Human Cell Modeling for Cardiovascular Diseases


Abstract
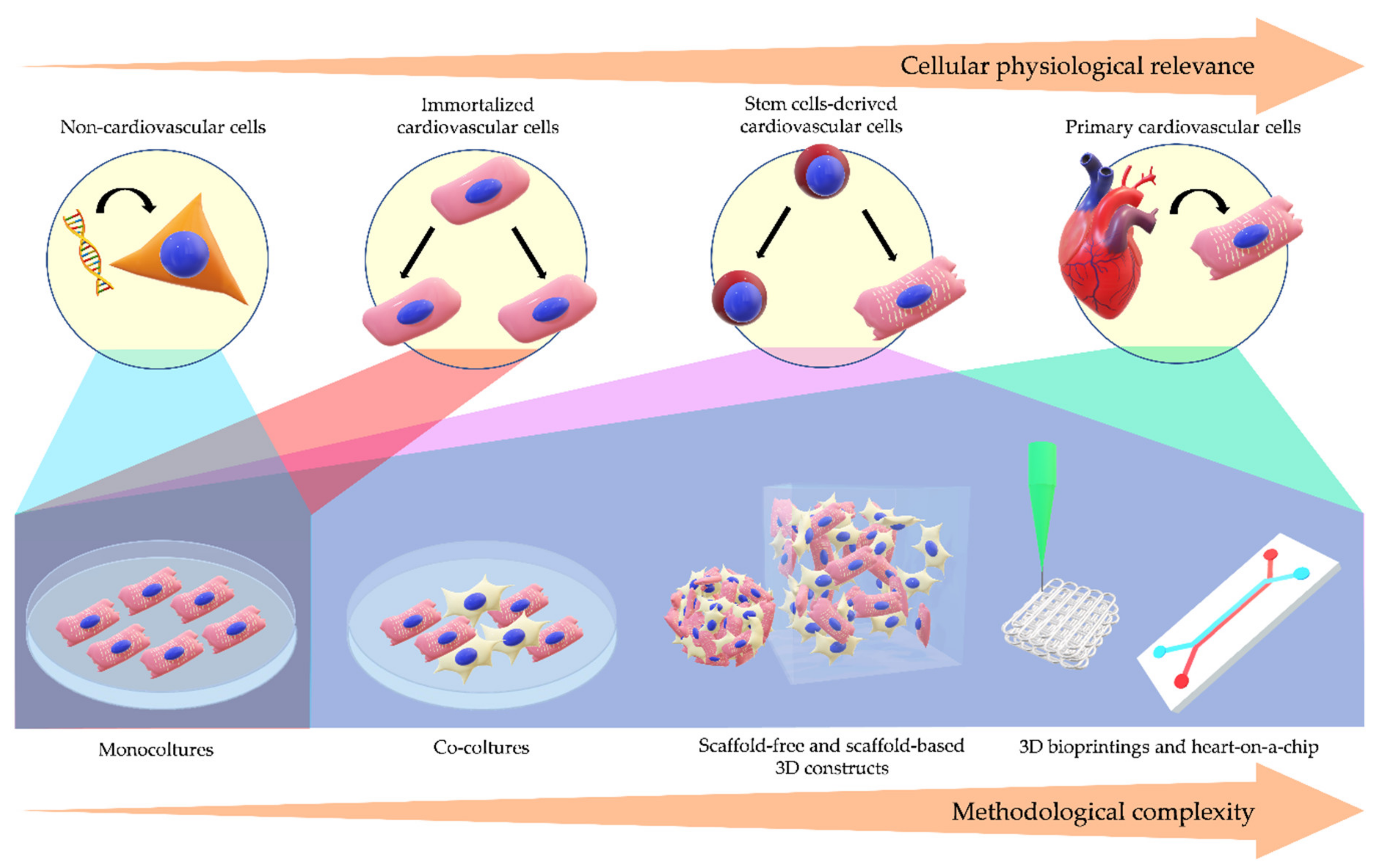
:1. Introduction
2. Non-Cardiovascular Cells
2.1. HEK 293
2.2. Buccal Mucosa Cells
3. Cardiovascular Cells
3.1. Primary Atrial and Ventricular Cardiomyocytes
3.2. Primary Cardiac Mesenchymal Cells
3.2.1. C-Kit Positive Cardiac Mesenchymal Cells
3.2.2. Fibro/Adipocyte Progenitors
3.3. Primary Endothelial Cells
3.4. Primary Vascular Smooth Muscle Cells
3.5. Cell Line AC 16
4. Stem Cells
4.1. Human Embryonic Stem Cells
4.2. Human Induced Pluripotent Stem Cells
4.2.1. Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes
4.2.2. Human Induced Pluripotent Stem Cells-Derived Endothelial Cells
4.2.3. Human Induced Pluripotent Stem Cells-Derived Smooth Muscle Cells
5. Multicellular Models
5.1. Co-Cultures
5.2. 3D Cultures
5.2.1. Scaffold-Free 3D Cultures
5.2.2. Scaffold-Based 3D Cultures
5.2.3. Printed 3D Cultures
5.2.4. 3D Organ-On-A-Chip
5.2.5. Examples of Applications of 3D Cultures
6. Discussion
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3D | three-dimensional |
| ACM | arrhythmogenic cardiomyopathy |
| AF | atrial fibrillation |
| Ang II | angiotensin II |
| ANP | atrial natriuretic peptide |
| BMC | buccal mucosa cell |
| BMP2 | bone morphogenetic protein 2 |
| BNP | B-type natriuretic peptide |
| CaMK II | Ca++/calmodulin-dependent protein kinase II |
| cAMP | cyclic adenosine monophosphate |
| CASM | smooth muscle cells from coronary artery |
| CAVD | calcific aortic valve disease |
| C-kit | KIT proto-oncogene receptor tyrosine-protein kinase |
| CM | cardiomyocyte |
| CMPC | cardiac mesenchymal progenitor cell |
| C-MSC | cardiac mesenchymal stromal cell |
| CRP | C-reactive protein |
| CTEPH | chronic thromboembolic pulmonary hypertension |
| CVD | cardiovascular disease |
| Cx40 | connexin 40 |
| Cx43 | connexin 43 |
| EC | endothelial cell |
| ECM | extracellular matrix |
| ERBB | erythroblastic oncogene B |
| ERK1/2 | extracellular signal-regulated kinase ½ |
| ESC | embryonic stem cell |
| FAP | fibro/adipocyte progenitor |
| FBS | fetal bovine serum |
| FD | Fabry disease |
| Gb3 | globotriaosylceramide |
| GLA | α-galactosidase A |
| GSK3β | glycogen synthase kinase 3β |
| GTPase RhoA | GTPase Ras homolog family member A |
| HAEC | human aortic endothelial cell |
| HA-VSMC | human aortic vascular smooth muscle cell |
| HCM | hypertrophic cardiomyopathy |
| HCN4 | potassium/sodium hyperpolarization-activated cyclic nucleotide-gated channel 4 |
| hCPC | human cardiac progenitor cell |
| HDL | high density lipoprotein |
| HEK | human embryonic kidney |
| hESC | human embryonic stem cell |
| hiPSC | human induced pluripotent stem cell |
| HLA-DR | human leukocyte antigen—DR isotype |
| HMGB1 | high-mobility group box-1 |
| HUVEC | human umbilical vein endothelial cell |
| ICAM1 | intercellular adhesion molecules 1 |
| IHF | ischemic heart failure |
| IK1 | inward rectifier K+ current |
| IKUR | ultra-rapid delayed rectifier K+ current |
| iPSC | induced pluripotent stem cell |
| IR | ischemia/reperfusion |
| Iso | isoproterenol hydrochloride |
| Ito | transient outward K+ current |
| JNK | c-Jun-N-terminal kinase |
| LDL | low-density lipoproteins |
| LOX1 | lectin-like oxLDL receptor 1 |
| LQT | long QT syndrome |
| MI | myocardial infarction |
| miR-133 | microRNA-133 |
| MMP | matrix metalloproteinase |
| MSC | mesenchymal stromal cell |
| mTOR | target of rapamycin |
| MYH11 | myosin heavy chain-11 |
| MYH7 | myosin heavy chain-7 |
| MYL2 | myosin light chain-2 |
| NANOG | homeobox protein NANOG |
| NCSC | neural crest stem cell |
| NF-κB | nuclear factor k-light-chain-enhancer of activated B cells |
| NRG | neuregulin |
| Oct-4 | octamer-binding transcription factor 4 |
| OOC | organ-on-a-chip |
| p38-MAPK | p38 mitogen-activated protein kinase |
| PAEC | pulmonary arterial endothelial cell |
| PASMC | pulmonary artery smooth muscle cells |
| PDGF | platelet-derived growth factor |
| PDGFRα | platelet-derived growth factor receptor α |
| PDMS | polydimethylsiloxane |
| PG | plakoglobin |
| PGA | polyglycolic acid |
| PKC-α | protein kinase C-α |
| PLA | polylactide acid |
| PMC | paraxial mesoderm cell |
| PQQ | pyrroloquinoline quinone |
| PRR | pro-renin receptor |
| RAS | renin-angiotensin system |
| ROS | reactive oxygen species |
| RyR | ryanodine receptor |
| S1PR1 | sphingosine-1-phosphate receptor 1 |
| Sox2 | sex determining region Y-box 2 |
| SSEA-4 | stage-specific embryonic antigen 4 |
| SVSM | smooth muscle cells from saphenous vein |
| TdP | torsade de pointes |
| TGF-β | transforming growth factor β |
| TNF-α | tumor necrosis factor-α |
| TNNT2 | troponin 2 |
| VCAM1 | vascular cell adhesion protein 1 |
| VSMC | vascular smooth muscle cell |
| WT | wild type |
| YAP1 | yes-associated protein 1 |
| β1AR | β1-adrenergic receptor |
| β-MHC | β-myosin heavy chain |
References
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report from the American Heart Association. Available online: https://pubmed.ncbi.nlm.nih.gov/30700139/ (accessed on 26 June 2020).
- Jimenez-Tellez, N.; Greenway, S.C. Cellular models for human cardiomyopathy: What is the best option? World J. Cardiol. 2019, 11, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Garbern, J.C.; Mummery, C.L.; Lee, R.T. Model Systems for Cardiovascular Regenerative Biology. Cold Spring Harb. Perspect. Med. 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommariva, E.; Stadiotti, I.; Perrucci, G.L.; Tondo, C.; Pompilio, G. Cell models of arrhythmogenic cardiomyopathy: Advances and opportunities. Dis. Model. Mech. 2017, 10, 823–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Han, J.; Li, H.; Zhang, X.; lan Liu, L.; Chen, F.; Zeng, B. Human Embryonic Kidney 293 Cells: A Vehicle for Biopharmaceutical Manufacturing, Structural Biology, and Electrophysiology. Cells Tissues Organs 2018, 205, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, R.; Baruscotti, M.; Gnecchi-Ruscone, T.; DiFrancesco, D. Familial Sinus Bradycardia Associated with a Mutation in the Cardiac Pacemaker Channel. Available online: https://www.nejm.org/doi/10.1056/NEJMoa052475?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub%3Dwww.ncbi.nlm.nih.gov (accessed on 2 December 2019).
- Barbuti, A.; Gravante, B.; Riolfo, M.; Milanesi, R.; Terragni, B.; DiFrancesco, D. Localization of Pacemaker Channels in Lipid Rafts Regulates Channel Kinetics. Circ. Res. 2004, 94, 1325–1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asimaki, A.; Syrris, P.; Wichter, T.; Matthias, P.; Saffitz, J.E.; McKenna, W.J. A Novel Dominant Mutation in Plakoglobin Causes Arrhythmogenic Right Ventricular Cardiomyopathy. Am. J. Hum. Genet. 2007, 81, 964–973. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, A.; Rengo, G.; Liccardo, D.; Pagano, G.; Zincarelli, C.; De Angelis, M.C.; Puglia, R.; Di Pietro, E.; Rabinowitz, J.E.; Barone, M.V.; et al. β1-Adrenergic Receptor and Sphingosine-1-Phosphate Receptor 1 Reciprocal Down-Regulation Influences Cardiac Hypertrophic Response and Progression Toward Heart Failure: Protective Role of S1PR1 Cardiac Gene Therapy. Circulation 2013, 128, 1612–1622. [Google Scholar] [CrossRef] [Green Version]
- Asimaki, A.; Protonotarios, A.; James, C.A.; Chelko, S.P.; Tichnell, C.; Murray, B.; Tsatsopoulou, A.; Anastasakis, A.; te Riele, A.; Kléber, A.G.; et al. Characterizing the Molecular Pathology of Arrhythmogenic Cardiomyopathy in Patient Buccal Mucosa Cells. Circ. Arrhythm. Electrophysiol. 2016, 9, e003688. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.S.M.; Huzen, J.; de Boer, R.A.; van Gilst, W.H.; van Veldhuisen, D.J.; van der Harst, P. Telomere Length of Circulating Leukocyte Subpopulations and Buccal Cells in Patients with Ischemic Heart Failure and Their Offspring. PLoS ONE 2011, 6, e23118. [Google Scholar] [CrossRef] [Green Version]
- Severs, N.J. The cardiac muscle cell. BioEssays News Rev. Mol. Cell. Dev. Biol. 2000, 22, 188–199. [Google Scholar] [CrossRef]
- Coppini, R.; Ferrantini, C.; Aiazzi, A.; Mazzoni, L.; Sartiani, L.; Mugelli, A.; Poggesi, C.; Cerbai, E. Isolation and Functional Characterization of Human Ventricular Cardiomyocytes from Fresh Surgical Samples. J. Vis. Exp. 2014, 86, e51116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuckelmann, D.J.; Näbauer, M.; Erdmann, E. Alterations of K+ currents in isolated human ventricular myocytes from patients with terminal heart failure. Circ. Res. 1993, 73, 379–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Wible, B.; Li, G.-R.; Wang, Z.; Nattel, S. Antisense Oligodeoxynucleotides Directed Against Kv1.5 mRNA Specifically Inhibit Ultrarapid Delayed Rectifier K+ Current in Cultured Adult Human Atrial Myocytes. Circ. Res. 1997, 80, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Ferrantini, C.; Pioner, J.M.; Mazzoni, L.; Gentile, F.; Tosi, B.; Rossi, A.; Belardinelli, L.; Tesi, C.; Palandri, C.; Matucci, R.; et al. Late sodium current inhibitors to treat exercise-induced obstruction in hypertrophic cardiomyopathy: An in vitro study in human myocardium. Br. J. Pharmacol. 2018, 175, 2635–2652. [Google Scholar] [CrossRef] [Green Version]
- Coppini, R.; Ferrantini, C.; Pioner, J.M.; Santini, L.; Wang, Z.J.; Palandri, C.; Scardigli, M.; Vitale, G.; Sacconi, L.; Stefàno, P.; et al. Electrophysiological and Contractile Effects of Disopyramide in Patients with Obstructive Hypertrophic Cardiomyopathy. JACC Basic Transl. Sci. 2019, 4, 795–813. [Google Scholar] [CrossRef]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Haniffa, M.A.; Collin, M.P.; Buckley, C.D.; Dazzi, F. Mesenchymal stem cells: The fibroblasts’ new clothes? Haematologica 2009, 94, 258–263. [Google Scholar] [CrossRef]
- Brown, R.D.; Ambler, S.K.; Mitchell, M.D.; Long, C.S. THE CARDIAC FIBROBLAST: Therapeutic Target in Myocardial Remodeling and Failure. Annu. Rev. Pharmacol. Toxicol. 2004, 45, 657–687. [Google Scholar] [CrossRef]
- Pilato, C.A.; Stadiotti, I.; Maione, A.S.; Saverio, V.; Catto, V.; Tundo, F.; Dello Russo, A.; Tondo, C.; Pompilio, G.; Casella, M.; et al. Isolation and Characterization of Cardiac Mesenchymal Stromal Cells from Endomyocardial Bioptic Samples of Arrhythmogenic Cardiomyopathy Patients. J. Vis. Exp. 2018, 132, e57263. [Google Scholar] [CrossRef]
- Rossini, A.; Frati, C.; Lagrasta, C.; Graiani, G.; Scopece, A.; Cavalli, S.; Musso, E.; Baccarin, M.; Di Segni, M.; Fagnoni, F.; et al. Human cardiac and bone marrow stromal cells exhibit distinctive properties related to their origin. Cardiovasc. Res. 2011, 89, 650–660. [Google Scholar] [CrossRef]
- Kang, I.S.; Suh, J.; Lee, M.-N.; Lee, C.; Jin, J.; Lee, C.; Yang, Y.I.; Jang, Y.; Oh, G.T. Characterization of human cardiac mesenchymal stromal cells and their extracellular vesicles comparing with human bone marrow derived mesenchymal stem cells. BMB Rep. 2020, 53, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Maggio, S.; Milano, G.; De Marchis, F.; D’Ambrosio, A.; Bertolotti, M.; Palacios, B.S.; Badi, I.; Sommariva, E.; Pompilio, G.; Capogrossi, M.C.; et al. Non-oxidizable HMGB1 induces cardiac fibroblasts migration via CXCR4 in a CXCL12-independent manner and worsens tissue remodeling after myocardial infarction. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2017, 1863, 2693–2704. [Google Scholar] [CrossRef] [PubMed]
- Sommariva, E.; Brambilla, S.; Carbucicchio, C.; Gambini, E.; Meraviglia, V.; Dello Russo, A.; Farina, F.M.; Casella, M.; Catto, V.; Pontone, G.; et al. Cardiac mesenchymal stromal cells are a source of adipocytes in arrhythmogenic cardiomyopathy. Eur. Heart J. 2016, 37, 1835–1846. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Structure and regulation of Kit protein-tyrosine kinase—The stem cell factor receptor. Biochem. Biophys. Res. Commun. 2005, 338, 1307–1315. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult Cardiac Stem Cells Are Multipotent and Support Myocardial Regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Chopra, H.; Hung, M.K.; Kwong, D.L.; Zhang, C.F.; Pow, E.H.N. Insights into Endothelial Progenitor Cells: Origin, Classification, Potentials, and Prospects. Stem Cells Int. 2018, 2018. [Google Scholar] [CrossRef]
- Gambini, E.; Pompilio, G.; Biondi, A.; Alamanni, F.; Capogrossi, M.C.; Agrifoglio, M.; Pesce, M. C-kit+ cardiac progenitors exhibit mesenchymal markers and preferential cardiovascular commitment. Cardiovasc. Res. 2011, 89, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Van Berlo, J.H.; Kanisicak, O.; Maillet, M.; Vagnozzi, R.J.; Karch, J.; Lin, S.-C.J.; Middleton, R.C.; Marbán, E.; Molkentin, J.D. c-kit+ Cells Minimally Contribute Cardiomyocytes to the Heart. Nature 2014, 509, 337–341. [Google Scholar] [CrossRef]
- Gambini, E.; Perrucci, G.L.; Bassetti, B.; Spaltro, G.; Campostrini, G.; Lionetti, M.C.; Pilozzi, A.; Martinelli, F.; Farruggia, A.; DiFrancesco, D.; et al. Preferential myofibroblast differentiation of cardiac mesenchymal progenitor cells in the presence of atrial fibrillation. Transl. Res. J. Lab. Clin. Med. 2018, 192, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y.; et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell Sci. 2011, 124, 3654–3664. [Google Scholar] [CrossRef] [Green Version]
- Stadiotti, I.; Catto, V.; Casella, M.; Tondo, C.; Pompilio, G.; Sommariva, E. Arrhythmogenic Cardiomyopathy: The Guilty Party in Adipogenesis. J. Cardiovasc. Transl. Res. 2017, 10, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Maione, A.S.; Pilato, C.A.; Casella, M.; Gasperetti, A.; Stadiotti, I.; Pompilio, G.; Sommariva, E. Fibrosis in Arrhythmogenic Cardiomyopathy: The Phantom Thread in the Fibro-Adipose Tissue. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardi, R.; Chen, S.N.; Ruggiero, A.; Gurha, P.; Czernuszewicz, G.Z.; Willerson, J.T.; Marian, A.J. Cardiac Fibro-Adipocyte Progenitors Express Desmosome Proteins and Preferentially Differentiate to Adipocytes Upon Deletion of the Desmoplakin Gene. Circ. Res. 2016, 119, 41–54. [Google Scholar] [CrossRef]
- Soliman, H.; Paylor, B.; Scott, R.W.; Lemos, D.R.; Chang, C.; Arostegui, M.; Low, M.; Lee, C.; Fiore, D.; Braghetta, P.; et al. Pathogenic Potential of Hic1-Expressing Cardiac Stromal Progenitors. Cell Stem Cell 2020, 26, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Pereira, I.A.; Borba, E.F. The role of inflammation, humoral and cell mediated autoimmunity in the pathogenesis of atherosclerosis. Swiss Med. Wkly. 2008, 138, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Medina-Leyte, D.J.; Domínguez-Pérez, M.; Mercado, I.; Villarreal-Molina, M.T.; Jacobo-Albavera, L. Use of Human Umbilical Vein Endothelial Cells (HUVEC) as a Model to Study Cardiovascular Disease: A Review. Appl. Sci. 2020, 10, 938. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Lan, M.; Wei, H.; Zhang, D.; Liu, J.; Teng, J. Downregulated microRNA-133a induces HUVECs injury: Potential role of the (pro) renin receptor in angiotensin II-dependent hypertension. Mol. Med. Rep. 2019, 20, 2796–2804. [Google Scholar] [CrossRef] [Green Version]
- Torres-Vázquez, J.; Kamei, M.; Weinstein, B.M. Molecular distinction between arteries and veins. Cell Tissue Res. 2003, 314, 43–59. [Google Scholar] [CrossRef]
- Deng, D.X.F.; Tsalenko, A.; Vailaya, A.; Ben-Dor, A.; Kundu, R.; Estay, I.; Tabibiazar, R.; Kincaid, R.; Yakhini, Z.; Bruhn, L.; et al. Differences in Vascular Bed Disease Susceptibility Reflect Differences in Gene Expression Response to Atherogenic Stimuli. Circ. Res. 2006, 98, 200–208. [Google Scholar] [CrossRef]
- Fan, X.; Chen, X.; Feng, Q.; Peng, K.; Wu, Q.; Passerini, A.G.; Simon, S.I.; Sun, C. Downregulation of GATA6 in mTOR-inhibited human aortic endothelial cells: Effects on TNF-α-induced VCAM-1 expression and monocytic cell adhesion. Am. J. Physiol.-Heart Circ. Physiol. 2018, 316, H408–H420. [Google Scholar] [CrossRef]
- Wynants, M.; Quarck, R.; Ronisz, A.; Alfaro-Moreno, E.; Raemdonck, D.V.; Meyns, B.; Delcroix, M. Effects of C-reactive protein on human pulmonary vascular cells in chronic thromboembolic pulmonary hypertension. Eur. Respir. J. 2012, 40, 886–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacakova, L.; Travnickova, M.; Filova, E.; Matějka, R.; Stepanovska, J.; Musilkova, J.; Zarubova, J.; Molitor, M. The Role of Vascular Smooth Muscle Cells in the Physiology and Pathophysiology of Blood Vessels. Muscle Cell Tissue Curr. Status Res. Field 2018, 229. [Google Scholar] [CrossRef] [Green Version]
- Red-Horse, K.; Siekmann, A.F. Veins and Arteries Build Hierarchical Branching Patterns Differently: Bottom-Up versus Top-Down. BioEssays News Rev. Mol. Cell. Dev. Biol. 2019, 41, e1800198. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lin, C.; Zhou, H.J.; Min, W. Smooth muscle cell differentiation: Mechanisms and models for vascular diseases. Front. Biol. 2017, 12, 392–405. [Google Scholar] [CrossRef]
- Cheng, Q.; Zhang, M.; Zhang, M.; Ning, L.; Chen, D. Long non-coding RNA LOC285194 regulates vascular smooth muscle cell apoptosis in atherosclerosis. Bioengineered 2020, 11, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Quarck, R.; Wynants, M.; Ronisz, A.; Sepulveda, M.R.; Wuytack, F.; Van Raemdonck, D.; Meyns, B.; Delcroix, M. Characterization of proximal pulmonary arterial cells from chronic thromboembolic pulmonary hypertension patients. Respir. Res. 2012, 13, 27. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-X.; Li, J.-F.; Yang, Y.-H.; Zhai, Z.-G.; Gu, S.; Liu, Y.; Miao, R.; Zhong, P.-P.; Wang, Y.; Huang, X.-X.; et al. Renin-angiotensin system regulates pulmonary arterial smooth muscle cell migration in chronic thromboembolic pulmonary hypertension. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 314, L276–L286. [Google Scholar] [CrossRef]
- Davidson, M.M.; Nesti, C.; Palenzuela, L.; Walker, W.F.; Hernandez, E.; Protas, L.; Hirano, M.; Isaac, N.D. Novel cell lines derived from adult human ventricular cardiomyocytes. J. Mol. Cell. Cardiol. 2005, 39, 133–147. [Google Scholar] [CrossRef]
- Wen, J.; Shen, J.; Zhou, Y.; Zhao, X.; Dai, Z.; Jin, Y. Pyrroloquinoline quinone attenuates isoproterenol hydrochloride-induced cardiac hypertrophy in AC16 cells by inhibiting the NF-κB signaling pathway. Int. J. Mol. Med. 2020, 45, 873–885. [Google Scholar] [CrossRef]
- Khan, K.; Makhoul, G.; Yu, B.; Schwertani, A.; Cecere, R. The cytoprotective impact of yes-associated protein 1 after ischemia-reperfusion injury in AC16 human cardiomyocytes. Exp. Biol. Med. 2019, 244, 802–812. [Google Scholar] [CrossRef]
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reubinoff, B.E.; Pera, M.F.; Fong, C.Y.; Trounson, A.; Bongso, A. Embryonic stem cell lines from human blastocysts: Somatic differentiation in vitro. Nat. Biotechnol. 2000, 18, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Levenberg, S.; Golub, J.S.; Amit, M.; Itskovitz-Eldor, J.; Langer, R. Endothelial cells derived from human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 4391–4396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Wilson, K.D.; Smith, B.; Kraft, D.L.; Jia, F.; Huang, M.; Xie, X.; Robbins, R.C.; Gambhir, S.S.; Weissman, I.L.; et al. Functional and Transcriptional Characterization of Human Embryonic Stem Cell-Derived Endothelial Cells for Treatment of Myocardial Infarction. PLoS ONE 2009, 4, e8443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsch, C.; Challet-Meylan, L.; Thoma, E.C.; Urich, E.; Heckel, T.; O’Sullivan, J.F.; Grainger, S.J.; Kapp, F.G.; Sun, L.; Christensen, K.; et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 2015, 17, 994–1003. [Google Scholar] [CrossRef]
- Dambrot, C.; Passier, R.; Atsma, D.; Mummery, C.L. Cardiomyocyte differentiation of pluripotent stem cells and their use as cardiac disease models. Biochem. J. 2011, 434, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Wagner, R.A.; Wilson, K.D.; Xie, X.; Fu, J.-D.; Drukker, M.; Lee, A.; Li, R.A.; Gambhir, S.S.; Weissman, I.L.; et al. Transcriptional and functional profiling of human embryonic stem cell-derived cardiomyocytes. PLoS ONE 2008, 3, e3474. [Google Scholar] [CrossRef] [Green Version]
- Blazeski, A.; Zhu, R.; Hunter, D.W.; Weinberg, S.H.; Boheler, K.R.; Zambidis, E.T.; Tung, L. Electrophysiological and contractile function of cardiomyocytes derived from human embryonic stem cells. Prog. Biophys. Mol. Biol. 2012, 110, 178–195. [Google Scholar] [CrossRef] [Green Version]
- Devalla, H.D.; Schwach, V.; Ford, J.W.; Milnes, J.T.; El-Haou, S.; Jackson, C.; Gkatzis, K.; Elliott, D.A.; Chuva de Sousa Lopes, S.M.; Mummery, C.L.; et al. Atrial-like cardiomyocytes from human pluripotent stem cells are a robust preclinical model for assessing atrial-selective pharmacology. EMBO Mol. Med. 2015, 7, 394–410. [Google Scholar] [CrossRef]
- Zhu, W.-Z.; Xie, Y.; Moyes, K.W.; Gold, J.D.; Askari, B.; Laflamme, M.A. Neuregulin/ErbB Signaling Regulates Cardiac Subtype Specification in Differentiating Human Embryonic Stem Cells. Circ. Res. 2010, 107, 776–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Földes, G.; Mioulane, M.; Wright, J.S.; Liu, A.Q.; Novak, P.; Merkely, B.; Gorelik, J.; Schneider, M.D.; Ali, N.N.; Harding, S.E. Modulation of human embryonic stem cell-derived cardiomyocyte growth: A testbed for studying human cardiac hypertrophy? J. Mol. Cell. Cardiol. 2011, 50, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.-Y.; Chien, C.-S.; Yarmishyn, A.A.; Chou, S.-J.; Yang, Y.-P.; Wang, M.-L.; Wang, C.-Y.; Leu, H.-B.; Yu, W.-C.; Chang, Y.-L.; et al. Generation of GLA-Knockout Human Embryonic Stem Cell Lines to Model Autophagic Dysfunction and Exosome Secretion in Fabry Disease-Associated Hypertrophic Cardiomyopathy. Cells 2019, 8, 327. [Google Scholar] [CrossRef] [Green Version]
- Gurdon, J.B. Nuclear Transplantation in Eggs and Oocytes. J. Cell Sci. 1986, 1986, 287–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, S. Strategies and New Developments in the Generation of Patient-Specific Pluripotent Stem Cells. Cell Stem Cell 2007, 1, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, J.; Kolanowski, T.J.; Kurpisz, M. Techniques for the induction of human pluripotent stem cell differentiation towards cardiomyocytes. J. Tissue Eng. Regen. Med. 2017, 11, 1658–1674. [Google Scholar] [CrossRef]
- Yang, X.; Pabon, L.; Murry, C.E. Engineering Adolescence. Circ. Res. 2014, 114, 511–523. [Google Scholar] [CrossRef] [Green Version]
- Barbuti, A.; Benzoni, P.; Campostrini, G.; Dell’Era, P. Human derived cardiomyocytes: A decade of knowledge after the discovery of induced pluripotent stem cells. Dev. Dyn. 2016, 245, 1145–1158. [Google Scholar] [CrossRef] [Green Version]
- Karakikes, I.; Ameen, M.; Termglinchan, V.; Wu, J.C. Human Induced Pluripotent Stem Cell–Derived Cardiomyocytes. Circ. Res. 2015, 117, 80–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Jiang, J.; Han, P.; Yuan, Q.; Zhang, J.; Zhang, X.; Xu, Y.; Cao, H.; Meng, Q.; Chen, L.; et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011, 21, 579–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, P.A.; Darche, F.F.; Ullrich, N.D.; Geschwill, P.; Greber, B.; Rivinius, R.; Seyler, C.; Müller-Decker, K.; Draguhn, A.; Utikal, J.; et al. Subtype-specific differentiation of cardiac pacemaker cell clusters from human induced pluripotent stem cells. Stem Cell Res. Ther. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzoni, P.; Campostrini, G.; Landi, S.; Bertini, V.; Marchina, E.; Iascone, M.; Ahlberg, G.; Olesen, M.S.; Crescini, E.; Mora, C.; et al. Human iPSC modelling of a familial form of atrial fibrillation reveals a gain of function of If and ICaL in patient-derived cardiomyocytes. Cardiovasc. Res. 2020, 116, 1147–1160. [Google Scholar] [CrossRef] [Green Version]
- Van Wagoner David, R.; Pond Amber, L.; Lamorgese, M.; Rossie Sandra, S.; McCarthy Patrick, M.; Nerbonne Jeanne, M. Atrial L-Type Ca2+ Currents and Human Atrial Fibrillation. Circ. Res. 1999, 85, 428–436. [Google Scholar] [CrossRef] [Green Version]
- Lan, F.; Lee, A.S.; Liang, P.; Sanchez-Freire, V.; Nguyen, P.K.; Wang, L.; Han, L.; Yen, M.; Wang, Y.; Sun, N.; et al. Abnormal Calcium Handling Properties Underlie Familial Hypertrophic Cardiomyopathy Pathology in Patient-Specific Induced Pluripotent Stem Cells. Cell Stem Cell 2013, 12, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Sala, L.; Gnecchi, M.; Schwartz, P.J. Long QT Syndrome Modelling with Cardiomyocytes Derived from Human-induced Pluripotent Stem Cells. Arrhythmia Electrophysiol. Rev. 2019, 8, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Peng, G.-Y.; Lin, Y.; Li, J.-J.; Wang, Y.; Huang, H.-Y.; Shen, Z.-Y. The Application of Induced Pluripotent Stem Cells in Pathogenesis Study and Gene Therapy for Vascular Disorders: Current Progress and Future Challenges. Stem Cells Int. 2019, 2019. [Google Scholar] [CrossRef]
- Sa, S.; Gu, M.; Chappell, J.; Shao, N.-Y.; Ameen, M.; Elliott, K.A.T.; Li, D.; Grubert, F.; Li, C.G.; Taylor, S.; et al. Induced Pluripotent Stem Cell Model of Pulmonary Arterial Hypertension Reveals Novel Gene Expression and Patient Specificity. Am. J. Respir. Crit. Care Med. 2016, 195, 930–941. [Google Scholar] [CrossRef]
- Hitomi, T.; Habu, T.; Kobayashi, H.; Okuda, H.; Harada, K.H.; Osafune, K.; Taura, D.; Sone, M.; Asaka, I.; Ameku, T.; et al. Downregulation of Securin by the variant RNF213 R4810K (rs112735431, G>A) reduces angiogenic activity of induced pluripotent stem cell-derived vascular endothelial cells from moyamoya patients. Biochem. Biophys. Res. Commun. 2013, 438, 13–19. [Google Scholar] [CrossRef]
- Ge, X.; Ren, Y.; Bartulos, O.; Lee, M.Y.; Yue, Z.; Kim, K.Y.; Li, W.; Amos, P.J.; Bozkulak, E.C.; Iyer, A.; et al. Modeling Supravalvular Aortic Stenosis Syndrome With Human Induced Pluripotent Stem Cells. Circulation 2012, 126, 1695–1704. [Google Scholar] [CrossRef]
- Jiao, J.; Xiong, W.; Wang, L.; Yang, J.; Qiu, P.; Hirai, H.; Shao, L.; Milewicz, D.; Chen, Y.E.; Yang, B. Differentiation defect in neural crest-derived smooth muscle cells in patients with aortopathy associated with bicuspid aortic valves. EBioMedicine 2016, 10, 282–290. [Google Scholar] [CrossRef] [Green Version]
- Paschos, N.K.; Brown, W.E.; Eswaramoorthy, R.; Hu, J.C.; Athanasiou, K.A. Advances in tissue engineering through stem cell-based co-culture. J. Tissue Eng. Regen. Med. 2015, 9, 488–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mummery, C.L.; Zhang, J.; Ng, E.S.; Elliott, D.A.; Elefanty, A.G.; Kamp, T.J. Differentiation of Human ES and iPS Cells to Cardiomyocytes: A Methods Overview. Circ. Res. 2012, 111, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, M.C.; White, S.L.P.; Zhou, W. Design and utilization of macrophage and vascular smooth muscle cell co-culture systems in atherosclerotic cardiovascular disease investigation. Vasc. Med. 2014, 19, 394–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maione, A.S.; Cipolletta, E.; Sorriento, D.; Borriello, F.; Soprano, M.; Rusciano, M.R.; D’Esposito, V.; Markabaoui, A.K.; De Palma, G.D.; Martino, G.; et al. Cellular subtype expression and activation of CaMKII regulate the fate of atherosclerotic plaque. Atherosclerosis 2017, 256, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Sebastião, M.J.; Serra, M.; Pereira, R.; Palacios, I.; Gomes-Alves, P.; Alves, P.M. Human cardiac progenitor cell activation and regeneration mechanisms: Exploring a novel myocardial ischemia/reperfusion in vitro model. Stem Cell Res. Ther. 2019, 10, 77. [Google Scholar] [CrossRef] [Green Version]
- Wallace, C.S.; Truskey, G.A. Direct-contact co-culture between smooth muscle and endothelial cells inhibits TNF-α-mediated endothelial cell activation. Am. J. Physiol.-Heart Circ. Physiol. 2010, 299, H338–H346. [Google Scholar] [CrossRef] [Green Version]
- Vunjak-Novakovic, G.; Lui, K.O.; Tandon, N.; Chien, K.R. Bioengineering Heart Muscle: A Paradigm for Regenerative Medicine. Annu. Rev. Biomed. Eng. 2011, 13, 245–267. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.S.T.; Macadangdang, J.; Leung, W.; Laflamme, M.A.; Kim, D.-H. Human iPSC-derived cardiomyocytes and tissue engineering strategies for disease modeling and drug screening. Biotechnol. Adv. 2017, 35, 77–94. [Google Scholar] [CrossRef]
- Vunjak-Novakovic, G.; Tandon, N.; Godier, A.; Maidhof, R.; Marsano, A.; Martens, T.P.; Radisic, M. Challenges in Cardiac Tissue Engineering. Tissue Eng. Part B Rev. 2010, 16, 169–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroll, K.; Chabria, M.; Wang, K.; Häusermann, F.; Schuler, F.; Polonchuk, L. Electro-mechanical conditioning of human iPSC-derived cardiomyocytes for translational research. Prog. Biophys. Mol. Biol. 2017, 130, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Van Duinen, V.; Trietsch, S.J.; Joore, J.; Vulto, P.; Hankemeier, T. Microfluidic 3D cell culture: From tools to tissue models. Curr. Opin. Biotechnol. 2015, 35, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prandi, F.; Piola, M.; Soncini, M.; Colussi, C.; D’Alessandra, Y.; Penza, E.; Agrifoglio, M.; Vinci, M.C.; Polvani, G.; Gaetano, C.; et al. Adventitial Vessel Growth and Progenitor Cells Activation in an Ex Vivo Culture System Mimicking Human Saphenous Vein Wall Strain after Coronary Artery Bypass Grafting. PLoS ONE 2015, 10, e0117409. [Google Scholar] [CrossRef] [Green Version]
- Hoes, M.F.; Bomer, N.; van der Meer, P. Concise Review: The Current State of Human In Vitro Cardiac Disease Modeling: A Focus on Gene Editing and Tissue Engineering. Stem Cells Transl. Med. 2019, 8, 66–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fennema, E.; Rivron, N.; Rouwkema, J.; van Blitterswijk, C.; de Boer, J. Spheroid culture as a tool for creating 3D complex tissues. Trends Biotechnol. 2013, 31, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Chabria, M.; Figtree, G.A.; Polonchuk, L.; Gentile, C. Stem Cell-Derived Cardiac Spheroids as 3D In Vitro Models of the Human Heart Microenvironment. In Stem Cell Niche: Methods and Protocols; Turksen, K., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 51–59. ISBN 978-1-4939-9508-0. [Google Scholar]
- Zamani, M.; Karaca, E.; Huang, N.F. Multicellular Interactions in 3D Engineered Myocardial Tissue. Front. Cardiovasc. Med. 2018, 5, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, S.; Shimizu, T. Three-dimensional cardiac tissue fabrication based on cell sheet technology. Adv. Drug Deliv. Rev. 2016, 96, 103–109. [Google Scholar] [CrossRef]
- Caspi, O.; Lesman, A.; Basevitch, Y.; Gepstein, A.; Arbel, G.; Habib, I.H.M.; Gepstein, L.; Levenberg, S. Tissue Engineering of Vascularized Cardiac Muscle From Human Embryonic Stem Cells. Circ. Res. 2007, 100, 263–272. [Google Scholar] [CrossRef]
- Kenar, H.; Kose, G.T.; Toner, M.; Kaplan, D.L.; Hasirci, V. A 3D aligned microfibrous myocardial tissue construct cultured under transient perfusion. Biomaterials 2011, 32, 5320–5329. [Google Scholar] [CrossRef] [Green Version]
- Acun, A.; Zorlutuna, P. Engineered myocardium model to study the roles of HIF-1α and HIF1A-AS1 in paracrine-only signaling under pathological level oxidative stress. Acta Biomater. 2017, 58, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Saludas, L.; Pascual-Gil, S.; Prósper, F.; Garbayo, E.; Blanco-Prieto, M. Hydrogel based approaches for cardiac tissue engineering. Int. J. Pharm. 2017, 523, 454–475. [Google Scholar] [CrossRef] [PubMed]
- Zia, S.; Mozafari, M.; Natasha, G.; Tan, A.; Cui, Z.; Seifalian, A.M. Hearts beating through decellularized scaffolds: Whole-organ engineering for cardiac regeneration and transplantation. Crit. Rev. Biotechnol. 2016, 36, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Gershlak, J.R.; Hernandez, S.; Fontana, G.; Perreault, L.R.; Hansen, K.J.; Larson, S.A.; Binder, B.Y.K.; Dolivo, D.M.; Yang, T.; Dominko, T.; et al. Crossing kingdoms: Using decellularized plants as perfusable tissue engineering scaffolds. Biomaterials 2017, 125, 13–22. [Google Scholar] [CrossRef]
- Wang, R.M.; Christman, K.L. Decellularized myocardial matrix hydrogels: In basic research and preclinical studies. Adv. Drug Deliv. Rev. 2016, 96, 77–82. [Google Scholar] [CrossRef]
- Borovjagin, A.V.; Ogle Brenda, M.; Berry, J.L.; Zhang, J. From Microscale Devices to 3D Printing. Circ. Res. 2017, 120, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cui, R.; Sun, L.; Aifantis, K.E.; Fan, Y.; Feng, Q.; Cui, F.; Watari, F. 3D-Printed Biopolymers for Tissue Engineering Application. Available online: https://www.hindawi.com/journals/ijps/2014/829145/ (accessed on 31 May 2020).
- Mosadegh, B.; Xiong, G.; Dunham, S.; Min, J.K. Current progress in 3D printing for cardiovascular tissue engineering. Biomed. Mater. 2015, 10, 034002. [Google Scholar] [CrossRef]
- Gao, L.; Kupfer, M.E.; Jung, J.P.; Yang, L.; Zhang, P.; Sie, Y.D.; Tran, Q.; Ajeti, V.; Freeman, B.T.; Fast, V.G.; et al. Myocardial Tissue Engineering With Cells Derived from Human Induced-Pluripotent Stem Cells and a Native-Like, High-Resolution, 3-Dimensionally Printed Scaffold. Circ. Res. 2017, 120, 1318–1325. [Google Scholar] [CrossRef] [Green Version]
- Gaetani, R.; Doevendans, P.A.; Metz, C.H.G.; Alblas, J.; Messina, E.; Giacomello, A.; Sluijter, J.P.G. Cardiac tissue engineering using tissue printing technology and human cardiac progenitor cells. Biomaterials 2012, 33, 1782–1790. [Google Scholar] [CrossRef]
- Pati, F.; Cho, D.-W. Bioprinting of 3D Tissue Models Using Decellularized Extracellular Matrix Bioink. In 3D Cell Culture: Methods and Protocols; Koledova, Z., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 381–390. ISBN 978-1-4939-7021-6. [Google Scholar]
- Lee, J.; Razu, M.E.; Wang, X.; Lacerda, C.; Kim, J. Biomimetic Cardiac Microsystems for Pathophysiological Studies and Drug Screens. J. Lab. Autom. 2015, 20, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Visone, R.; Talò, G.; Occhetta, P.; Cruz-Moreira, D.; Lopa, S.; Pappalardo, O.A.; Redaelli, A.; Moretti, M.; Rasponi, M. A microscale biomimetic platform for generation and electro-mechanical stimulation of 3D cardiac microtissues. APL Bioeng. 2018, 2. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, C.G.M.; Brandt, M.M.; Poulis, N.; Anten, J.; van der Moolen, M.; Kramer, L.; Homburg, E.F.G.A.; Louzao-Martinez, L.; Pei, J.; Krebber, M.M.; et al. A new microfluidic model that allows monitoring of complex vascular structures and cell interactions in a 3D biological matrix. Lab Chip 2020, 20, 1827–1844. [Google Scholar] [CrossRef]
- Lam, S.F.; Shirure, V.S.; Chu, Y.E.; Soetikno, A.G.; George, S.C. Microfluidic device to attain high spatial and temporal control of oxygen. PLoS ONE 2018, 13, e0209574. [Google Scholar] [CrossRef] [Green Version]
- Pesce, M.; Santoro, R. Feeling the right force: How to contextualize the cell mechanical behavior in physiologic turnover and pathologic evolution of the cardiovascular system. Pharmacol. Ther. 2017, 171, 75–82. [Google Scholar] [CrossRef]
- Marsano, A.; Conficconi, C.; Lemme, M.; Occhetta, P.; Gaudiello, E.; Votta, E.; Cerino, G.; Redaelli, A.; Rasponi, M. Beating heart on a chip: A novel microfluidic platform to generate functional 3D cardiac microtissues. Lab Chip 2016, 16, 599–610. [Google Scholar] [CrossRef]
- Campisi, M.; Shin, Y.; Osaki, T.; Hajal, C.; Chiono, V.; Kamm, R.D. 3D self-organized microvascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes. Biomaterials 2018, 180, 117–129. [Google Scholar] [CrossRef]
- Giacomelli, E.; Meraviglia, V.; Campostrini, G.; Cochrane, A.; Cao, X.; van Helden, R.W.J.; Krotenberg Garcia, A.; Mircea, M.; Kostidis, S.; Davis, R.P.; et al. Human-iPSC-Derived Cardiac Stromal Cells Enhance Maturation in 3D Cardiac Microtissues and Reveal Non-cardiomyocyte Contributions to Heart Disease. Cell Stem Cell 2020, 26, 862–879. [Google Scholar] [CrossRef]
- Kawatou, M.; Masumoto, H.; Fukushima, H.; Morinaga, G.; Sakata, R.; Ashihara, T.; Yamashita, J.K. Modelling Torsade de Pointes arrhythmias in vitro in 3D human iPS cell-engineered heart tissue. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Vunjak-Novakovic, G. Human Tissue-Engineered Model of Myocardial Ischemia–Reperfusion Injury. Tissue Eng. Part A 2019, 25, 711–724. [Google Scholar] [CrossRef]
- Robert, J.; Weber, B.; Frese, L.; Emmert, M.Y.; Schmidt, D.; von Eckardstein, A.; Rohrer, L.; Hoerstrup, S.P. A Three-Dimensional Engineered Artery Model for In Vitro Atherosclerosis Research. PLoS ONE 2013, 8, e79821. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Koo, S.; Finnegan, M.A.; Loskill, P.; Huebsch, N.; Marks, N.C.; Conklin, B.R.; Grigoropoulos, C.P.; Healy, K.E. Three Dimension Filamentous Human Cardiac Tissue Model. Biomaterials 2014, 35, 1367–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Valk, D.C.; Van der Ven, C.F.T.; Blaser, M.C.; Grolman, J.M.; Wu, P.-J.; Fenton, O.S.; Lee, L.H.; Tibbitt, M.W.; Andresen, J.L.; Wen, J.R.; et al. Engineering a 3D-Bioprinted Model of Human Heart Valve Disease Using Nanoindentation-Based Biomechanics. Nanomaterials 2018, 8, 296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zangi, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with iPSC and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Mastikhina, O.; Moon, B.-U.; Williams, K.; Hatkar, R.; Gustafson, D.; Mourad, O.; Sun, X.; Koo, M.; Lam, A.Y.L.; Sun, Y.; et al. Human cardiac fibrosis-on-a-chip model recapitulates disease hallmarks and can serve as a platform for drug testing. Biomaterials 2020, 233, 119741. [Google Scholar] [CrossRef]
- Claycomb, W.C.; Lanson, N.A.; Stallworth, B.S.; Egeland, D.B.; Delcarpio, J.B.; Bahinski, A.; Izzo, N.J. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc. Natl. Acad. Sci. USA 1998, 95, 2979–2984. [Google Scholar] [CrossRef] [Green Version]
- Marczenke, M.; Fell, J.; Piccini, I.; Röpke, A.; Seebohm, G.; Greber, B. Generation and cardiac subtype-specific differentiation of PITX2-deficient human iPS cell lines for exploring familial atrial fibrillation. Stem Cell Res. 2017, 21, 26–28. [Google Scholar] [CrossRef]
- Karbassi, E.; Fenix, A.; Marchiano, S.; Muraoka, N.; Nakamura, K.; Yang, X.; Murry, C.E. Cardiomyocyte maturation: Advances in knowledge and implications for regenerative medicine. Nat. Rev. Cardiol. 2020, 17, 341–359. [Google Scholar] [CrossRef]
- Santoro, R.; Perrucci, G.L.; Gowran, A.; Pompilio, G. Unchain My Heart: Integrins at the Basis of iPSC Cardiomyocyte Differentiation. Stem Cells Int. 2019, 2019, 8203950. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Arneri, A.; Bersini, S.; Shin, S.-R.; Zhu, K.; Goli-Malekabadi, Z.; Aleman, J.; Colosi, C.; Busignani, F.; Dell’Erba, V.; et al. Bioprinting 3D microfibrous scaffolds for engineering endothelialized myocardium and heart-on-a-chip. Biomaterials 2016, 110, 45–59. [Google Scholar] [CrossRef] [Green Version]
- Vunjak-Novakovic, G.; Bhatia, S.; Chen, C.; Hirschi, K. HeLiVa platform: Integrated heart-liver-vascular systems for drug testing in human health and disease. Stem Cell Res. Ther. 2013, 4, S8. [Google Scholar] [CrossRef] [Green Version]
- Rajan, S.A.P.; Aleman, J.; Wan, M.; Pourhabibi Zarandi, N.; Nzou, G.; Murphy, S.; Bishop, C.E.; Sadri-Ardekani, H.; Shupe, T.; Atala, A.; et al. Probing prodrug metabolism and reciprocal toxicity with an integrated and humanized multi-tissue organ-on-a-chip platform. Acta Biomater. 2020, 106, 124–135. [Google Scholar] [CrossRef]
- Skardal, A.; Murphy, S.V.; Devarasetty, M.; Mead, I.; Kang, H.-W.; Seol, Y.-J.; Shrike Zhang, Y.; Shin, S.-R.; Zhao, L.; Aleman, J.; et al. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Antman, E.M.; Loscalzo, J. Precision medicine in cardiology. Nat. Rev. Cardiol. 2016, 13, 591–602. [Google Scholar] [CrossRef]
- Frolov, A.V.; Vaikhanskaya, T.G.; Melnikova, O.P.; Vorobiev, A.P.; Guel, L.M. Risk stratification personalised model for prediction of life-threatening ventricular tachyarrhythmias in patients with chronic heart failure. Kardiol. Pol. 2017, 75, 682–688. [Google Scholar] [CrossRef]
- Akhtar, M.; Elliott, P.M. Risk Stratification for Sudden Cardiac Death in Non-Ischaemic Dilated Cardiomyopathy. Curr. Cardiol. Rep. 2019, 21, 155. [Google Scholar] [CrossRef] [Green Version]
- Kotchen, T.A.; Cowley, A.W.; Liang, M. Ushering Hypertension into a New Era of Precision Medicine. JAMA 2016, 315, 343–344. [Google Scholar] [CrossRef]
- Brown, S.-A.; Pereira, N. Pharmacogenomic Impact of CYP2C19 Variation on Clopidogrel Therapy in Precision Cardiovascular Medicine. J. Pers. Med. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Gregorich, Z.R.; Zhu, W.; Mattapally, S.; Oduk, Y.; Lou, X.; Kannappan, R.; Borovjagin, A.V.; Walcott, G.P.; Pollard, A.E.; et al. Large Cardiac-muscle Patches Engineered from Human Induced-pluripotent Stem-cell–derived Cardiac Cells Improve Recovery from Myocardial Infarction in Swine. Circulation 2018, 137, 1712–1730. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, R.Y.; Park, B.-W.; Lee, S.; Choi, S.W.; Park, J.-H.; Choi, J.J.; Kim, S.-W.; Jang, J.; Cho, D.-W.; et al. Dual stem cell therapy synergistically improves cardiac function and vascular regeneration following myocardial infarction. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Anderson, C.W.; Sewanan, L.R.; Kural, M.H.; Huang, Y.; Luo, J.; Gui, L.; Riaz, M.; Lopez, C.A.; Ng, R.; et al. Modular design of a tissue engineered pulsatile conduit using human induced pluripotent stem cell-derived cardiomyocytes. Acta Biomater. 2020, 102, 220–230. [Google Scholar] [CrossRef]
- Li, Z.; Han, Z.; Wu, J.C. Transplantation of human embryonic stem cell-derived endothelial cells for vascular diseases. J. Cell. Biochem. 2009, 106, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Qin, N.; Lu, X.-A.; Li, J.; Han, X.; Ni, X.; Ye, L.; Shen, Z.; Chen, W.; Zhao, Z.-A.; et al. Human embryonic stem cell-derived cardiomyocyte therapy in mouse permanent ischemia and ischemia-reperfusion models. Stem Cell Res. Ther. 2019, 10, 167. [Google Scholar] [CrossRef] [Green Version]
- Kawamura, M.; Miyagawa, S.; Miki, K.; Saito, A.; Fukushima, S.; Higuchi, T.; Kawamura, T.; Kuratani, T.; Daimon, T.; Shimizu, T.; et al. Feasibility, Safety, and Therapeutic Efficacy of Human Induced Pluripotent Stem Cell-Derived Cardiomyocyte Sheets in a Porcine Ischemic Cardiomyopathy Model. Circulation 2012, 126, S29–S37. [Google Scholar] [CrossRef] [Green Version]
- Miura, K.; Okada, Y.; Aoi, T.; Okada, A.; Takahashi, K.; Okita, K.; Nakagawa, M.; Koyanagi, M.; Tanabe, K.; Ohnuki, M.; et al. Variation in the safety of induced pluripotent stem cell lines. Nat. Biotechnol. 2009, 27, 743–745. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Cell Models | Cardiovascular Diseases | Clinical Relevance | References |
|---|---|---|---|
| HEK 293 | Familial sinus bradycardia | Pathogenic mechanism | [6] |
| Arrhythmogenic Cardiomyopathy | Pathogenic mechanism | [8] | |
| BMCs | Arrhythmogenic Cardiomyopathy | Pathogenic mechanism | [10] |
| Heart failure | - | [11] | |
| CMs | Heart failure | Pathogenic mechanism | [14] |
| Hypertrophic Cardiomyopathy | Drug discovery | [16,17] | |
| C-MSCs | Myocardial ischemia | Pathogenic mechanism | [24] |
| Arrhythmogenic Cardiomyopathy | Pathogenic mechanism/Involvement in disease pathogenesis | [25] | |
| c-kit + C-MSCs | Atrial Fibrillation | Pathogenic mechanism | [31] |
| FAPs | Arrhythmogenic Cardiomyopathy | Involvement in disease pathogenesis | [35] |
| ECs | Hypertension | Pathogenic mechanism | [39,43] |
| Atherosclerosis | Pathogenic mechanism/Involvement in disease pathogenesis | [41] | |
| Pathogenic mechanism | [42] | ||
| VSMCs | Hypertension | Involvement in disease pathogenesis | [48] |
| Pathogenic mechanism | [49] | ||
| Atherosclerosis | Differentiation phenotypes | [46] | |
| Pathogenic mechanism/Drug discovery | [47] | ||
| AC 16 | Hypertrophic Cardiomyopathy | Drug discovery | [51] |
| Myocardial ischemia | Pathogenic mechanism | [52] | |
| hESC-CMs | Hypertrophic Cardiomyopathy | Pathogenic mechanism | [64,65] |
| hiPSC-CMs | Atrial Fibrillation | Pathogenic mechanism | [76] |
| Hypertrophic Cardiomyopathy | Drug discovery | [78] | |
| Long QT Syndrome | Pathogenic mechanism/Drug discovery | [79] | |
| hiPSC-ECs | Hypertension | Drug discovery | [81] |
| Moyamoya disease | Pathogenic mechanism | [82] | |
| hiPSC-SMCs | Supravalvular aortic stenosis syndrome | Pathogenic mechanism | [83] |
| Bicuspid Aortic Valve-related Thoracic Aortic Aneurysm | Pathogenic mechanism/Drug discovery | [84] | |
| VSMCs and CD14+ cells co-culture | Atherosclerosis | Pathogenic mechanism | [88] |
| hCPCs and hiPSC-CMs co-culture | Myocardial ischemia | Regenerative therapy mechanisms | [89] |
| ECs and VSMCs co-culture | Atherosclerosis | Pathogenic mechanism | [90] |
| hiPSC-CMs, hiPSC-ECs and hiPSC-cardiac fibroblasts: scaffold-free 3D microtissue | Arrhythmogenic Cardiomyopathy | Pathogenic mechanism | [122] |
| hiPSC-CMs and MSCs: cell sheet | Torsade de pointes | Pathogenic mechanism/Drug response | [123] |
| hiPSC-CMs: scaffold-based 3D tissue | Myocardial ischemia | Pathogenic mechanism | [124] |
| ECs: scaffold-based 3D tissue | Atherosclerosis | Pathogenic mechanism | [125] |
| hiPSC-CMs: scaffold-based 3D tissue | Long QT syndrome | Pathogenic mechanism/Drug response | [126] |
| 3D-bioprinted valve interstitial cells | Calcific aortic valve disease | Pathogenic mechanism | [127] |
| hiPSC-CMs: heart-on-a-chip | Mitochondrial cardiomyopathy of Barth syndrome | Pathogenic mechanism | [128] |
| hiPSC-CMs and cardiac fibroblasts: heart-on-a-chip | Cardiac fibrosis | Drug discovery | [129] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lippi, M.; Stadiotti, I.; Pompilio, G.; Sommariva, E. Human Cell Modeling for Cardiovascular Diseases. Int. J. Mol. Sci. 2020, 21, 6388. https://doi.org/10.3390/ijms21176388
Lippi M, Stadiotti I, Pompilio G, Sommariva E. Human Cell Modeling for Cardiovascular Diseases. International Journal of Molecular Sciences. 2020; 21(17):6388. https://doi.org/10.3390/ijms21176388
Chicago/Turabian StyleLippi, Melania, Ilaria Stadiotti, Giulio Pompilio, and Elena Sommariva. 2020. "Human Cell Modeling for Cardiovascular Diseases" International Journal of Molecular Sciences 21, no. 17: 6388. https://doi.org/10.3390/ijms21176388
APA StyleLippi, M., Stadiotti, I., Pompilio, G., & Sommariva, E. (2020). Human Cell Modeling for Cardiovascular Diseases. International Journal of Molecular Sciences, 21(17), 6388. https://doi.org/10.3390/ijms21176388