathogenesis Independently of the Octapeptide Repeat Region. Mol. Neurobiol. 2020, 57, 1203–1216. [Google Scholar] [CrossRef]
- Prusiner, S.B. Genetic and infectious prion diseases. Arch. Neurol. 1993, 50, 1129–1153. [Google Scholar] [CrossRef]
- Brown, P.; Gibbs, C.J., Jr.; Rodgers-Johnson, P.; Asher, D.M.; Sulima, M.P.; Bacote, A.; Goldfarb, L.G.; Gajdusek, D.C. Human spongiform encephalopathy: The National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann. Neurol. 1994, 35, 513–529. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Miyata, H.; Uchiyama, K.; Ootsuyama, A.; Inubushi, S.; Mori, T.; Muramatsu, N.; Katamine, S.; Sakaguchi, S. Biological and biochemical characterization of mice expressing prion protein devoid of the octapeptide repeat region after infection with prions. PLoS ONE 2012, 7, e43540. [Google Scholar] [CrossRef]
- Hara, H.; Miyata, H.; Das, N.R.; Chida, J.; Yoshimochi, T.; Uchiyama, K.; Watanabe, H.; Kondoh, G.; Yokoyama, T.; Sakaguchi, S. Prion Protein Devoid of the Octapeptide Repeat Region Delays Bovine Spongiform Encephalopathy Pathogenesis in Mice. J. Virol. 2017, 92, e01368-17. [Google Scholar] [CrossRef]
- Chiesa, R.; Piccardo, P.; Ghetti, B.; Harris, D.A. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 1998, 21, 1339–1351. [Google Scholar] [CrossRef]
- Chiesa, R.; Drisaldi, B.; Quaglio, E.; Migheli, A.; Piccardo, P.; Ghetti, B.; Harris, D.A. Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 5574–5579. [Google Scholar] [CrossRef] [PubMed]
- Biasini, E.; Seegulam, M.E.; Patti, B.N.; Solforosi, L.; Medrano, A.Z.; Christensen, H.M.; Senatore, A.; Chiesa, R.; Williamson, R.A.; Harris, D.A. Non-infectious aggregates of the prion protein react with several PrPSc-Directed antibodies. J. Neurochem. 2008, 105, 2190–2204. [Google Scholar] [CrossRef] [PubMed]
- Castilla, J.; Gutierrez-Adan, A.; Brun, A.; Pintado, B.; Salguero, F.J.; Parra, B.; Segundo, F.D.; Ramirez, M.A.; Rabano, A.; Cano, M.J.; et al. Transgenic mice expressing bovine PrP with a four extra repeat octapeptide insert mutation show a spontaneous, non-transmissible, neurodegenerative disease and an expedited course of BSE infection. FEBS Lett. 2005, 579, 6237–6246. [Google Scholar] [CrossRef]
- Castilla, J.; Gutierrez-Adan, A.; Brun, A.; Pintado, B.; Parra, B.; Ramirez, M.A.; Salguero, F.J.; Diaz San Segundo, F.; Rabano, A.; Cano, M.J.; et al. Different behavior toward bovine spongiform encephalopathy infection of bovine prion protein transgenic mice with one extra repeat octapeptide insert mutation. J. Neurosci. 2004, 24, 2156–2164. [Google Scholar] [CrossRef]
- Eigenbrod, S.; Frick, P.; Bertsch, U.; Mitteregger-Kretzschmar, G.; Mielke, J.; Maringer, M.; Piening, N.; Hepp, A.; Daude, N.; Windl, O.; et al. Substitutions of PrP N-terminal histidine residues modulate scrapie disease pathogenesis and incubation time in transgenic mice. PLoS ONE 2017, 12, e0188989. [Google Scholar] [CrossRef]
- Hsiao, K.K.; Scott, M.; Foster, D.; Groth, D.F.; DeArmond, S.J.; Prusiner, S.B. Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science 1990, 250, 1587–1590. [Google Scholar] [CrossRef]
- Hsiao, K.K.; Groth, D.; Scott, M.; Yang, S.L.; Serban, H.; Rapp, D.; Foster, D.; Torchia, M.; Dearmond, S.J.; Prusiner, S.B. Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc. Natl. Acad. Sci. USA 1994, 91, 9126–9130. [Google Scholar] [CrossRef]
- Yang, W.; Cook, J.; Rassbach, B.; Lemus, A.; DeArmond, S.J.; Mastrianni, J.A. A New Transgenic Mouse Model of Gerstmann-Sträussler-Scheinker Syndrome Caused by the A117V Mutation of PRNP. J. Neurosci. 2009, 29, 10072–10080. [Google Scholar] [CrossRef]
- Fischer, M.; Rulicke, T.; Raeber, A.; Sailer, A.; Moser, M.; Oesch, B.; Brandner, S.; Aguzzi, A.; Weissmann, C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996, 15, 1255–1264. [Google Scholar] [CrossRef]
- Flechsig, E.; Shmerling, D.; Hegyi, I.; Raeber, A.J.; Fischer, M.; Cozzio, A.; von Mering, C.; Aguzzi, A.; Weissmann, C. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 2000, 27, 399–408. [Google Scholar] [CrossRef]
- Weissmann, C.; Flechsig, E. PrP knock-out and PrP transgenic mice in prion research. Br. Med. Bull. 2003, 66, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [PubMed]
- Wille, H.; Bian, W.; McDonald, M.; Kendall, A.; Colby, D.W.; Bloch, L.; Ollesch, J.; Borovinskiy, A.L.; Cohen, F.E.; Prusiner, S.B.; et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proc. Natl. Acad. Sci. USA 2009, 106, 16990–16995. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Fernandez, E.; Vos, M.R.; Afanasyev, P.; Cebey, L.; Sevillano, A.M.; Vidal, E.; Rosa, I.; Renault, L.; Ramos, A.; Peters, P.J.; et al. The Structural Architecture of an Infectious Mammalian Prion Using Electron Cryomicroscopy. PLoS Pathog. 2016, 12, e1005835. [Google Scholar] [CrossRef] [PubMed]
- Baskakov, I.V.; Caughey, B.; Requena, J.R.; Sevillano, A.M.; Surewicz, W.K.; Wille, H. The prion 2018 round tables (I): The structure of PrPSc. Prion 2019, 13, 46–52. [Google Scholar] [CrossRef]
- Spagnolli, G.; Rigoli, M.; Orioli, S.; Sevillano, A.M.; Faccioli, P.; Wille, H.; Biasini, E.; Requena, J.R. Full atomistic model of prion structure and conversion. PLoS Pathog. 2019, 15, e1007864. [Google Scholar] [CrossRef]
- Caughey, B.; Raymond, G.J.; Ernst, D.; Race, R.E. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): Implications regarding the site of conversion of PrP to the protease-resistant state. J. Virol. 1991, 65, 6597–6603. [Google Scholar] [CrossRef]
- Borchelt, D.R.; Taraboulos, A.; Prusiner, S.B. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J. Biol. Chem. 1992, 267, 16188–16199. [Google Scholar]
- Walmsley, A.R.; Zeng, F.; Hooper, N.M. The N-terminal region of the prion protein ectodomain contains a lipid raft targeting determinant. J. Biol. Chem. 2003, 278, 37241–37248. [Google Scholar] [CrossRef]
- Yu, S.; Yin, S.; Li, C.; Wong, P.; Chang, B.; Xiao, F.; Kang, S.C.; Yan, H.; Xiao, G.; Tien, P.; et al. Aggregation of prion protein with insertion mutations is proportional to the number of inserts. Biochem. J. 2007, 403, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Thakur, A.K.; Srivastava, A.K.; Srinivas, V.; Chary, K.V.; Rao, C.M. Copper alters aggregation behavior of prion protein and induces novel interactions between its N- and C-terminal regions. J. Biol. Chem. 2011, 286, 38533–38545. [Google Scholar] [CrossRef] [PubMed]
- Giachin, G.; Mai, P.T.; Tran, T.H.; Salzano, G.; Benetti, F.; Migliorati, V.; Arcovito, A.; Della Longa, S.; Mancini, G.; D’Angelo, P.; et al. The non-octarepeat copper binding site of the prion protein is a key regulator of prion conversion. Sci. Rep. 2015, 5, 15253. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.S.; Mastrianni, J.A.; Scott, M.R.; DeFea, K.A.; Tremblay, P.; Torchia, M.; DeArmond, S.J.; Prusiner, S.B.; Lingappa, V.R. A transmembrane form of the prion protein in neurodegenerative disease. Science 1998, 279, 827–834. [Google Scholar] [CrossRef]
- Hegde, R.S.; Tremblay, P.; Groth, D.; DeArmond, S.J.; Prusiner, S.B.; Lingappa, V.R. Transmissible and genetic prion diseases share a common pathway of neurodegeneration. Nature 1999, 402, 822–826. [Google Scholar] [CrossRef]
- Shmerling, D.; Hegyi, I.; Fischer, M.; Blattler, T.; Brandner, S.; Gotz, J.; Rulicke, T.; Flechsig, E.; Cozzio, A.; von Mering, C.; et al. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell 1998, 93, 203–214. [Google Scholar] [CrossRef]
- Li, A.; Christensen, H.M.; Stewart, L.R.; Roth, K.A.; Chiesa, R.; Harris, D.A. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007, 26, 548–558. [Google Scholar] [CrossRef]
- Westergard, L.; Turnbaugh, J.A.; Harris, D.A. A nine amino acid domain is essential for mutant prion protein toxicity. J. Neurosci. 2011, 31, 14005–14017. [Google Scholar] [CrossRef]
- Turnbaugh, J.A.; Westergard, L.; Unterberger, U.; Biasini, E.; Harris, D.A. The N-terminal, polybasic region is critical for prion protein neuroprotective activity. PLoS ONE 2011, 6, e25675. [Google Scholar] [CrossRef]
- Solomon, I.H.; Khatri, N.; Biasini, E.; Massignan, T.; Huettner, J.E.; Harris, D.A. An N-terminal polybasic domain and cell surface localization are required for mutant prion protein toxicity. J. Biol. Chem. 2011, 286, 14724–14736. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; McDonald, A.J.; Markham, K.; Rich, C.B.; McHugh, K.P.; Tatzelt, J.; Colby, D.W.; Millhauser, G.L.; Harris, D.A. The N-terminus of the prion protein is a toxic effector regulated by the C-terminus. Elife 2017, 6, e23473. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Disease Type | PrPs | Amino Acid Sequence of the Polybasic Region (Residues 23–31) 1 | Susceptibility to Prions | References |
|---|---|---|---|---|
| Acquired prion disease | WT PrP | KKRPKPGGW | • Normal. | |
| PrP∆23–31 | − − − − − − − − − | • Markedly reduced to RML scrapie prions. | [63] | |
| PrP∆25–50 | KK− − − − − − − | • Not reduced to RML and 22L scrapie prions | [64] | |
| PrP∆23–26 | − − − −KPGGW | • Only slightly or not reduced to 127S and LA19K scrapie prions and BSE prions. | [65] | |
| PrP-M | KQHPHPGGW | • Markedly reduced to 127S and LA19K prions and BSE prions | [65] | |
| PrP3K3A | AARPAPGGW | • Markedly reduced to RML and 22L scrapie prions. | [66] |
| Disease Type | PrPs | Number of the OR Sequence | Clinicopathological Features | References |
| Hereditary prion disease | PG14 | 14 1 | • Spontaneously develop cerebellar neurodegeneration. • Accumulate very slightly but substantially PK-resistant PrPScPG14 in the brain. • No prion infectivity associated with PrPScPG14. | [71,72,73] |
| Bo10OR-PrP | 10 2 | • Spontaneously develop cerebellar neurodegeneration. • Accumulate insoluble and slightly PK-resistant 10OR-PrPSc in their brains. • No prion infectivity associated with 10OR-PrPSc. | [74] | |
| Disease Type | PrPs | Number of the OR Sequence | Susceptibility to Prions | References |
| Acquired prion disease | PrP∆OR | 0 1 | • Reduced to BSE prions, but not to RML and 22L scrapie prions. | [70] |
| Bo7OR-PrP | 7 2 | • Increased to BSE prions. | [75] | |
| Bo10OR-PrP | 10 2 | • Increased to BSE prions. | [74] | |
| PrP(TetraH>G) | 51 (with 4 histidine residues mutated to glycine residues) | • Reduced to RML prions. | [76] |
| Disease Type | PrPs | The Post-OR Sequence | Clinicopathological Features | References |
| Hereditary prion disease | PrP-P101L | Proline residue at position 101 mutated to leucine residue in mouse PrP | • Spontaneously develop prion disease-like diseases. • Accumulate weakly protease-resistant PrP-P101L in the brain. • Accumulate prion infectivity associated with weakly protease-resistant PrP-P101L. | [77,78] |
| PrP-A116V | Alanine residue at position 116 mutated to valine residue in mouse PrP | • Spontaneously developed prion disease-like diseases. • Accumulate partly insoluble and weakly protease-resistant PrP-A116V in the brain. • No data available as to infectivity associated with protease-resistant PrP-A116V. | [79] | |
| Disease Type | PrPs | The Post-OR Sequence | Susceptibility to Prions | References |
| Acquired prion disease | PrP∆32–80 | Intact | • Fully susceptible to RML scrapie prions. | [80] |
| PrP∆32–93 | The post-OR residues 91–93 deleted | • Partially reduced to RML scrapie prions. | [81] | |
| PrP∆32–106 | The post-OR residues 91–106 deleted | • Resistant to RML scrapie prions. | [82] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hara, H.; Sakaguchi, S. N-Terminal Regions of Prion Protein: Functions and Roles in Prion Diseases. Int. J. Mol. Sci. 2020, 21, 6233. https://doi.org/10.3390/ijms21176233
Hara H, Sakaguchi S. N-Terminal Regions of Prion Protein: Functions and Roles in Prion Diseases. International Journal of Molecular Sciences. 2020; 21(17):6233. https://doi.org/10.3390/ijms21176233
Chicago/Turabian StyleHara, Hideyuki, and Suehiro Sakaguchi. 2020. "N-Terminal Regions of Prion Protein: Functions and Roles in Prion Diseases" International Journal of Molecular Sciences 21, no. 17: 6233. https://doi.org/10.3390/ijms21176233
APA StyleHara, H., & Sakaguchi, S. (2020). N-Terminal Regions of Prion Protein: Functions and Roles in Prion Diseases. International Journal of Molecular Sciences, 21(17), 6233. https://doi.org/10.3390/ijms21176233