Molecular Basis of Inflammation in the Pathogenesis of Cardiomyopathies

 , ,
, ,  ,
,  ,
,  ,
, 

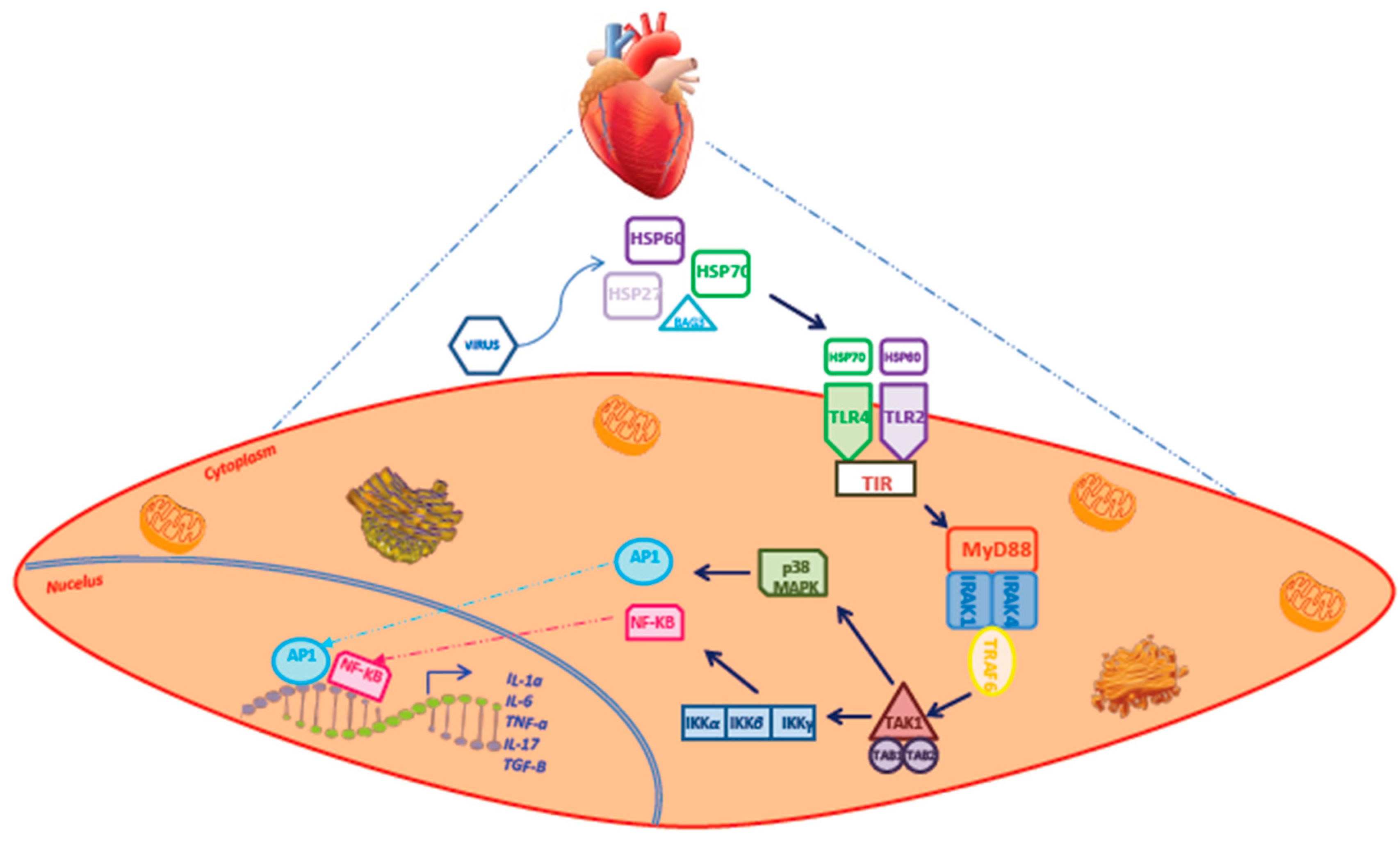{kind=link}
{kind=link}
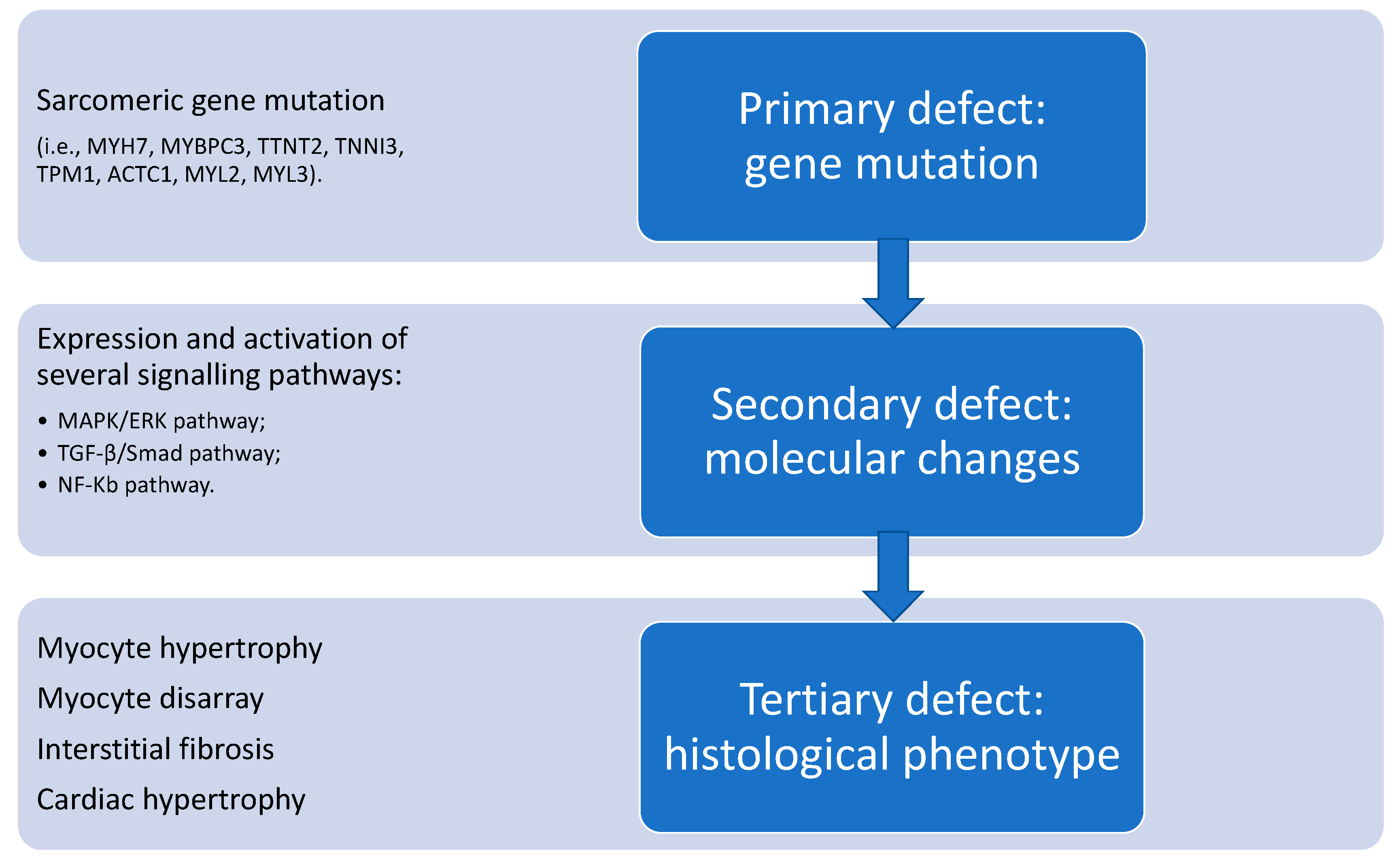{kind=link}
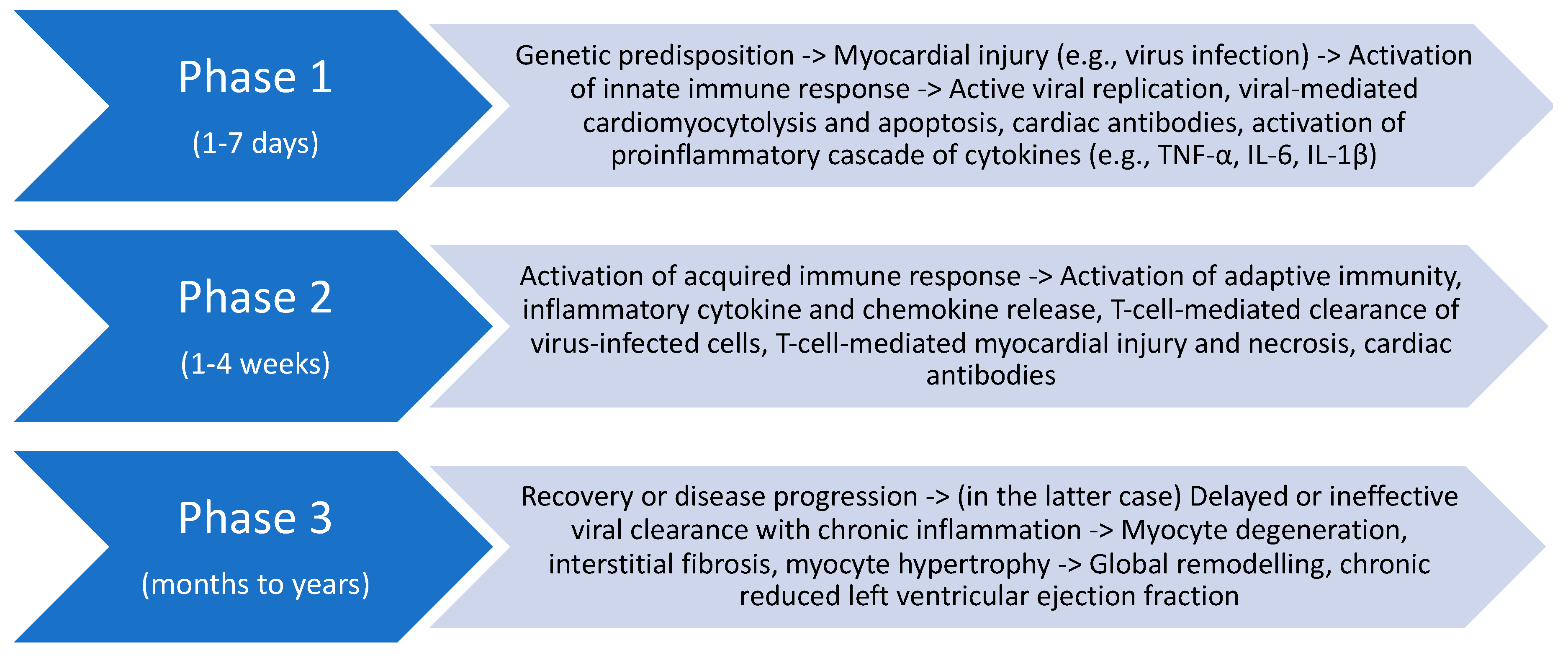{kind=link}
Abstract
:1. Introduction
2. Cardiac Inflammation and Fibrosis: “The Vicious Cycle”
3. Inflammation Mechanisms in Specific Cardiomyopathies
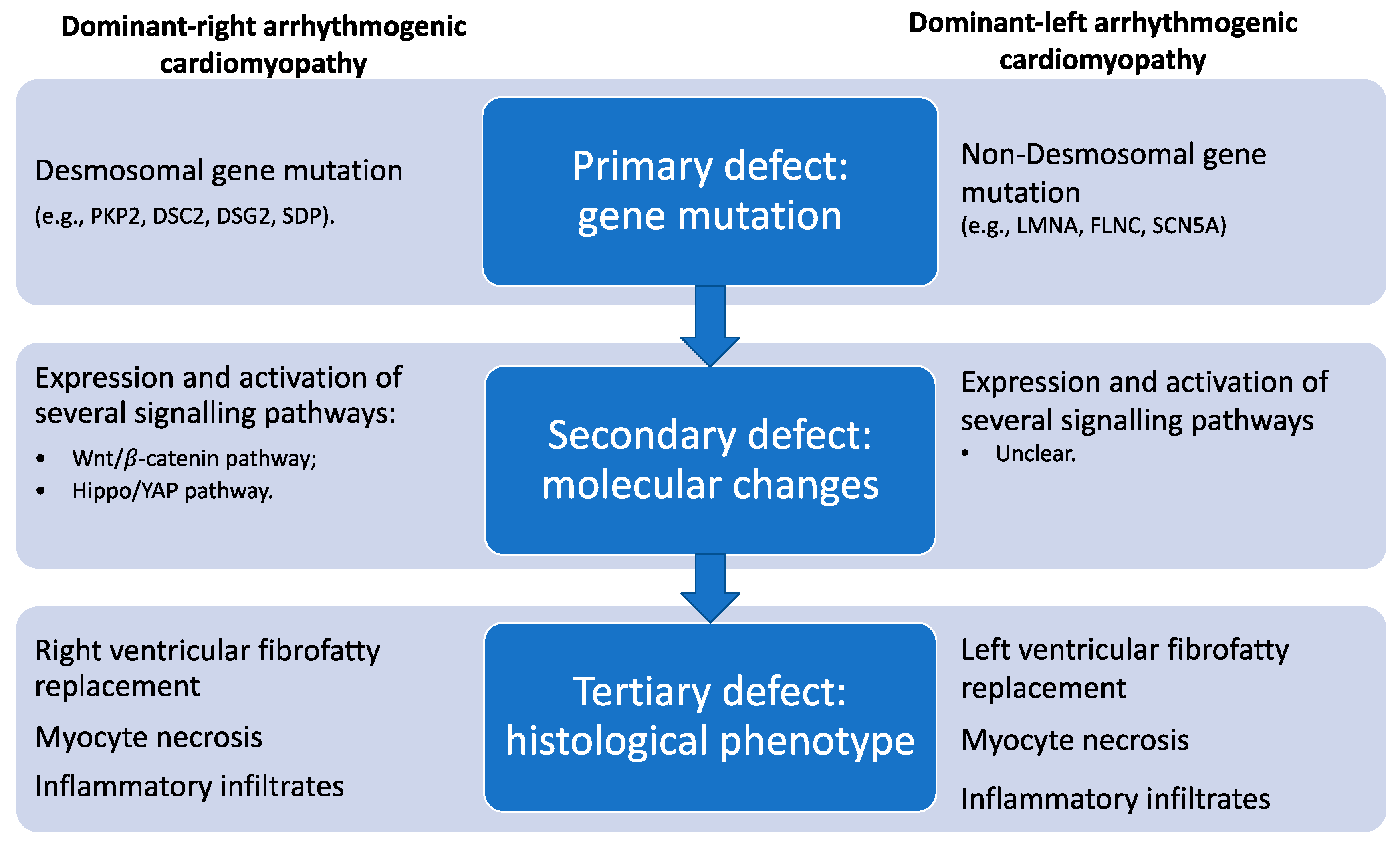
3.1. Arrhythmogenic Cardiomyopathy
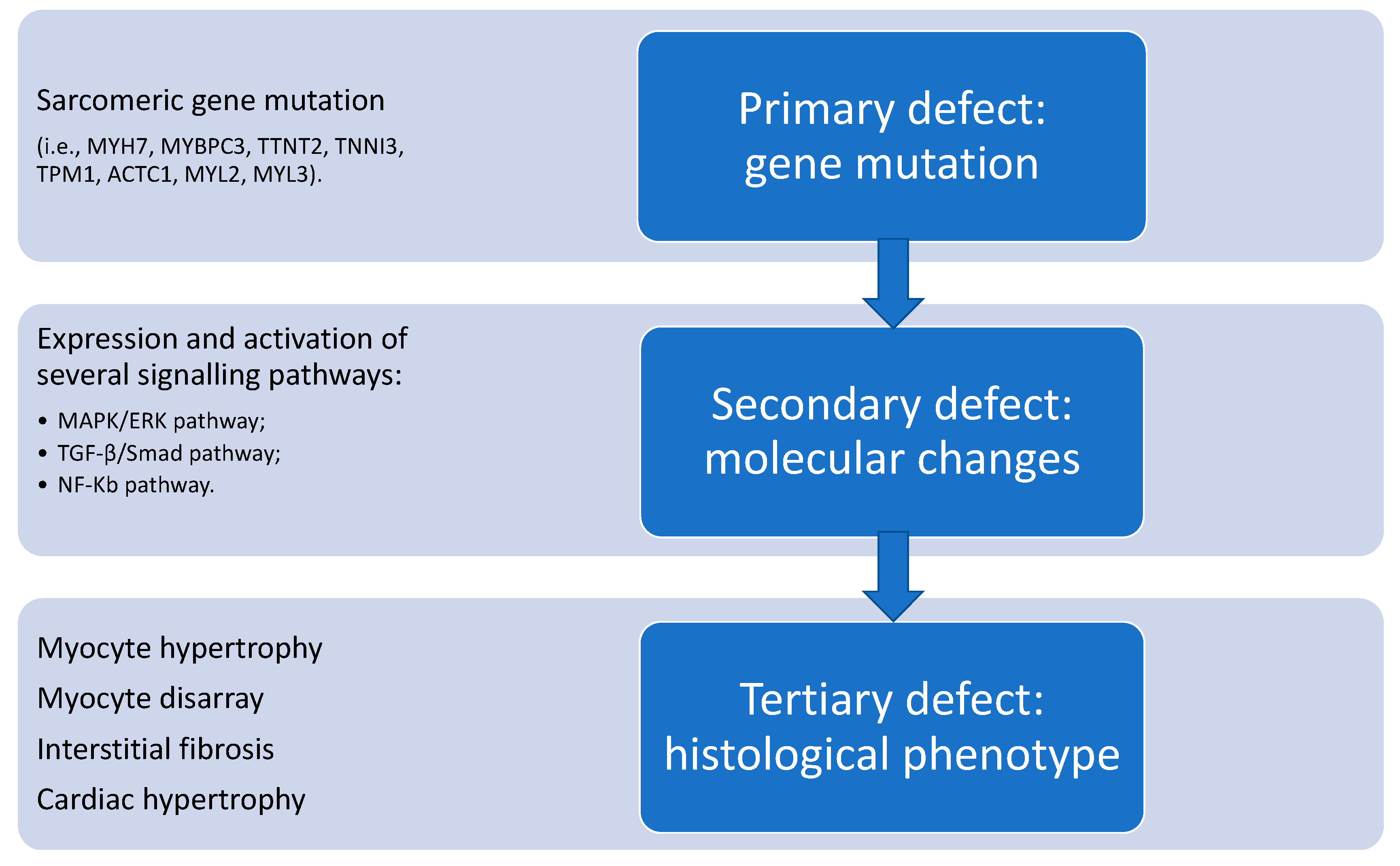
3.2. Hypertrophic Cardiomyopathy
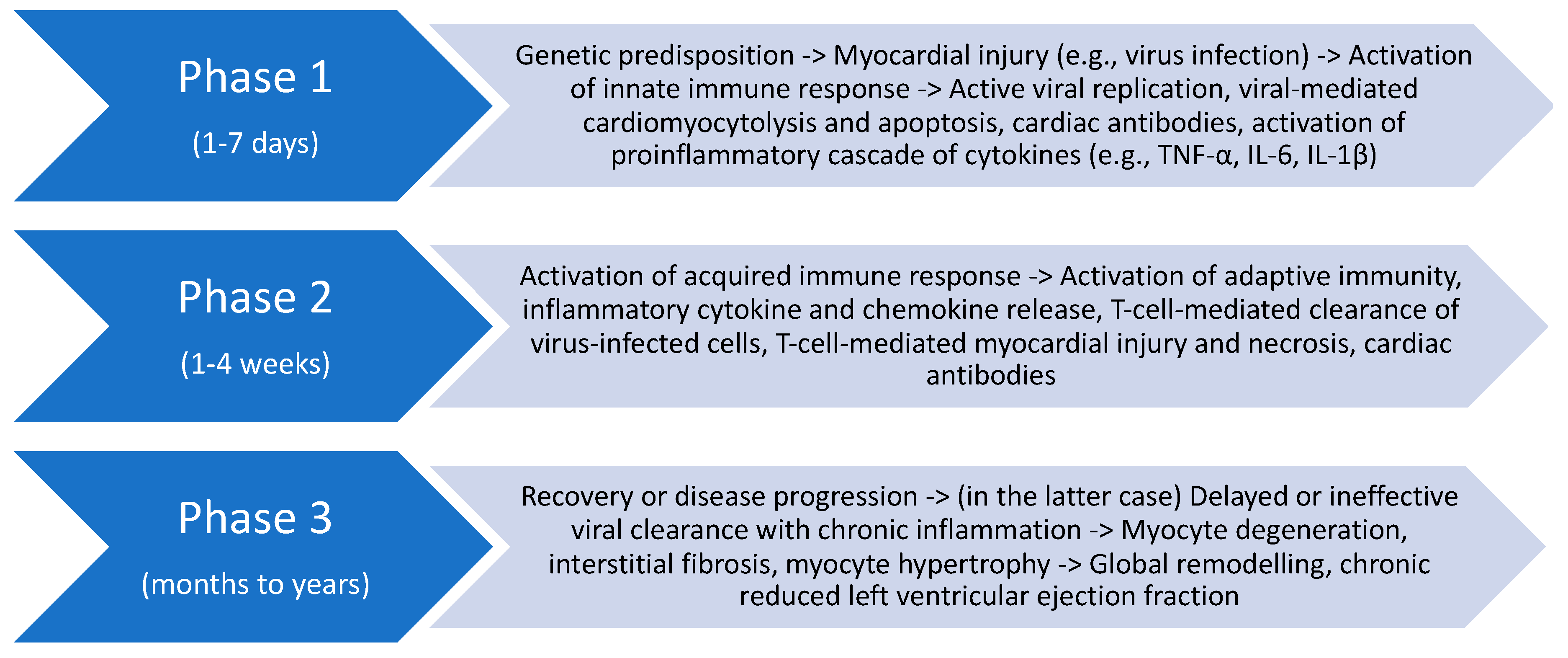
3.3. Dilated Cardiomyopathy
4. Future Inflammation-Based Therapy in CMPs
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elliott, P.M.; Andersson, B.; Arbustini, E.; Bilinska, Z.; Cecchi, F.; Charron, P.; Dubourg, O.; Kühl, U.; Maisch, B.; McKenna, W.J.; et al. Classification of the cardiomyopathies: A position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 2008, 29, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monda, E.; Limongelli, G. The hospitalizations in hypertrophic cardiomyopathy: “The dark side of the moon”. Int. J. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Thiene, G.; Corrado, D.; Nava, A.; Rossi, L.; Poletti, A.; Boffa, G.M.; Daliento, L.; Pennelli, N. Right ventricular cardiomyopathy: Is there evidence of an inflammatory aetiology? Eur. Hear. J. 1991, 12 (Suppl. D), 22–25. [Google Scholar] [CrossRef]
- Matsumori, A.; Yamada, T.; Suzuki, H.; Matoba, Y.; Sasayama, S. Increased circulating cytokines in patients with myocarditis and cardiomyopathy. Br. Heart J. 1994, 72, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J.; McKenna, W.J. Dilated cardiomyopathy: An introduction to pathology and pathogenesis. Br. Heart J. 1994, 72, S24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L., Jr. Inflammation in Heart Failure: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 1324–1340. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; de Boer, R.A. From Inflammation to Fibrosis-Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Kalluri, R. Mechanistic connection between inflammation and fibrosis. Kidney Int. Suppl. 2010, 100, S22–S26. [Google Scholar] [CrossRef] [Green Version]
- Coggins, M.; Rosenzweig, A. The Fire Within: Cardiac Inflammatory Signaling in Health and Disease. Circ. Res. 2012, 110, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Liu, P.P.; Mann, D.L. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 2015, 15, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Y.; Jia, L.; Han, Y.; Cheng, J.; Li, H.; Qi, Y.; Du, J. Macrophage-Stimulated Cardiac Fibroblast Production of IL-6 Is Essential for TGF β/Smad Activation and Cardiac Fibrosis Induced by Angiotensin II. PLoS ONE 2012, 7, e35144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000 Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.A.; Mughal, R.S.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Mechanism of TNFalpha-induced IL-1alpha, IL1beta and IL-6 expression in human cardiac fibroblasts: Effects of statins and thiazolidinediones. Cardiovasc. Res. 2007, 76, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. TLR signaling pathways. Semin. Immunol. 2004, 16, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Monaco, C.; Gregan, S.M.; Navin, T.J.; Foxwell, B.M.; Davies, A.H.; Feldmann, M. Toll-Like Receptor-2 Mediates Inflammation and Matrix Degradation in Human Atherosclerosis. Circulation 2009, 120, 2462–2469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piek, A.; de Boer, R.A.; Silljé, H.H.W. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, F.I.; McKenna, W.J.; Sherrill, D.; Basso, C.; Bauce, B.; Bluemke, D.A.; Calkins, H.; Corrado, D.; Cox, M.G.; Daubert, J.P.; et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: Proposed modification of the Task Force Criteria. Eur. Heart J. 2010, 31, 806–814. [Google Scholar] [CrossRef] [Green Version]
- Limongelli, G.; Nunziato, M.; Mazzaccara, C.; Intrieri, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Di Maggio, F.; Frisso, G.; Salvatore, F. Genotype-Phenotype Correlation: A Triple DNA Mutational Event in a Boy Entering Sport Conveys an Additional Pathogenicity Risk. Genes Basel 2020, 11, 524. [Google Scholar] [CrossRef]
- Patel, D.M.; Dubash, A.D.; Kreitzer, G.; Green, K.J. Disease mutations in desmoplakin inhibit Cx43 membrane targeting mediated by desmoplakin–EB1 interactions. J. Cell Biol. 2014, 206, 779–797. [Google Scholar] [CrossRef] [Green Version]
- Zhurinsky, J.; Shtutman, M.; Ben-Ze’Ev, A. Plakoglobin and beta-catenin: Protein interactions, regulation and biological roles. J. Cell Sci. 2000, 113, 3127–3139. [Google Scholar] [PubMed]
- Hu, Y.; Pu, W.T. Hippo activation in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 402–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H.T. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef] [PubMed]
- Asimaki, A.; Kapoor, S.; Plovie, E.; Arndt, A.K.; Adams, E.; Liu, Z.; James, C.A.; Judge, D.P.; Calkins, H.; Churko, J.; et al. Identification of a New Modulator of the Intercalated Disc in a Zebrafish Model of Arrhythmogenic Cardiomyopathy. Sci. Transl. Med. 2014, 6, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelko, S.P.; Asimaki, A.; Andersen, P.; Bedja, D.; Amat-Alarcon, N.; DeMazumder, D.; Jasti, R.; MacRae, C.A.; Leber, R.; Kleber, A.G.; et al. Central role for GSK3beta in the pathogenesis of arrhythmogenic cardiomyopathy. JCI Insight 2016, 1, 85923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Baldinger, S.H.; Gandjbakhch, E.; Maury, P.; Sellal, J.M.; Androulakis, A.F.; Waintraub, X.; Charron, P.; Rollin, A.; Richard, P.; et al. Long-Term Arrhythmic and Nonarrhythmic Outcomes of Lamin A/C Mutation Carriers. J. Am. Coll. Cardiol. 2016, 68, 2299–2307. [Google Scholar] [CrossRef]
- Van Der Zwaag, P.A.; van Rijsingen, I.A.; Asimaki, A.; Jongbloed, J.D.; van Veldhuisen, D.J.; Wiesfeld, A.C.; Cox, M.G.; van Lochem, L.T.; de Boer, R.A.; Hofstra, R.M.; et al. Phospholamban R14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur. J. Heart Fail. 2012, 14, 1199–1207. [Google Scholar] [CrossRef]
- Ortiz-Genga, M.F.; Cuenca, S.; Ferro, M.D.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Towbin, J.A.; McKenna, W.J.; Abrams, D.J.; Ackerman, M.J.; Calkins, H.; Darrieux, F.C.C.; Daubert, J.P.; de Chillou, C.; DePasquale, E.C.; Desai, M.Y.; et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019, 16, e301–e372. [Google Scholar] [CrossRef] [Green Version]
- Bowles, N.E.; Ni, J.; Marcus, F.; Towbin, J.A. The detection of cardiotropic viruses in the myocardium of patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy. J. Am. Coll. Cardiol. 2002, 39, 892–895. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, O.; Fernández-Falgueras, A.; Sarquella-Brugada, G.; Sanchez, O.; Cesar, S.; Mademont, I.; Allegue, C.; Mates, J.; Pérez-Serra, A.; Coll, M.; et al. A Genetically Vulnerable Myocardium May Predispose to Myocarditis. J. Am. Coll. Cardiol. 2015, 66, 2913–2914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campian, M.E.; Verberne, H.J.; Hardziyenka, M.; de Groot, E.A.; van Moerkerken, A.F.; Van Eck-Smit, B.L.; Tan, H.L. Assessment of inflammation in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 2079–2085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavrogeni, S.I.; Markousis-Mavrogenis, G.; Aggeli, C.; Tousoulis, D.; Kitas, G.D.; Kolovou, G.; Iliodromitis, E.K.; Sfikakis, P.P. Arrhythmogenic Inflammatory Cardiomyopathy in Autoimmune Rheumatic Diseases: A Challenge for Cardio-Rheumatology. Diagn. Basel 2019, 9, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asimaki, A.; Tandri, H.; Duffy, E.R.; Winterfield, J.R.; Mackey-Bojack, S.; Picken, M.M.; Cooper, L.T.; Wilber, D.J.; Marcus, F.I.; Basso, C.; et al. Altered desmosomal proteins in granulomatous myocarditis and potential pathogenic links to arrhythmogenic right ventricular cardiomyopathy. Circ. Arrhythm. Electrophysiol. 2011, 4, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonny, A.; Lellouche, N.; Ditah, I.; Hidden-Lucet, F.; Yitemben, M.T.; Granger, B.; Larrazet, F.; Frank, R.; Fontaine, G. C-Reactive Protein in Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy and Relationship with Ventricular Tachycardia. Cardiol. Res. Pract. 2010, 2010, 919783. [Google Scholar] [CrossRef] [Green Version]
- Miles, C.; Finocchiaro, G.; Papadakis, M.; Gray, B.; Westaby, J.; Ensam, B.; Basu, J.; Parry-Williams, G.; Papatheodorou, S.; Paterson, C.; et al. Sudden Death and Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy. Circulation 2019, 139, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Charron, P.; Elliott, P.M.; Gimeno, J.R.; Caforio, A.L.; Kaski, J.P.; Tavazzi, L.; Tendera, M.; Maupain, C.; Laroche, C.; Rubiś, P.; et al. The Cardiomyopathy Registry of the EURObservational Research Programme of the European Society of Cardiology: Baseline data and contemporary management of adult patients with cardiomyopathies. Eur. Heart J. 2018, 39, 1784–1793. [Google Scholar] [CrossRef] [Green Version]
- Mavrogeni, S.; Protonotarios, N.; Tsatsopoulou, A.; Papachristou, P.; Sfendouraki, E.; Papadopoulos, G. Naxos disease evolution mimicking acute myocarditis: The role of cardiovascular magnetic resonance imaging. Int. J. Cardiol. 2013, 166, e14–e15. [Google Scholar] [CrossRef]
- Smith, E.D.; Lakdawala, N.K.; Papoutsidakis, N.; Aubert, G.; Mazzanti, A.; McCanta, A.C.; Agarwal, P.P.; Arscott, P.; Dellefave-Castillo, L.M.; Vorovich, E.E.; et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation 2020, 141, 1872–1884. [Google Scholar] [CrossRef]
- Elliott, P.M.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyo-pathy: The Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar]
- Esposito, A.; Monda, E.; Gragnano, F.; De Simone, F.; Cesaro, A.; Natale, F.; Concilio, C.; Moscarella, E.; Caiazza, M.; Pazzanese, V.; et al. Prevalence and clinical implications of hyperhomocysteinaemia in patients with hypertrophic cardiomyopathy and MTHFR C6777T polymorphism. Eur. J. Prev. Cardiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Monda, E.; Tramonte, S.; Gragnano, F.; Masarone, D.; Frisso, G.; Esposito, A.; Gravino, R.; Ammendola, E.; Salerno, G.; et al. Prevalence and clinical significance of red flags in patients with hypertrophic cardiomyopathy. Int. J. Cardiol. 2020, 299, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Limongelli, G.; Nunziato, M.; D’Argenio, V.; Esposito, M.V.; Monda, E.; Mazzaccara, C.; Caiazza, M.; D’Aponte, A.; D’Andrea, A.; Bossone, E.; et al. Yield and clinical significance of genetic screening in elite and amateur athletes. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Monda, E.; Sarubbi, B.; Russo, M.G.; Caiazza, M.; Mazzaccara, C.; Magrelli, J.; Rubino, M.; Esposito, A.; Perna, A.; Passariello, A.; et al. Unexplained sudden cardiac arrest in children: Clinical and genetic characteristics of survivors. Eur. J. Prev. Cardiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Caiazza, M.; Rubino, M.; Monda, E.; Passariello, A.; Fusco, A.; Cirillo, A.; Esposito, A.; Pierno, A.; De Fazio, F.; Pacileo, R.; et al. Combined PTPN11 and MYBPC3 Gene Mutations in an Adult Patient with Noonan Syndrome and Hypertrophic Cardiomyopathy. Genes 2020, 11, 947. [Google Scholar] [CrossRef] [PubMed]
- Belfiore, M.P.; Iacobellis, F.; Acampora, E.; Caiazza, M.; Rubino, M.; Monda, E.; Magaldi, M.R.; Tarallo, A.; Sasso, M.; De Pasquale, V.; et al. Aortopathies in mouse models of Pompe, Fabry and Mucopolysaccharidosis IIIB lysosomal storage diseases. PLoS ONE 2020, 15, e233050. [Google Scholar] [CrossRef]
- Marian, A.J.; Braunwald, E. Hypertrophic Cardiomyopathy: Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 749–770. [Google Scholar] [CrossRef]
- Finocchiaro, G.; Magavern, E.; Sinagra, G.; Ashley, E.; Papadakis, M.; Tome-Esteban, M.T.; Sharma, S.; Olivotto, I. Impact of Demographic Features, Lifestyle, and Comorbidities on the Clinical Expression of Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2017, 6, e7161. [Google Scholar] [CrossRef] [Green Version]
- Zen, K.; Irie, H.; Doue, T.; Takamiya, M.; Yamano, T.; Sawada, T.; Azuma, A.; Matsubara, H. Analysis of circulating apoptosis mediators and proinflammatory cytokines in patients with idiopathic hypertrophic cardiomyopathy: Comparison between nonobstructive and dilated-phase hypertrophic cardiomyopathy. Int. Heart J. 2005, 46, 231–244. [Google Scholar] [CrossRef] [Green Version]
- Högye, M.; Mándi, Y.; Csanády, M.; Sepp, R.; Buzas, K. Comparison of circulating levels of interleukin-6 and tumor necrosis factor-alpha in hypertrophic cardiomyopathy and in idiopathic dilated cardiomyopathy. Am. J. Cardiol. 2004, 94, 249–251. [Google Scholar] [CrossRef]
- Kuusisto, J.; Kärjä, V.; Sipola, P.; Kholová, I.; Peuhkurinen, K.; Jääskeläinen, P.; Naukkarinen, A.; Ylä-Herttuala, S.; Punnonen, K.; Laakso, M. Low-grade inflammation and the phenotypic expression of myocardial fibrosis in hypertrophic cardiomyopathy. Heart 2012, 98, 1007–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Ellims, A.H.; Beale, A.L.; Taylor, A.J.; Murphy, A.; Dart, A.M. Systemic inflammation is associated with myocardial fibrosis, diastolic dysfunction, and cardiac hypertrophy in patients with hypertrophic cardiomyopathy. Am. J. Transl. Res. 2017, 9, 5063–5073. [Google Scholar] [PubMed]
- Bryant, D.; Becker, L.; Richardson, J.; Shelton, J.; Franco, F.; Peshock, R.; Thompson, M.; Giroir, B. Cardiac Failure in Transgenic Mice With Myocardial Expression of Tumor Necrosis Factor-α. Circulation 1998, 97, 1375–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Cheng, G.; Jin, R.; Afzal, M.R.; Samanta, A.; Xuan, Y.-T.; Girgis, M.; Elias, H.; Zhu, Y.; Davani, A.; et al. Deletion of Interleukin-6 Attenuates Pressure Overload-Induced Left Ventricular Hypertrophy and Dysfunction. Circ. Res. 2016, 118, 1918–1929. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.C.; Owens, A.P., III; Sadayappan, S. Tissue-level inflammation and ventricular remodeling in hypertrophic cardiomyopathy. J. Thromb. Thrombolysis 2020, 49, 177–183. [Google Scholar] [CrossRef]
- Brill, B.S.A.G.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Mariero, L.H.; Torp, M.; Heiestad, C.M.; Baysa, A.; Li, Y.; Valen, G.; Vaage, J.; Stensløkken, K.-O. Inhibiting nucleolin reduces inflammation induced by mitochondrial DNA in cardiomyocytes exposed to hypoxia and reoxygenation. Br. J. Pharmacol. 2019, 176, 4360–4372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Serra, A.; Toro, R.; Sarquella-Brugada, G.; De Gonzalo-Calvo, D.; Cesar, S.; Carro, E.; Llorente-Cortés, V.; Iglesias, A.; Brugada, J.; Brugada, R.; et al. Genetic basis of dilated cardiomyopathy. Int. J. Cardiol. 2016, 224, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, B.; D’Argenio, V.; Monda, E.; Vitale, A.; Caiazza, M.; Sacchetti, L.; Pastore, L.; Limongelli, G.; Frisso, G.; Mazzaccara, C. Genetic analysis resolves differential diagnosis of a familial syndromic dilated cardiomyopathy: A new case of Alström syndrome. Mol. Genet. Genom. Med. 2020, 8, e1260. [Google Scholar] [CrossRef]
- Imanaka-Yoshida, K. Inflammation in myocardial disease: From myocarditis to dilated cardiomyopathy. Pathol. Int. 2020, 70, 1–11. [Google Scholar] [CrossRef]
- Maisch, B. Cardio-Immunology of Myocarditis: Focus on Immune Mechanisms and Treatment Options. Front. Cardiovasc. Med. 2019, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachtenberg, B.H.; Hare, J.M. Inflammatory Cardiomyopathic Syndromes. Circ. Res. 2017, 121, 803–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caforio, A.L.P.; Malipiero, G.; Marcolongo, R.; Iliceto, S. Myocarditis: A Clinical Overview. Curr. Cardiol. Rep. 2017, 19, 63. [Google Scholar] [CrossRef] [PubMed]
- Bracamonte-Baran, W.; Čiháková, D. Cardiac Autoimmunity: Myocarditis. Adv. Exp. Med. Biol. 2017, 1003, 187–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mason, J.W. Myocarditis and dilated cardiomyopathy: An inflammatory link. Cardiovasc. Res. 2003, 60, 5–10. [Google Scholar] [CrossRef]
- Leslie, K.O.; Schwarz, J.; Simpson, K.; Huber, S.A. Progressive interstitial collagen deposition in Coxsackievirus B3-induced murine myocarditis. Am. J. Pathol. 1990, 136, 683–693. [Google Scholar]
- Wojnicz, R.; Nowalany-Kozielska, E.; Wojciechowska, C.; Glanowska, G.; Wilczewski, P.; Niklewski, T.; Zembala, M.; Polonski, L.; Rozek, M.M.; Wodniecki, J. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: Two-year follow-up results. Circulation 2001, 104, 39–45. [Google Scholar] [CrossRef] [Green Version]
- Frustaci, A.; Russo, M.A.; Chimenti, C. Randomized study on the efficacy of immunosuppressive therapy in patients with virus-negative inflammatory cardiomyopathy: The TIMIC study. Eur. Heart J. 2009, 30, 1995–2002. [Google Scholar] [CrossRef]
- Frustaci, A.; Chimenti, C.; Calabrese, F.; Pieroni, M.; Thiene, G.; Maseri, A. Immunosuppressive therapy for active lymphocytic myocarditis: Virological and immunologic profile of responders versus nonresponders. Circulation 2003, 107, 857–863. [Google Scholar] [CrossRef]
- Liu, P.; Aitken, K.; Kong, Y.-Y.; Opavsky, M.A.; Martino, T.; Dawood, F.; Wen, W.-H.; Kozieradzki, I.; Bachmaier, K.; Straus, D.; et al. The tyrosine kinase p56lck is essential in coxsackievirus B3-mediated heart disease. Nat. Med. 2000, 6, 429–434. [Google Scholar] [CrossRef]
- Van Tassell, B.W.; Abouzaki, N.A.; Erdle, C.O.; Carbone, S.; Trankle, C.R.; Melchior, R.D.; Turlington, J.S.; Thurber, C.J.; Christopher, S.; Dixon, D.L.; et al. Interleukin-1 Blockade in Acute Decompensated Heart Failure: A Randomized, Double-Blinded, Placebo-Controlled Pilot Study. J. Cardiovasc. Pharmacol. 2016, 67, 544–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tassell, B.W.; Canada, J.M.; Carbone, S.; Trankle, C.; Buckley, L.; Erdle, C.O.; Abouzaki, N.A.; Dixon, D.L.; Kadariya, D.; Christopher, S.; et al. Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ. Heart Fail. 2017, 10, e4373. [Google Scholar] [CrossRef] [PubMed]
- Everett, B.M.; Cornel, J.H.; Lainscak, M.; Anker, S.D.; Abbate, A.; Thuren, T.; Libby, P.; Glynn, R.J.; Ridker, P.M. Anti-Inflammatory Therapy with Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation 2019, 139, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Wallukat, G.; Dandel, M.; Bieda, H.; Brandes, K.; Spiegelsberger, S.; Nissen, E.; Kunze, R.; Hetzer, R. Immunoglobulin Adsorption in Patients with Idiopathic Dilated Cardiomyopathy. Circulation 2000, 101, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Dennert, R.; Velthuis, S.; Schalla, S.; Eurlings, L.; Van Suylen, R.-J.; Van Paassen, P.; Tervaert, J.W.C.; Wolffs, P.F.G.; Goossens, V.J.; Bruggeman, C.; et al. Intravenous immunoglobulin therapy for patients with idiopathic cardiomyopathy and endomyocardial biopsy-proven high PVB19 viral load. Antivir. Ther. 2010, 15, 193–201. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monda, E.; Palmiero, G.; Rubino, M.; Verrillo, F.; Amodio, F.; Di Fraia, F.; Pacileo, R.; Fimiani, F.; Esposito, A.; Cirillo, A.; et al. Molecular Basis of Inflammation in the Pathogenesis of Cardiomyopathies. Int. J. Mol. Sci. 2020, 21, 6462. https://doi.org/10.3390/ijms21186462
Monda E, Palmiero G, Rubino M, Verrillo F, Amodio F, Di Fraia F, Pacileo R, Fimiani F, Esposito A, Cirillo A, et al. Molecular Basis of Inflammation in the Pathogenesis of Cardiomyopathies. International Journal of Molecular Sciences. 2020; 21(18):6462. https://doi.org/10.3390/ijms21186462
Chicago/Turabian StyleMonda, Emanuele, Giuseppe Palmiero, Marta Rubino, Federica Verrillo, Federica Amodio, Francesco Di Fraia, Roberta Pacileo, Fabio Fimiani, Augusto Esposito, Annapaola Cirillo, and et al. 2020. "Molecular Basis of Inflammation in the Pathogenesis of Cardiomyopathies" International Journal of Molecular Sciences 21, no. 18: 6462. https://doi.org/10.3390/ijms21186462
APA StyleMonda, E., Palmiero, G., Rubino, M., Verrillo, F., Amodio, F., Di Fraia, F., Pacileo, R., Fimiani, F., Esposito, A., Cirillo, A., Fusco, A., Moscarella, E., Frisso, G., Russo, M. G., Pacileo, G., Calabrò, P., Scudiero, O., Caiazza, M., & Limongelli, G. (2020). Molecular Basis of Inflammation in the Pathogenesis of Cardiomyopathies. International Journal of Molecular Sciences, 21(18), 6462. https://doi.org/10.3390/ijms21186462