Hsp70 and NF-kB Mediated Control of Innate Inflammatory Responses in a Canine Macrophage Cell Line

 ,
,
Abstract
:1. Introduction
2. Results
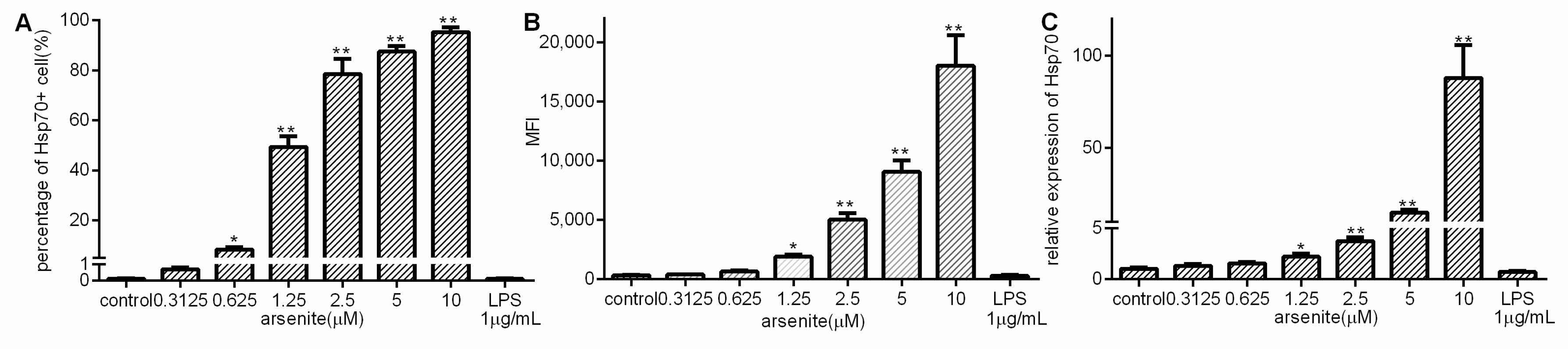
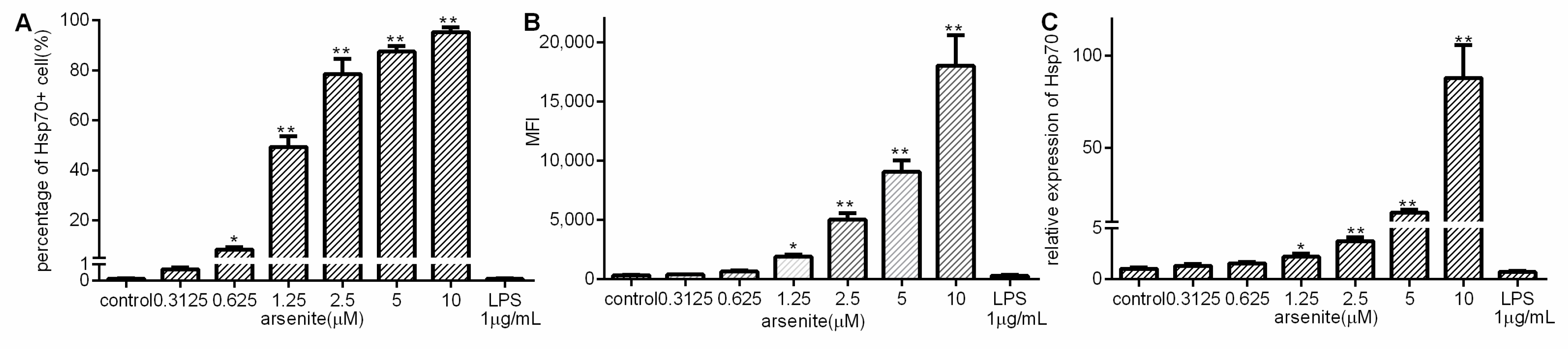
2.1. Induction of Hsp70 in Arsenite-Stressed 030D Cells
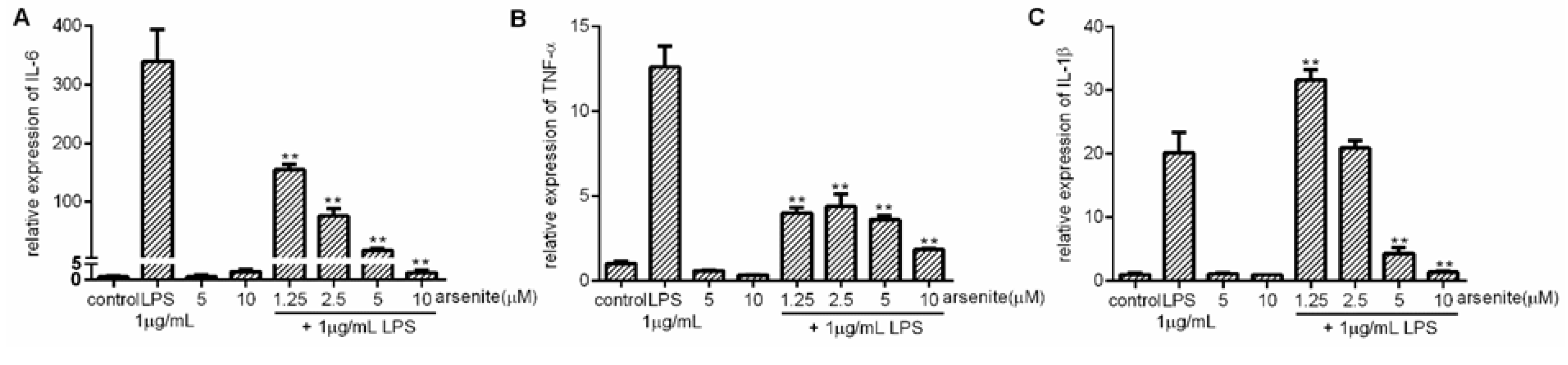
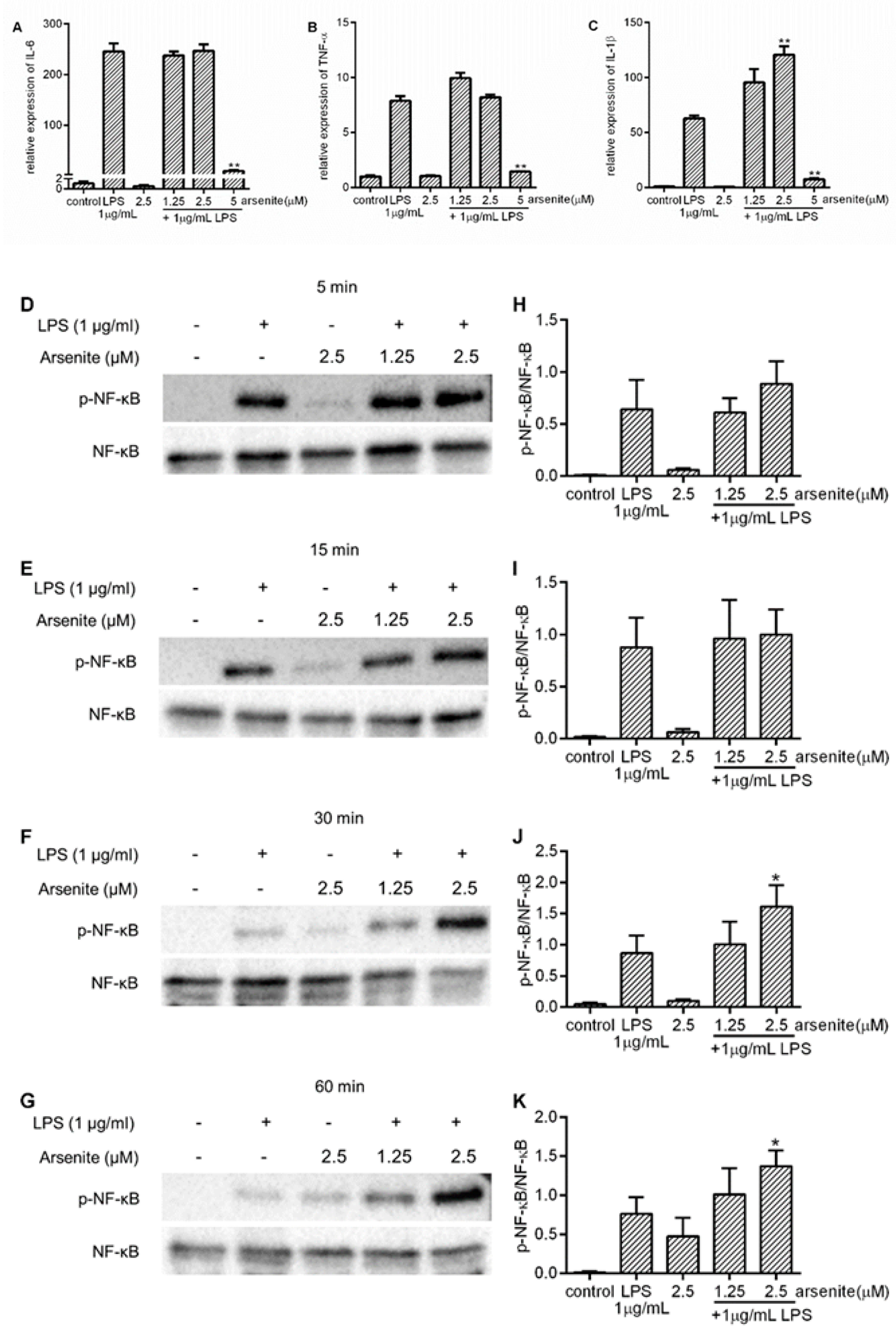
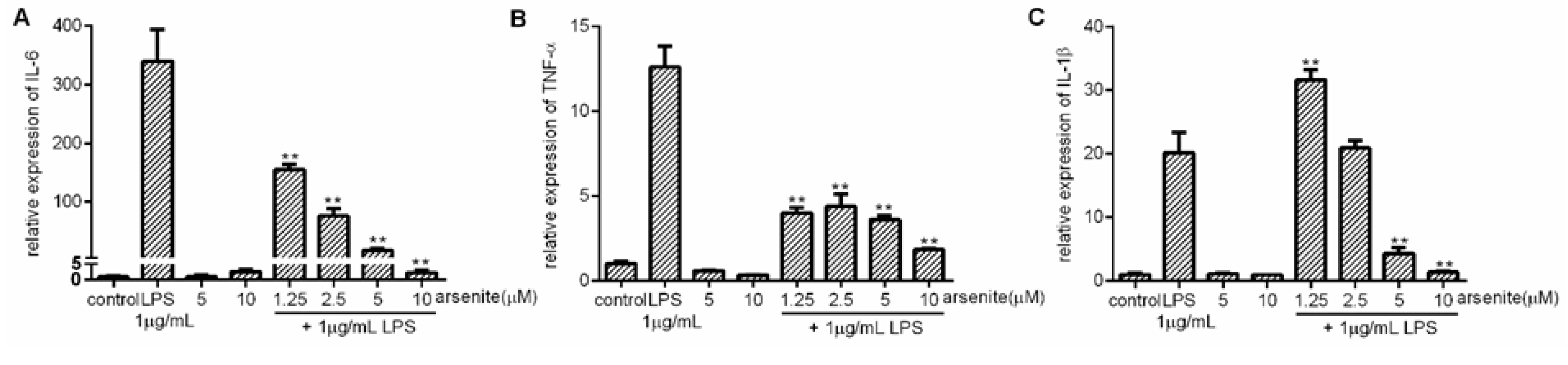
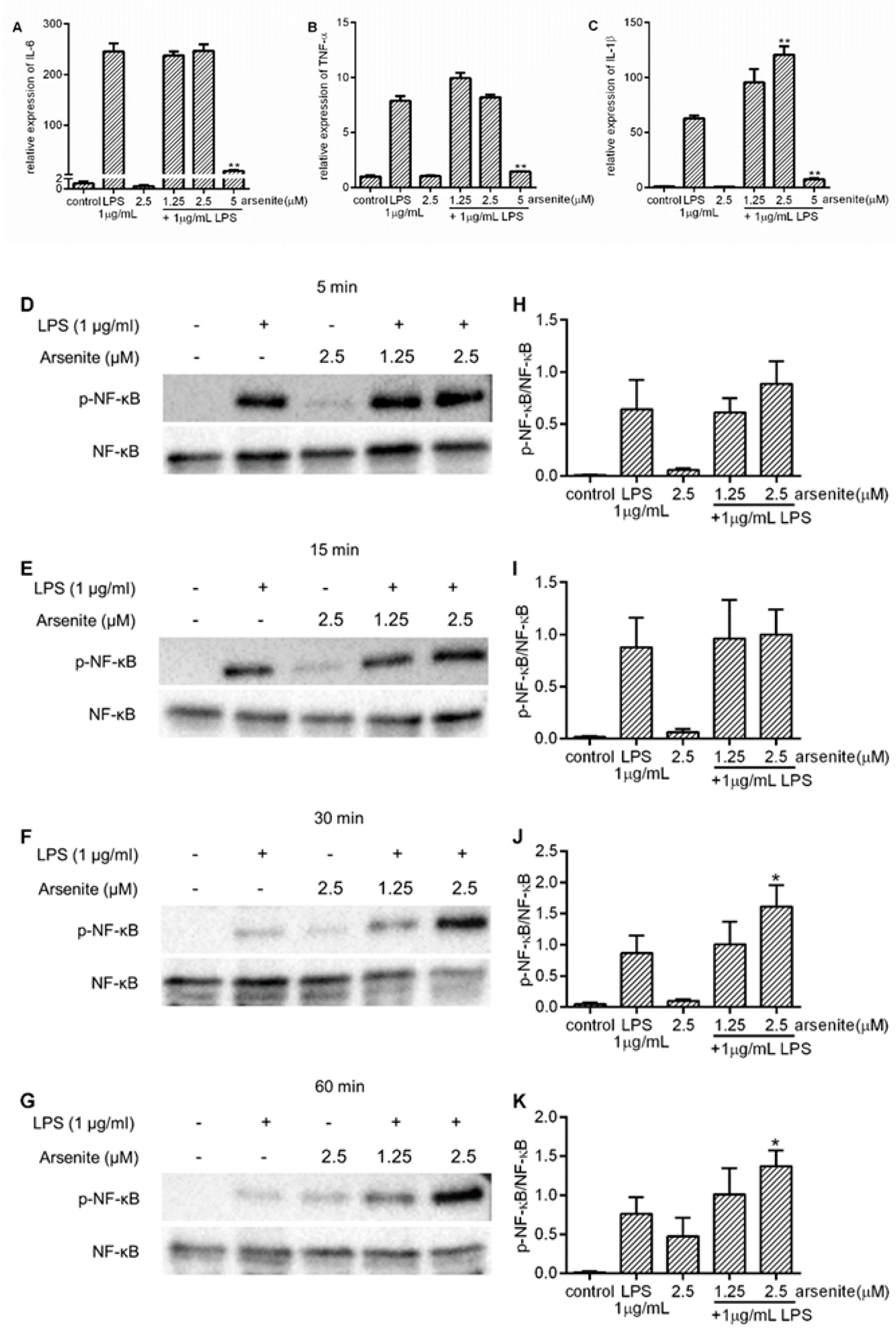
2.2. Inhibition of Expression of LPS-Induced Pro-Inflammatory Cytokines in Stressed 030D Cells
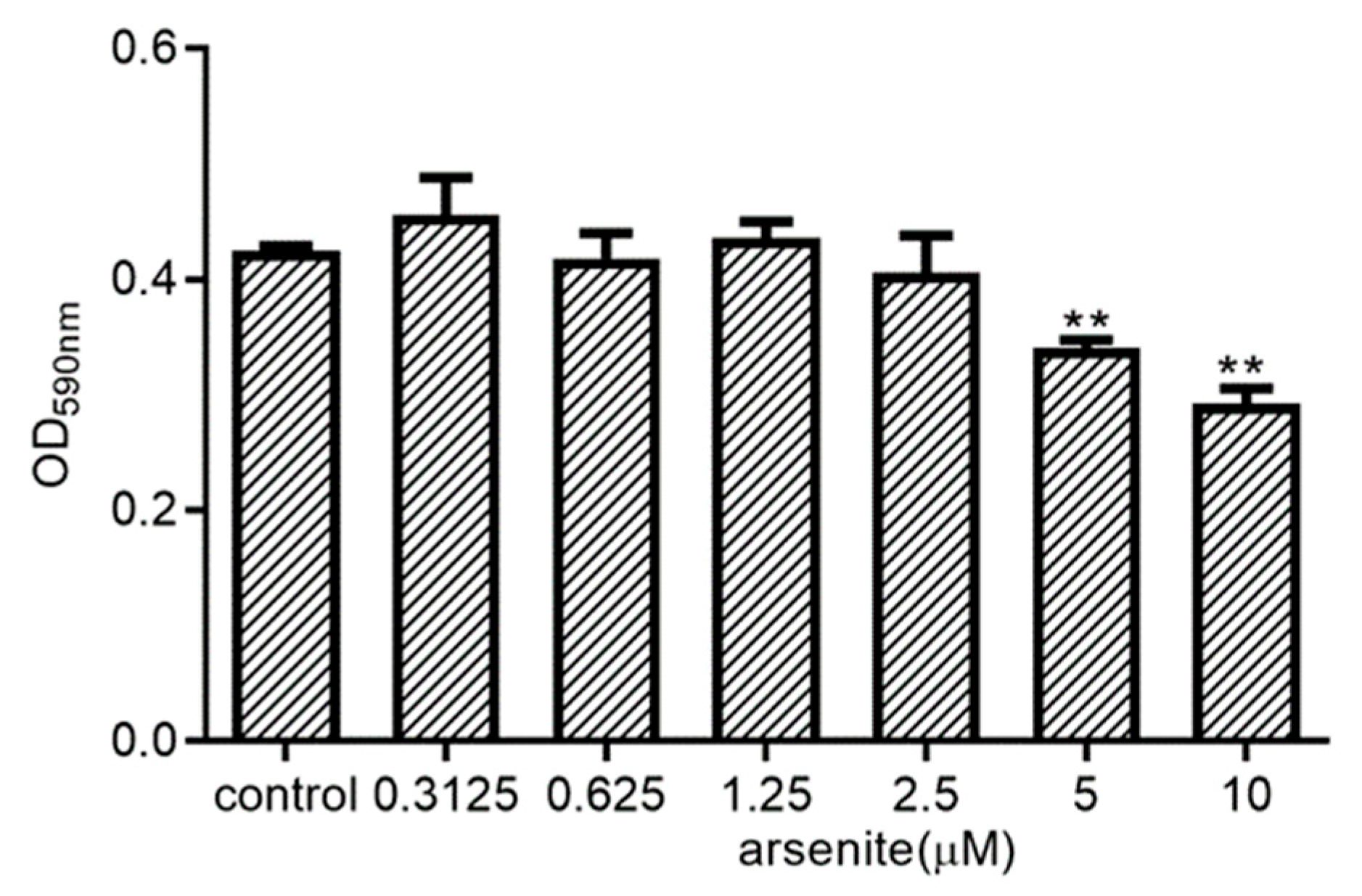
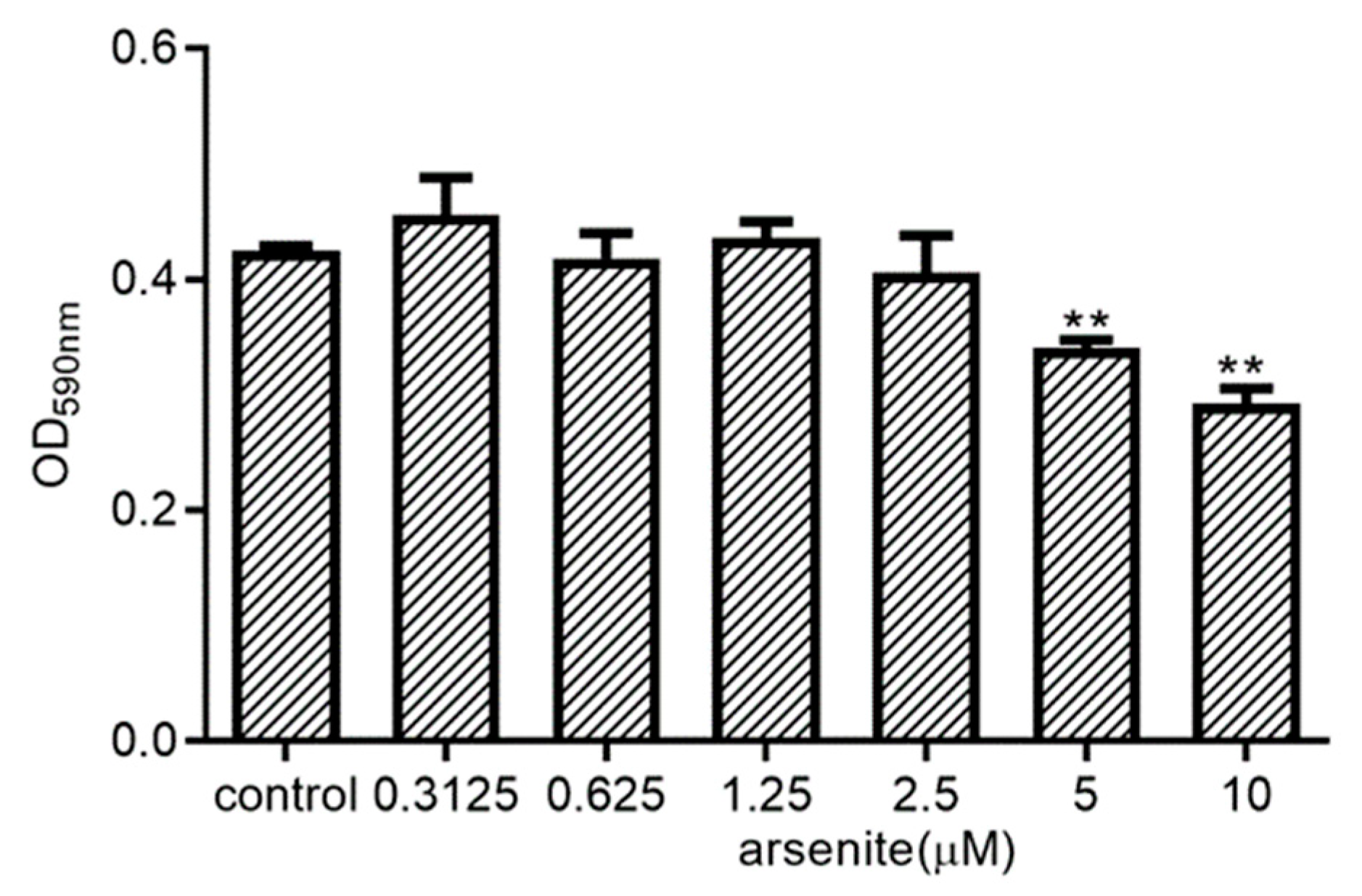
2.3. Effect of Arsenite on 030D Cell Viability
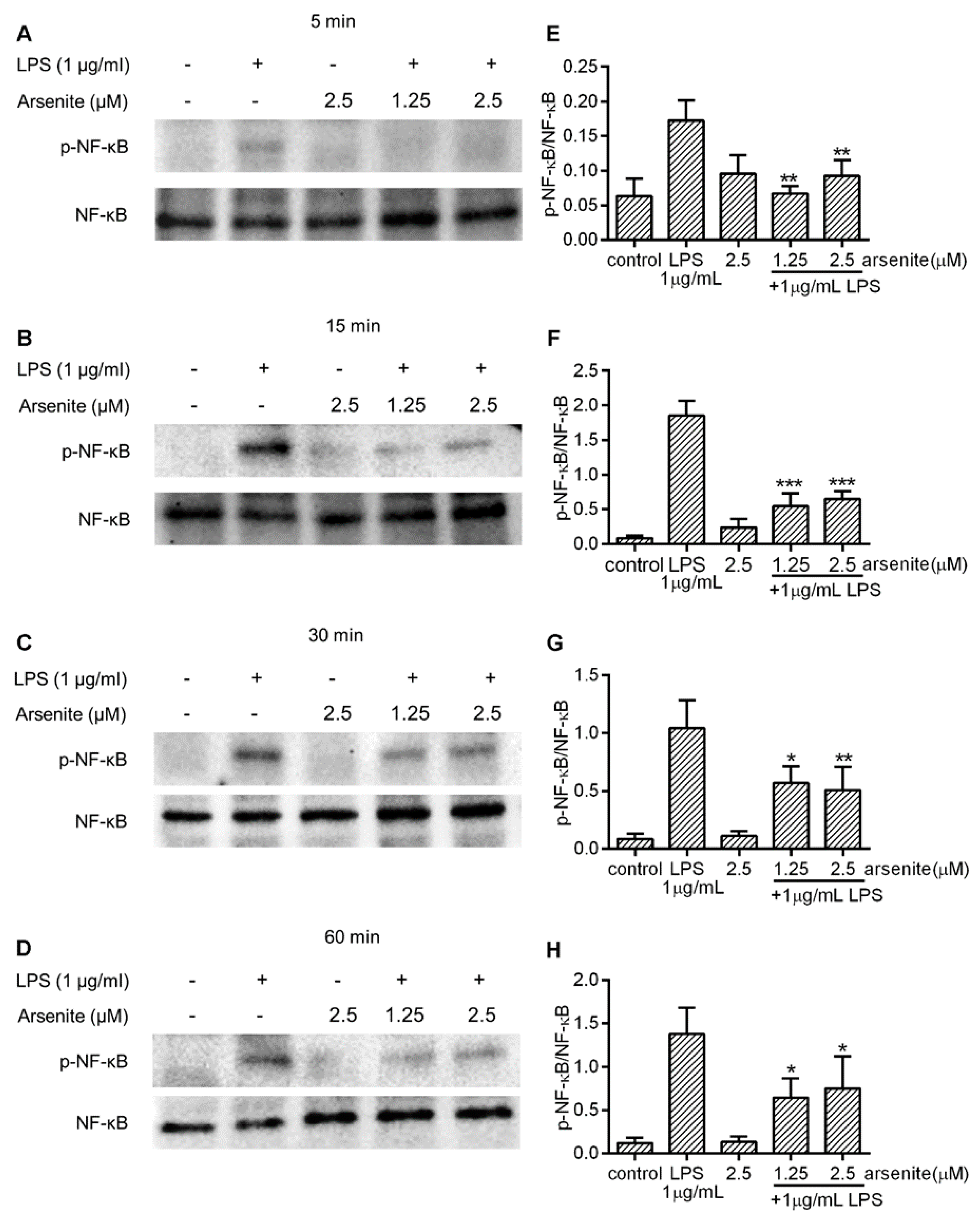
2.4. Effects of Cell Stress on NF-κB Phosphorylation in LPS-Stimulated 030D Cells
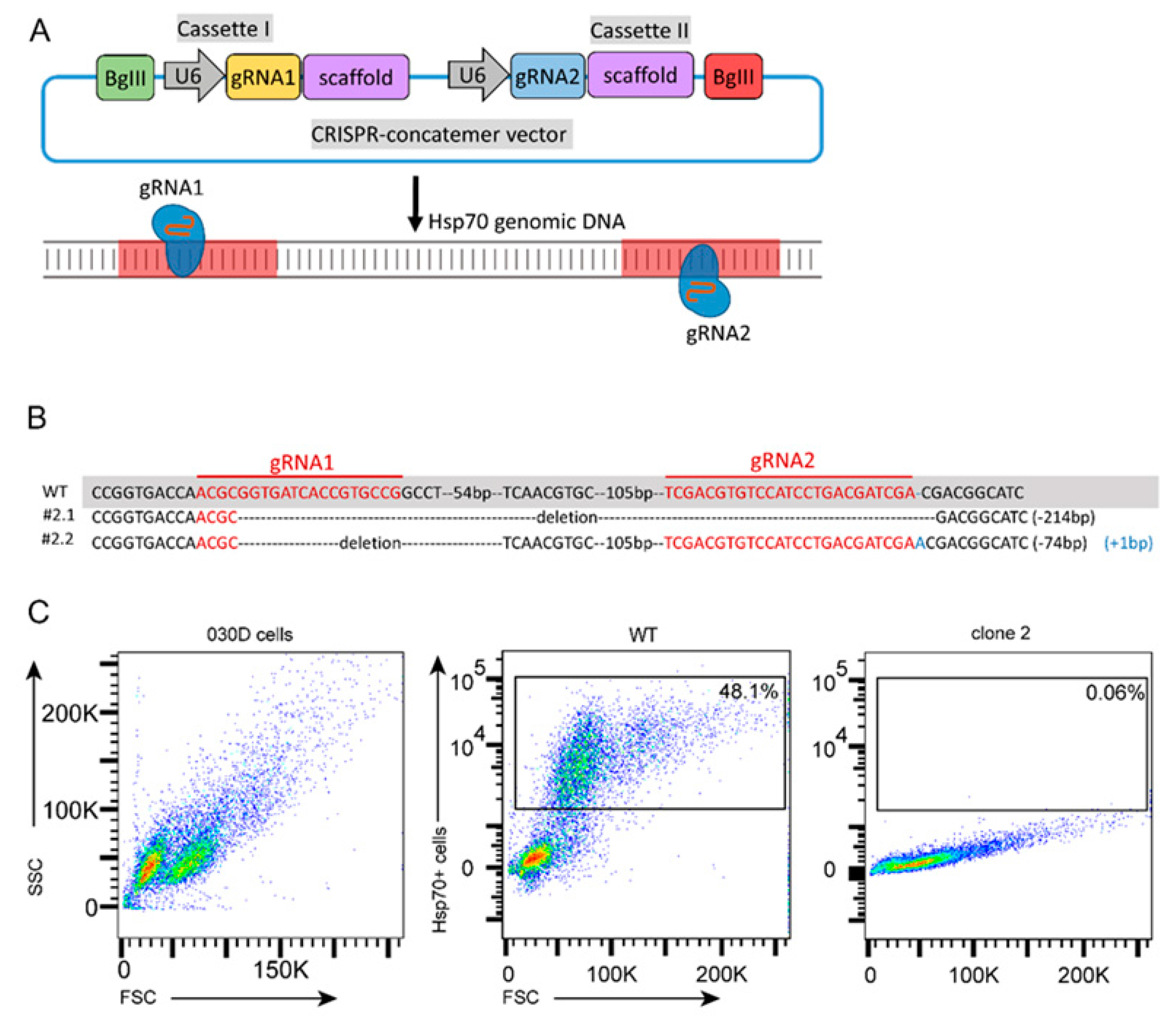
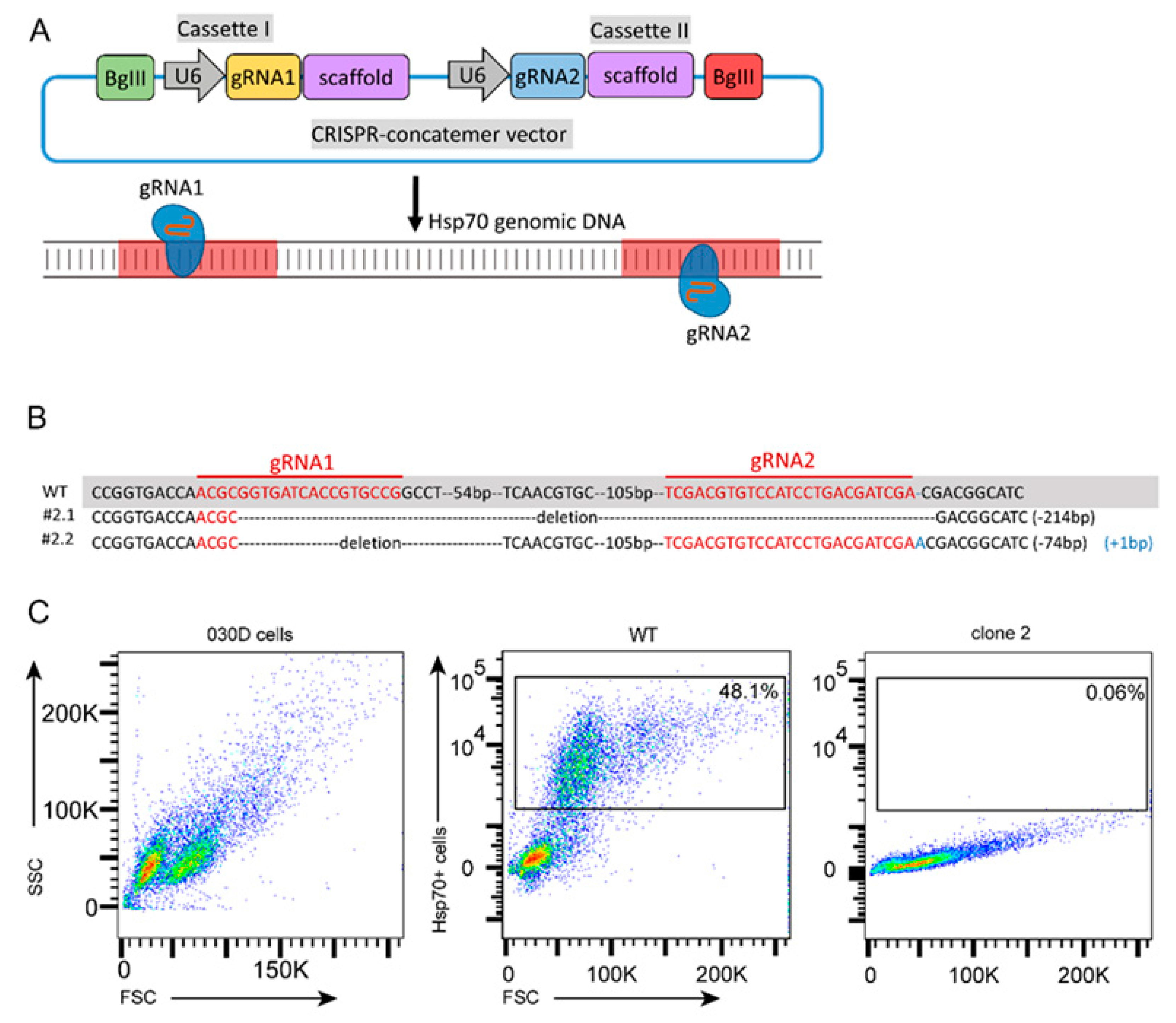
2.5. Generation and Validation of Hsp70 Knockout of a 030D Cell Line
2.6. The Effect of Hsp70 on Pro-Inflammatory Cytokine Expression and NF-κB Phosphorylation
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Analysis of Hsp70 Expression in 030D Cells by Flow Cytometry
4.3. Analysis of IL-6, IL-1β, TNF-α and Hsp70 Expression by Real-Time PCR
4.4. Cell Viability Assay
4.5. Assessment of NF-κB Activation by Western Blot
4.6. Design of Guide RNA for Hsp70 Knockout and Cloning
4.7. Establishment of a Hsp70 Knockout 030D Cell Line and Validation
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Hsp70 | Heat shock protein70 |
| NF-κB | nuclear factor kappa light chain enhancer of activated B cells |
| TNF-α | Tumor necrosis factor α |
| CRISPR | Clustered regularly interspaced short palindromic repeat |
| Cas | CRISPR-associated |
| LPS | Lipopolysaccharide |
| qPCR | Quantitative polymerase chain reaction |
| IL-6 | Interleukin 6 |
| gRNA | Guide ribonucleic acid |
| IκB | Inhibitor of nuclear factor kappa B |
| TLR | toll-like receptor |
| NO | Nitric oxide |
References
- Perdiguero, E.G.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; De Bruijn, M.F.; Geissmann, F. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage polarization: Different gene signatures in M1 (LPS+) vs. classically and M2 (LPS–) vs. alternatively activated macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Kroner, A. Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 2011, 12, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; Freeman, T.C. Transcriptomic analysis of mononuclear phagocyte differentiation and activation. Immunol. Rev. 2014, 262, 74–84. [Google Scholar] [CrossRef]
- Kasraie, S.; Werfel, T. Role of macrophages in the pathogenesis of atopic dermatitis. Mediat. Inflamm. 2013, 2013. [Google Scholar] [CrossRef] [Green Version]
- Na, Y.R.; Stakenborg, M.; Seok, S.H.; Matteoli, G. Macrophages in intestinal inflammation and resolution: A potential therapeutic target in IBD. Nat. Rev. Gastroenterol. Hepatol. 2019, 1. [Google Scholar] [CrossRef]
- Udalova, I.A.; Mantovani, A.; Feldmann, M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 472. [Google Scholar] [CrossRef]
- He, W.; Yuan, T.; Maedler, K. Macrophage-associated pro-inflammatory state in human islets from obese individuals. Nutr. Diabetes 2019, 9, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Ta, W.; Chawla, A.; Pollard, J. Origins and hallmarks of macrophages: Development, homeostasis, and disease. Nature 2013, 496, 445–455. [Google Scholar]
- Beinke, S.; Ley, S.C. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem. J. 2004, 382, 393–409. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hambleton, J.; Weinstein, S.L.; Lem, L.; DeFranco, A.L. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc. Natl. Acad. Sci. USA 1996, 93, 2774–2778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherle, P.A.; Jones, E.A.; Favata, M.F.; Daulerio, A.J.; Covington, M.B.; Nurnberg, S.A.; Magolda, R.L.; Trzaskos, J.M. Inhibition of MAP kinase kinase prevents cytokine and prostaglandin E2 production in lipopolysaccharide-stimulated monocytes. J. Immunol. 1998, 161, 5681–5686. [Google Scholar] [PubMed]
- Sakai, J.; Cammarota, E.; Wright, J.A.; Cicuta, P.; Gottschalk, R.A.; Li, N.; Fraser, I.D.; Bryant, C.E. Lipopolysaccharide-induced NF-κB nuclear translocation is primarily dependent on MyD88, but TNFα expression requires TRIF and MyD88. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köller, M.; Hensler, T.; König, B.; Prévost, G.; Alouf, J.; Konig, W. Induction of heat-shock proteins by bacterial toxins, lipid mediators and cytokines in human leukocytes. Zent. Bakteriol. 1993, 278, 365–376. [Google Scholar] [CrossRef]
- Teshima, S.; Rokutan, K.; Takahashi, M.; Nikawa, T.; Kishi, K. Induction of heat shock proteins and their possible roles in macrophages during activation by macrophage colony-stimulating factor. Biochem. J. 1996, 315, 497–504. [Google Scholar] [CrossRef]
- Jiang, B.; Xiao, W.; Shi, Y.; Liu, M.; Xiao, X. Heat shock pretreatment inhibited the release of Smac/DIABLO from mitochondria and apoptosis induced by hydrogen peroxide in cardiomyocytes and C2C12 myogenic cells. Cell Stress Chaperones 2005, 10, 252. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.C.; Liao, L.X.; Lv, H.N.; Liu, D.; Dong, W.; Zhu, J.; Chen, J.F.; Shi, M.L.; Fu, G.; Song, X.M.; et al. Highly Selective Activation of Heat Shock Protein 70 by Allosteric Regulation Provides an Insight into Efficient Neuroinflammation Inhibition. EBioMedicine 2017, 23, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Paimela, T.; Hyttinen, J.M.; Viiri, J.; Ryhanen, T.; Salminen, A.; Kaarniranta, K. Celastrol Regulates Innate Immunity Response Via Nf-B And Hsp70 In Human Retinal Pigment Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2011, 52, 864. [Google Scholar] [CrossRef]
- Wang, C.-H.; Chou, P.-C.; Chung, F.-T.; Lin, H.-C.; Huang, K.-H.; Kuo, H.-P. Heat shock protein70 is implicated in modulating NF-κB activation in alveolar macrophages of patients with active pulmonary tuberculosis. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Ding, X.Z.; Fernandez-Prada, C.M.; Bhattacharjee, A.K.; Hoover, D.L. Over-expression of hsp-70 inhibits bacterial lipopolysaccharide-induced production of cytokines in human monocyte-derived macrophages. Cytokine 2001, 16, 210–219. [Google Scholar] [CrossRef]
- Sheppard, P.W.; Sun, X.; Khammash, M.; Giffard, R.G. Overexpression of heat shock protein 72 attenuates NF-κB activation using a combination of regulatory mechanisms in microglia. PLoS Comput. Biol. 2014, 10, e1003471. [Google Scholar] [CrossRef]
- Bhagat, L.; Singh, V.P.; Dawra, R.K.; Saluja, A.K. Sodium arsenite induces heat shock protein 70 expression and protects against secretagogue-induced trypsinogen and NF-κB activation. J. Cell. Physiol. 2008, 215, 37–46. [Google Scholar] [CrossRef]
- Chen, H.; Wu, Y.; Zhang, Y.; Jin, L.; Luo, L.; Xue, B.; Lu, C.; Zhang, X.; Yin, Z. Hsp70 inhibits lipopolysaccharide-induced NF-κB activation by interacting with TRAF6 and inhibiting its ubiquitination. FEBS Lett. 2006, 580, 3145–3152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, Q.; Ludwig, I.S.; Kooten, P.J.; Sijts, A.J.; Rutten, V.P.; Van Eden, W.; Broere, F. Leucinostatin acts as a co-inducer for heat shock protein 70 in cultured canine retinal pigment epithelial cells. Cell Stress Chaperones 2020, 25, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Stringfield, T.M.; Shi, X.; Chen, Y. Arsenite induces a cell stress-response gene, RTP801, through reactive oxygen species and transcription factors Elk-1 and CCAAT/enhancer-binding protein. Biochem. J. 2005, 392, 93–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, M.; Lahiri, D.K. Significance of NF-κB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer’s disease and multiple sclerosis. Expert Opin. Ther. Targets 2015, 19, 471–487. [Google Scholar] [CrossRef] [Green Version]
- Papoutsopoulou, S.; Burkitt, M.D.; Bergey, F.; England, H.; Hough, R.; Schmidt, L.; Spiller, D.G.; White, M.R.H.; Paszek, P.; Jackson, D.A. Macrophage-specific NF-kB activation dynamics can segregate inflammatory bowel disease patients. Front. Immunol. 2019, 10, 2168. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Santani, D. Role of NF-κB in the pathogenesis of diabetes and its associated complications. Pharmacol. Rep. 2009, 61, 595–603. [Google Scholar] [CrossRef]
- Lee, C.-T.; Repasky, E.A. Opposing roles for heat and heat shock proteins in macrophage functions during inflammation: A function of cell activation state? Front. Immunol. 2012, 3, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferat-Osorio, E.; Sánchez-Anaya, A.; Gutiérrez-Mendoza, M.; Boscó-Gárate, I.; Wong-Baeza, I.; Pastelin-Palacios, R.; Pedraza-Alva, G.; Bonifaz, L.C.; Cortés-Reynosa, P.; Pérez-Salazar, E. Heat shock protein 70 down-regulates the production of toll-like receptor-induced pro-inflammatory cytokines by a heat shock factor-1/constitutive heat shock element-binding factor-dependent mechanism. J. Inflamm. 2014, 11, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daneri Becerra, C.D.R.; Galigniana, M.D. Regulatory role of heat-shock proteins in autoimmune and inflammatory diseases. Integr. Mol. Med. 2016. [Google Scholar] [CrossRef]
- Zheng, Z.; Kim, J.Y.; Ma, H.; Lee, J.E.; Yenari, M.A. Anti-inflammatory effects of the 70 kDa heat shock protein in experimental stroke. J. Cereb. Blood Flow Metab. 2008, 28, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Chen, D.; Du, B.; Pan, J. Heat shock response inhibits NF-κB activation and cytokine production in murine Kupffer cells. J. Surg. Res. 2005, 129, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.-C.; Goode, J.; Miething, C.; Göktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S. NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKβ. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [Green Version]
- Zasłona, Z.; Pålsson-McDermott, E.M.; Menon, D.; Haneklaus, M.; Flis, E.; Prendeville, H.; Corcoran, S.E.; Peters-Golden, M.; O’Neill, L.A. The induction of pro–IL-1β by lipopolysaccharide requires endogenous prostaglandin E2 production. J. Immunol. 2017, 198, 3558–3564. [Google Scholar] [CrossRef] [Green Version]
- Dokladny, K.; Lobb, R.; Wharton, W.; Ma, T.Y.; Moseley, P.L. LPS-induced cytokine levels are repressed by elevated expression of HSP70 in rats: Possible role of NF-κB. Cell Stress Chaperones 2010, 15, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Pahl, H.L. Activators and target genes of Rel/NF-κB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [Green Version]
- Perkins, N.D. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 2006, 25, 6717–6730. [Google Scholar] [CrossRef] [Green Version]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryo, A.; Suizu, F.; Yoshida, Y.; Perrem, K.; Liou, Y.-C.; Wulf, G.; Rottapel, R.; Yamaoka, S.; Lu, K.P. Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell 2003, 12, 1413–1426. [Google Scholar] [CrossRef]
- Yang, F.; Tang, E.; Guan, K.; Wang, C.-Y. IKKβ plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J. Immunol. 2003, 170, 5630–5635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, M.; West, A.; Ghosh, S. NF-κ B and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Yue, L.; Song, J.; Wu, Q.; Li, N.; Luo, L.; Lan, L.; Yin, Z. Inducible HSP70 antagonizes IL-1β cytocidal effects through inhibiting NF-kB activation via destabilizing TAK1 in HeLa cells. PLoS ONE 2012, 7, e50059. [Google Scholar] [CrossRef] [PubMed]
- Rüdiger, S.; Germeroth, L.; Schneider-Mergener, J.; Bukau, B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997, 16, 1501–1507. [Google Scholar] [CrossRef] [Green Version]
- Mayer, M.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.R.; Gillies, A.T.; Chang, L.; Thompson, A.D.; Gestwicki, J.E. Molecular chaperones DnaK and DnaJ share predicted binding sites on most proteins in the E. coli proteome. Mol. Biosyst. 2012, 8, 2323–2333. [Google Scholar] [CrossRef] [Green Version]
- Stein, K.C.; Kriel, A.; Frydman, J. Nascent polypeptide domain topology and elongation rate direct the cotranslational hierarchy of Hsp70 and TRiC/CCT. Mol. Cell 2019, 75, 1117–1130. [Google Scholar] [CrossRef]
- Guzhova, I.V.; Darieva, Z.A.; Melo, A.R.; Margulis, B.A. Major stress protein Hsp70 interacts with NF-kB regulatory complex in human T-lymphoma cells. Cell Stress Chaperones 1997, 2, 132. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.-Q.; Liu, G.-T. Induction of overexpression of the 27-and 70-kDa heat shock proteins by bicyclol attenuates concanavalin A-induced liver injury through suppression of nuclear factor-κB in mice. Mol. Pharmacol. 2009, 75, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Ran, R.; Lu, A.; Zhang, L.; Tang, Y.; Zhu, H.; Xu, H.; Feng, Y.; Han, C.; Zhou, G.; Rigby, A.C. Hsp70 promotes TNF-mediated apoptosis by binding IKKγ and impairing NF-κB survival signaling. Genes Dev. 2004, 18, 1466–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhard, D.; Levy, M.; Ley, D. Isolation and Characterization of Functionally and Phenotypically Distinct Continuous Monocytoid Cell Lines from a Canine Malignant Histiocytosis. In Proceedings of the Fourth International Veterinary Immunology Symposium, Davis, CA, USA, 16–21 July 1995. [Google Scholar]
- Merenda, A.; Andersson-Rolf, A.; Mustata, R.C.; Li, T.; Kim, H.; Koo, B.-K. A protocol for multiple gene knockout in mouse small intestinal organoids using a CRISPR-concatemer. J. Vis. Exp. 2017, 125, e55916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhao, J.; Zheng, X.; Cai, K.; Mao, Q.; Xia, H. Establishment of a novel hepatic steatosis cell model by Cas9/sgRNA-mediated DGKθ gene knockout. Mol. Med. Rep. 2018, 17, 2169–2176. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cassette 1 | Cassette 2 | |
|---|---|---|
| Forward sequence (5′-3′) | CACCGG[TCCATCCTGACGATCGACGA]GT | ACCGG[CGGCACGGTGATCACCGCGT]G |
| Reverse sequence (5′-3′) | TAAAAC[TCGTCGATCGTCAGGATGGA]CC | AAAAC[ACGCGGTGATCACCGTGCCG]C |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lyu, Q.; Wawrzyniuk, M.; Rutten, V.P.M.G.; van Eden, W.; Sijts, A.J.A.M.; Broere, F. Hsp70 and NF-kB Mediated Control of Innate Inflammatory Responses in a Canine Macrophage Cell Line. Int. J. Mol. Sci. 2020, 21, 6464. https://doi.org/10.3390/ijms21186464
Lyu Q, Wawrzyniuk M, Rutten VPMG, van Eden W, Sijts AJAM, Broere F. Hsp70 and NF-kB Mediated Control of Innate Inflammatory Responses in a Canine Macrophage Cell Line. International Journal of Molecular Sciences. 2020; 21(18):6464. https://doi.org/10.3390/ijms21186464
Chicago/Turabian StyleLyu, Qingkang, Magdalena Wawrzyniuk, Victor P. M. G. Rutten, Willem van Eden, Alice J. A. M. Sijts, and Femke Broere. 2020. "Hsp70 and NF-kB Mediated Control of Innate Inflammatory Responses in a Canine Macrophage Cell Line" International Journal of Molecular Sciences 21, no. 18: 6464. https://doi.org/10.3390/ijms21186464
APA StyleLyu, Q., Wawrzyniuk, M., Rutten, V. P. M. G., van Eden, W., Sijts, A. J. A. M., & Broere, F. (2020). Hsp70 and NF-kB Mediated Control of Innate Inflammatory Responses in a Canine Macrophage Cell Line. International Journal of Molecular Sciences, 21(18), 6464. https://doi.org/10.3390/ijms21186464