Cerebrospinal Fluid Mitochondrial DNA in Rapid and Slow Progressive Forms of Alzheimer’s Disease


 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
2.1. Characteristics of Study Cohort: AD Biomarkers in Patients and Neurological Disease Controls
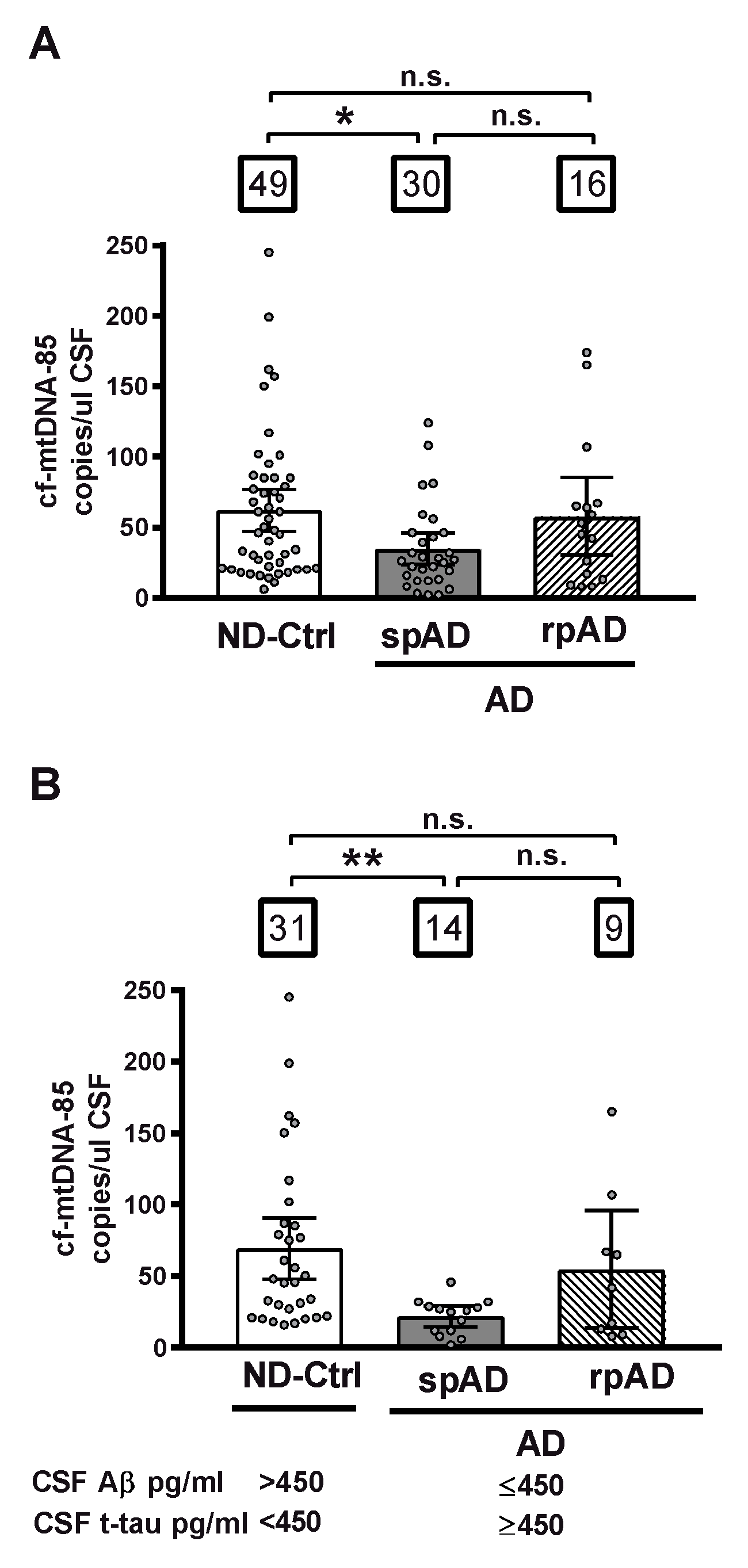
2.2. cf-mtDNA in AD Disease Progression
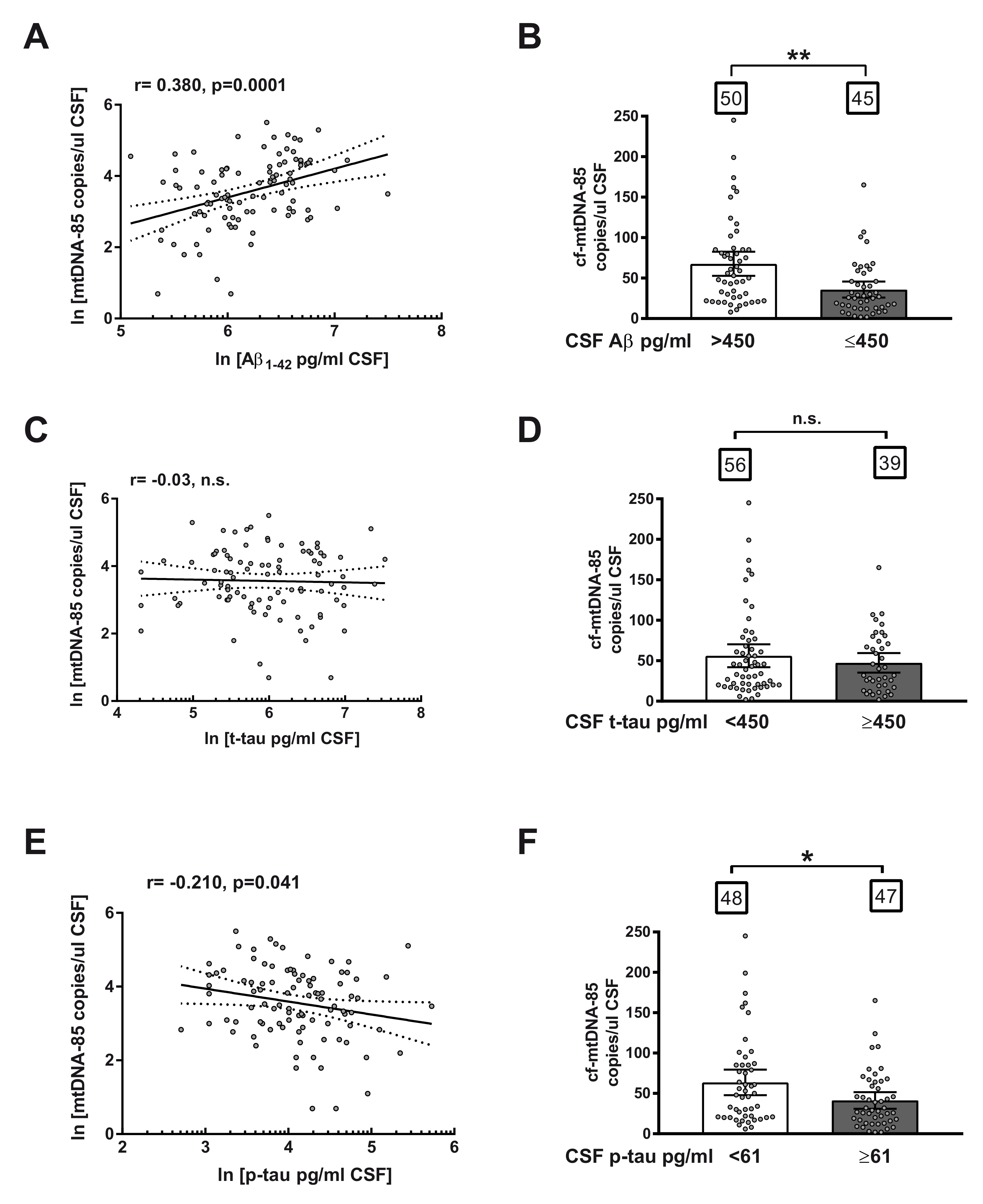
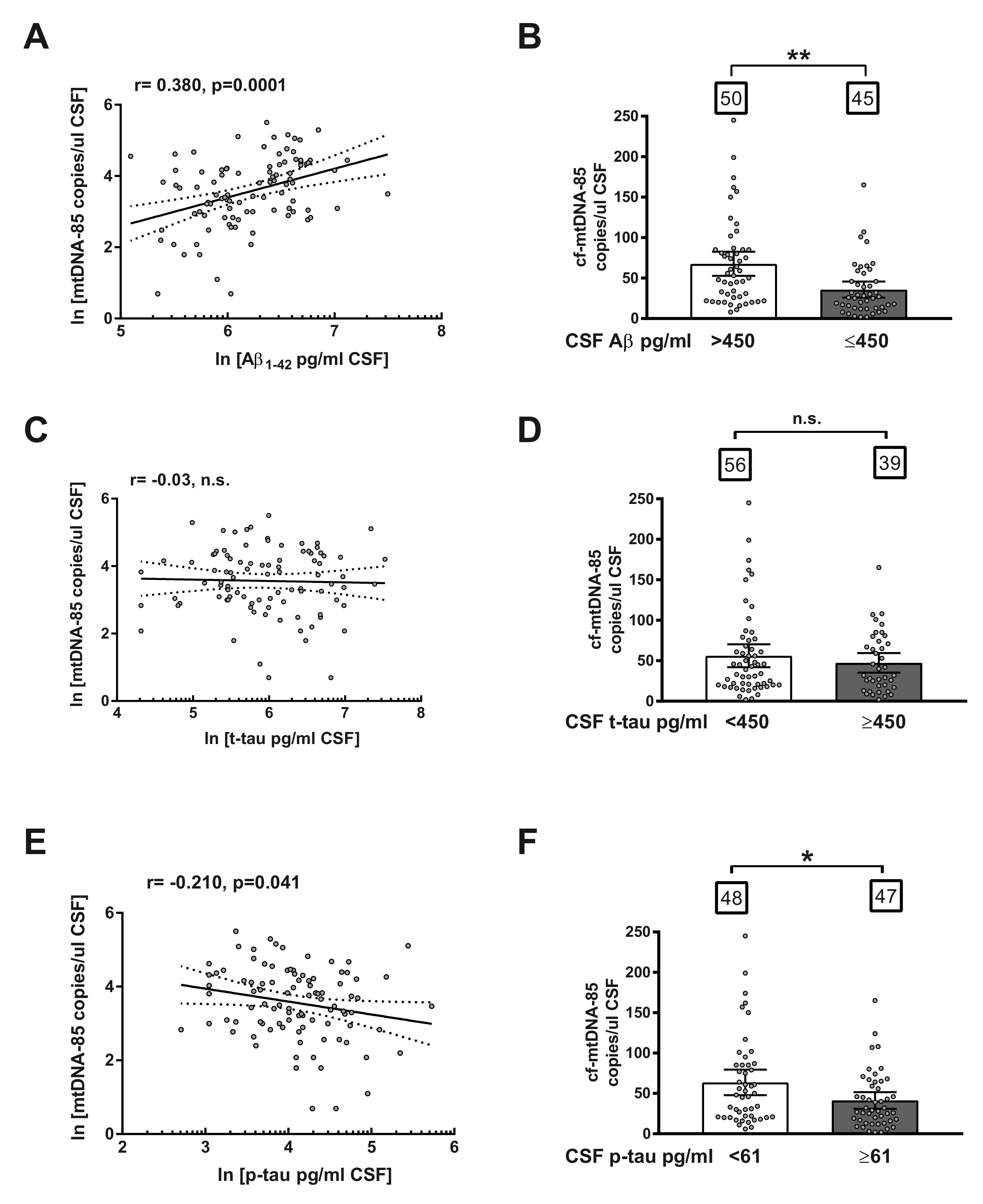
2.3. Relationship between cf-mtDNA-85 and Core CSF Biomarkers of AD
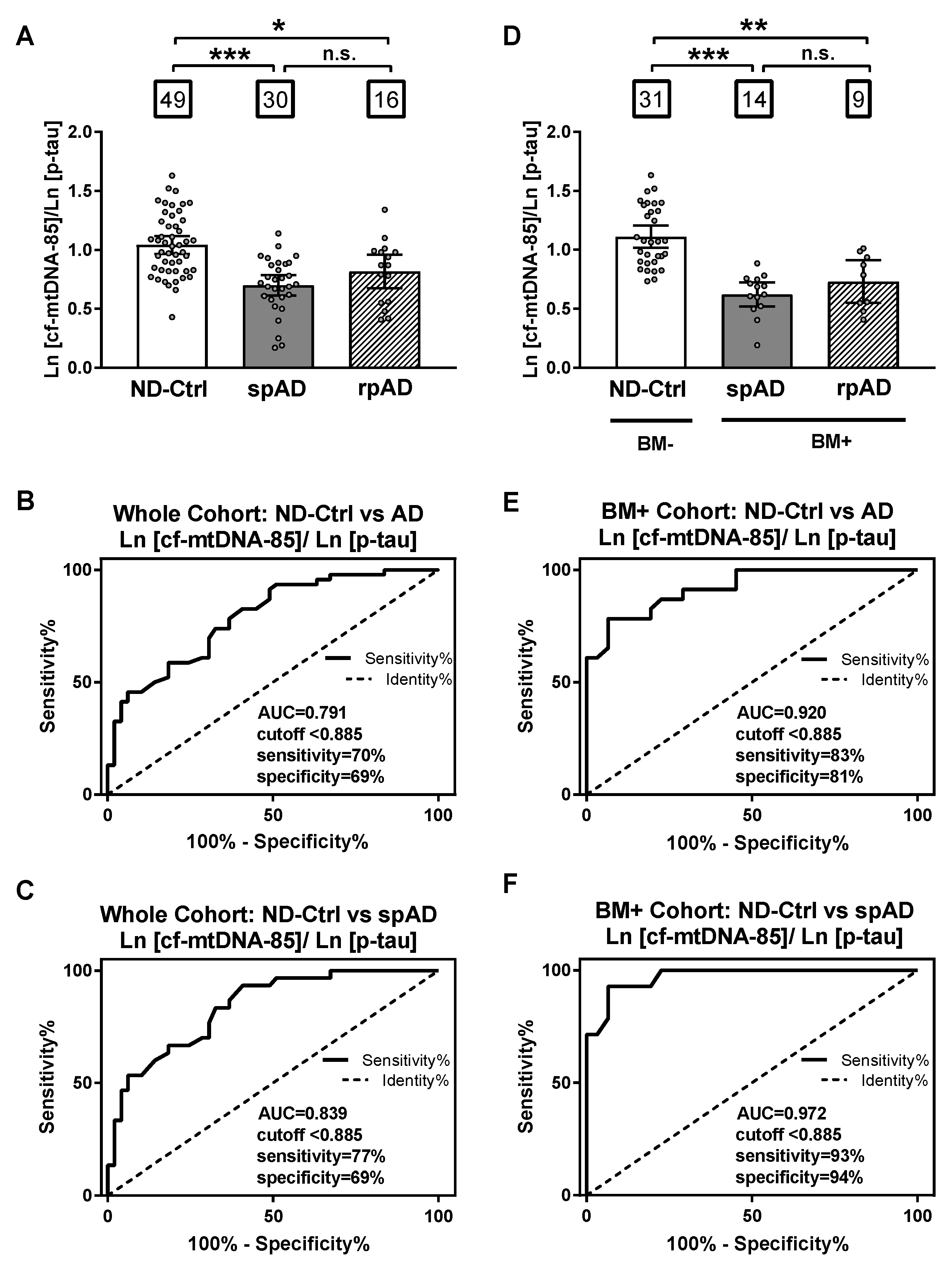
2.4. Ratio of cf-mtDNA-85 over p-tau in AD Progression
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. AD Biomarker Analysis in CSF Samples
4.3. Droplet Digital PCR
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid Beta peptide 1-42 |
| AD | Alzheimer’s disease |
| CSF | Cerebrospinal fluid |
| cf-mtDNA | Cell-free mitochondrial DNA |
| CJD | Creutzfeldt-Jakob disease |
| dPCR | Digital Polymerase Chain Reaction |
| ND-Ctrl | Control patients diagnosed with neurological diseases without dementia |
| rpAD | Alzheimer’s disease of rapid clinical progression |
| spAD | Alzheimer’s disease of slow clinical progression |
| p-tau | Phosphorylated tau protein |
| t-tau | Total amount of tau protein |
References
- Komarova, N.L.; Thalhauser, C.J. High degree of heterogeneity in Alzheimer’s disease progression patterns. PLoS Comput. Biol. 2011, 7, e1002251. [Google Scholar] [CrossRef]
- Mayeux, R.; Stern, Y.; Spanton, S. Heterogeneity in dementia of the Alzheimer type: Evidence of subgroups. Neurology 1985, 35, 453–461. [Google Scholar] [CrossRef]
- Thalhauser, C.J.; Komarova, N.L. Alzheimer’s disease: Rapid and slow progression. J. R. Soc. Interface 2012, 9, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Wilkosz, P.A.; Seltman, H.J.; Devlin, B.; Weamer, E.A.; Lopez, O.L.; DeKosky, S.T.; Sweet, R.A. Trajectories of cognitive decline in Alzheimer’s disease. Int. Psychogeriatr. 2010, 22, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Wolff, M.; Weitz, M.; Bartlau, T.; Korth, C.; Zerr, I. Rapidly progressive Alzheimer disease. Arch. Neurol. 2011, 68, 1124–1130. [Google Scholar] [CrossRef] [Green Version]
- Soto, M.E.; Andrieu, S.; Arbus, C.; Ceccaldi, M.; Couratier, P.; Dantoine, T.; Dartigues, J.F.; Gillette-Guyonnet, S.; Nourhashemi, F.; Ousset, P.J.; et al. Rapid cognitive decline in Alzheimer’s disease. Consensus paper. J. Nutr. Health Aging 2008, 12, 703–713. [Google Scholar]
- Buccione, I.; Perri, R.; Carlesimo, G.A.; Fadda, L.; Serra, L.; Scalmana, S.; Caltagirone, C. Cognitive and behavioural predictors of progression rates in Alzheimer’s disease. Eur. J. Neurol. 2007, 14, 440–446. [Google Scholar] [CrossRef]
- Dumont, C.; Voisin, T.; Nourhashemi, F.; Andrieu, S.; Koning, M.; Vellas, B. Predictive factors for rapid loss on the mini-mental state examination in Alzheimer’s disease. J. Nutr. Health. Aging. 2005, 9, 163–167. [Google Scholar]
- Josephs, K.A.; Ahlskog, J.E.; Parisi, J.E.; Boeve, B.F.; Crum, B.A.; Giannini, C.; Petersen, R.C. Rapidly progressive neurodegenerative dementias. Arch. Neurol. 2009, 66, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012, 2, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Wolff, M.; von Ahsen, N.; Zerr, I. Alzheimer’s disease: Genetic polymorphisms and rate of decline. Dement. Geriatr. Cogn. Disord. 2012, 33, 84–89. [Google Scholar] [CrossRef]
- Hansson, O.; Seibyl, J.; Stomrud, E.; Zetterberg, H.; Trojanowski, J.Q.; Bittner, T.; Lifke, V.; Corradini, V.; Eichenlaub, U.; Batrla, R.; et al. Alzheimer’s Disease Neuroimaging, I., CSF biomarkers of Alzheimer’s disease concord with amyloid-beta PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018, 14, 1470–1481. [Google Scholar] [CrossRef]
- Kester, M.I.; van der Vlies, A.E.; Blankenstein, M.A.; Pijnenburg, Y.A.; van Elk, E.J.; Scheltens, P.; van der Flier, W.M. CSF biomarkers predict rate of cognitive decline in Alzheimer disease. Neurology 2009, 73, 1353–1358. [Google Scholar] [CrossRef]
- Wallin, A.K.; Blennow, K.; Zetterberg, H.; Londos, E.; Minthon, L.; Hansson, O. CSF biomarkers predict a more malignant outcome in Alzheimer disease. Neurology 2010, 74, 1531–1537. [Google Scholar] [CrossRef]
- Abu-Rumeileh, S.; Capellari, S.; Parchi, P. Rapidly Progressive Alzheimer’s Disease: Contributions to Clinical-Pathological Definition and Diagnosis. J. Alzheimers Dis. 2018, 63, 887–897. [Google Scholar] [CrossRef]
- Schmidt, C.; Redyk, K.; Meissner, B.; Krack, L.; von Ahsen, N.; Roeber, S.; Kretzschmar, H.; Zerr, I. Clinical features of rapidly progressive Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2010, 29, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Cohen, M.L.; Kim, C.; Haldiman, T.; ElHag, M.; Mehndiratta, P.; Pichet, T.; Lissemore, F.; Shea, M.; Cohen, Y.; Chen, W.; et al. Rapidly progressive Alzheimer’s disease features distinct structures of amyloid-beta. Brain 2015, 138, 1009–1022. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Zetterberg, H. Biomarkers for Alzheimer’s disease: Current status and prospects for the future. J. Intern. Med. 2018, 284, 643–663. [Google Scholar] [CrossRef] [Green Version]
- Drummond, E.; Nayak, S.; Faustin, A.; Pires, G.; Hickman, R.A.; Askenazi, M.; Cohen, M.; Haldiman, T.; Kim, C.; Han, X.; et al. Proteomic differences in amyloid plaques in rapidly progressive and sporadic Alzheimer’s disease. Acta. Neuropathol. 2017, 133, 933–954. [Google Scholar] [CrossRef]
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann. Neurol. 2013, 74, 655–668. [Google Scholar] [CrossRef]
- Podlesniy, P.; Llorens, F.; Golanska, E.; Sikorska, B.; Liberski, P.; Zerr, I.; Trullas, R. Mitochondrial DNA differentiates Alzheimer’s disease from Creutzfeldt-Jakob disease. Alzheimers Dement. 2016, 12, 546–555. [Google Scholar] [CrossRef]
- Cervera-Carles, L.; Alcolea, D.; Estanga, A.; Ecay-Torres, M.; Izagirre, A.; Clerigue, M.; Garcia-Sebastian, M.; Villanua, J.; Escalas, C.; Blesa, R.; et al. Cerebrospinal fluid mitochondrial DNA in the Alzheimer’s disease continuum. Neurobiol Aging 2017, 53, 192.e1–192.e4. [Google Scholar] [CrossRef]
- Leuzy, A.; Chiotis, K.; Lemoine, L.; Gillberg, P.G.; Almkvist, O.; Rodriguez-Vieitez, E.; Nordberg, A. Tau PET imaging in neurodegenerative tauopathies-still a challenge. Mol. Psychiatry 2019, 24, 1112–1134. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.A.; Aisen, P.S.; Beckett, L.A.; Bennett, D.A.; Craft, S.; Fagan, A.M.; Iwatsubo, T.; Jack, C.R., Jr.; Kaye, J.; Montine, T.J.; et al. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 280–292. [Google Scholar] [CrossRef] [Green Version]
- Buchhave, P.; Minthon, L.; Zetterberg, H.; Wallin, A.K.; Blennow, K.; Hansson, O. Cerebrospinal fluid levels of beta-amyloid 1–42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 2012, 69, 98–106. [Google Scholar] [CrossRef]
- Fagan, A.M.; Xiong, C.; Jasielec, M.S.; Bateman, R.J.; Goate, A.M.; Benzinger, T.L.; Ghetti, B.; Martins, R.N.; Masters, C.L.; Mayeux, R.; et al. Longitudinal change in CSF biomarkers in autosomal-dominant Alzheimer’s disease. Sci. Transl. Med. 2014, 6, 226ra30. [Google Scholar] [CrossRef] [Green Version]
- Beach, T.G.; Monsell, S.E.; Phillips, L.E.; Kukull, W. Accuracy of the clinical diagnosis of Alzheimer disease at National Institute on Aging Alzheimer Disease Centers, 2005–2010. J. Neuropathol. Exp. Neurol. 2012, 71, 266–273. [Google Scholar] [CrossRef]
- Schmidt, C.; Artjomova, S.; Hoeschel, M.; Zerr, I. CSF prion protein concentration and cognition in patients with Alzheimer disease. Prion 2013, 7, 229–234. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Haik, S.; Satoh, K.; Rabano, A.; Martinez-Martin, P.; Roeber, S.; Brandel, J.P.; Calero-Lara, M.; de Pedro-Cuesta, J.; Laplanche, J.L.; et al. Rapidly progressive Alzheimer’s disease: A multicenter update. J. Alzheimers Dis. 2012, 30, 751–756. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef] [Green Version]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P.; et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Podlesniy, P.; Trullas, R. Biomarkers in Cerebrospinal Fluid: Analysis of Cell-Free Circulating Mitochondrial DNA by Digital PCR. Methods Mol. Biol. 2018, 1768, 111–126. [Google Scholar] [PubMed]
- Huggett, J.F. The Digital MIQE Guidelines Update: Minimum Information for Publication of Quantitative Digital PCR Experiments for 2020. Clin. Chem 2020, 66, 1012–1029. [Google Scholar] [CrossRef]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The digital MIQE guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| n | Gender (f/m) | Age | cf-mtDNA-85 (copies/uL CSF) | t-tau (pg/mL CSF) | Ab1-42 (pg/mL CSF) | p-tau (pg/mL CSF) | |
|---|---|---|---|---|---|---|---|
| ND-Controls | 49 | 26/23 | 69 (66,72) | 62 (47,77) | 323 (260,387) | 646 (564,729) | 46 (38,53) |
| Absence of AD Biomarker (Ab1-42 > 450 & t-tau < 450 (pg/mL)) | 31 | 16/15 | 71 (67,74) | 69 (48,91) | 243 (210,277) | 772 (680,864) | 37 (33,42) |
| Presence AD Biomarker (Ab1-42 ≤ 450 or t-tau ≥ 450 (pg/mL) | 18 | 10/8 | 66 (62,70) | 49 (33,65) | 460 (309,612) * | 430 (327,533) * | 61 (42,79) * |
| AD | 46 | 25/21 | 68 (65,71) | 43 (31,55) # | 649 ± (538,759) # | 444 (389,499) # | 99 (84,113) # |
| Presence of AD Biomarkers (Ab1-42 ≤ 450 & t-tau ≥ 450 (pg/mL)) | 23 | 13/10 | 66 (62,70) | 35 (19,51) | 846 (680,1012) * | 338 (306,370) * | 117 (91,143) * |
| spAD | 14 | 7/7 | 66 (60,73) | 22 (15,29) | 753 (572,934) | 336 (297,375) | 106 (71,142) |
| rpAD | 9 | 6/3 | 65 (60,70) | 55 (14,96) | 991 (642,1340) | 341 (272,410) | 133 (88,177) |
| Absence of AD Biomarker (Ab1-42 > 450 or t-tau < 450 (pg/mL) | 23 | 12/11 | 70 (66,74) | 51 (33,69) | 451 (349,553) | 550 (463,638) | 81 (70,92) |
| spAD | 16 | 9/7 | 69 (64,74) | 46 (26,66) | 476 (346,606) | 518 (418,619) | 85 (73,97) |
| rpAD | 7 | 3/4 | 71 (62,80) | 61 (12,111) | 394 (187,602) | 623 (412,835) | 71 (40,102) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Podlesniy, P.; Llorens, F.; Puigròs, M.; Serra, N.; Sepúlveda-Falla, D.; Schmidt, C.; Hermann, P.; Zerr, I.; Trullas, R. Cerebrospinal Fluid Mitochondrial DNA in Rapid and Slow Progressive Forms of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 6298. https://doi.org/10.3390/ijms21176298
Podlesniy P, Llorens F, Puigròs M, Serra N, Sepúlveda-Falla D, Schmidt C, Hermann P, Zerr I, Trullas R. Cerebrospinal Fluid Mitochondrial DNA in Rapid and Slow Progressive Forms of Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(17):6298. https://doi.org/10.3390/ijms21176298
Chicago/Turabian StylePodlesniy, Petar, Franc Llorens, Margalida Puigròs, Nuria Serra, Diego Sepúlveda-Falla, Christian Schmidt, Peter Hermann, Inga Zerr, and Ramon Trullas. 2020. "Cerebrospinal Fluid Mitochondrial DNA in Rapid and Slow Progressive Forms of Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 17: 6298. https://doi.org/10.3390/ijms21176298
APA StylePodlesniy, P., Llorens, F., Puigròs, M., Serra, N., Sepúlveda-Falla, D., Schmidt, C., Hermann, P., Zerr, I., & Trullas, R. (2020). Cerebrospinal Fluid Mitochondrial DNA in Rapid and Slow Progressive Forms of Alzheimer’s Disease. International Journal of Molecular Sciences, 21(17), 6298. https://doi.org/10.3390/ijms21176298