3. Discussion
Affordable blood-based surrogate biomarker assays that can reliably detect AD associated pathological changes at an early disease stage have enormous potential to support early and preclinical diagnosis and the development of novel, disease modifying treatments. Recent studies have shown that specific forms of the Aβ peptide can be detected in human blood plasma and represent highly promising AD biomarker candidates. For example, a multimer detection system employing two antibodies with overlapping N-terminal Aβ-epitopes for measuring Aβ oligomerization after spiking synthetic Aβ into blood plasma samples has been developed [
27]. Correlations between the plasma oligomerized Aβ and CSF Aβ
42, amyloid-positron-emission tomography (PET) results [
28], and measures of cognitive functions [
29,
30] were reported. Other research groups discovered that the plasma ratios Aβ
42/Aβ
40, Aβ
1–40/Aβ
1–42, and APP
669–711/Aβ
1-42 (i.e., Aβ
−3–40/Aβ
1–42), as measured by immunoprecipitation followed by mass spectrometry, predicted brain amyloid pathology with high accuracy [
19,
20,
23]. To the best of our knowledge, so far, commercial antibodies against N-terminally elongated Aβ
−3–X or immunoassays for the specific measurement of Aβ
−3–40 have not been available.
To set the groundwork for studying the biology of Aβ
−3–40 in more detail in future studies and for assessing Aβ
−3–40 as a potential reference for accentuating the AD-associated selective decrease in Aβ
42 in blood plasma and possibly other biological fluids, we have characterized two novel antibodies raised against the Aβ
−3–X N-terminus and developed a highly selective sandwich immunoassay for measuring Aβ
−3–40. Our observations show that the monoclonal antibodies 101-1-1 and 14-2-4 can detect Aβ
−3–40 without appreciable cross-reactivity with the canonical Aβ
1–40 or N-terminally truncated Aβ-variants in CIEF immunoassay and urea-SDS-PAGE/Western blot analysis. As it turned out, neither of the two antibodies are specific for the free N-terminal valine. Both also recognize amyloid precursor protein (APP), provided it does not carry the Swedish (K670N/M671L) double mutation located directly N-terminal to the β-secretase cleavage site. Whether or not the mAbs 101-1-1 and 14-2-4 display preferences for monomeric, oligomeric, or aggregated forms of the N-terminally elongated Aβ
−3–40 has not been assessed explicitly here. Published in vitro experiments including Thioflavin-T aggregation assay, size exclusion chromatography, and circular dichroism suggested a substantially lower self-aggregation tendency of Aβ
−3–40 than Aβ
1–42 [
23]. To the best of our knowledge, Aβ
−3–40 has not been reported in amyloid plaques so far, and essentially no information regarding its potential role in AD pathogenesis is available. In brain slices from APP
Ld transgenic mice analyzed in our present study, mAb 101-1-1 appeared to bind neuronal cell bodies and to dystrophic neurites resembling abnormal neuronal processes, but not to plaque cores. Presumably, the observed signals represent cellular APP.
MAb 101-1-1 was selected to serve as the capture antibody in the finalized version of our novel Aβ−3–40 immunoassay. The LLOD of the assay for measuring Aβ−3–40 was approximately 11 pg/mL, and the quantitative assay range was approximately 22 pg/mL–7.5 ng/mL. We did not observe appreciable cross reactivity with Aβ1–40, Aβ1–42, Aβ−3–38, and sAPPα. In contrast, at high concentrations, synthetic Aβ−3–42 produced measurable signals, indicating low cross reactivity at a level of <0.5%. The technical intra-assay repeatability passed the predefined acceptance limit of <15% CV, and we did not find evidence for systematic spatial bias. The inter-assay CV of the calculated Aβ−3–40 concentration in a QC IP eluate that was measured in five independent experiments within the assay validation campaign was 16.3%, and thus outside of the predefined 15% CV acceptance range. It appeared that the large scatter was owing to a single experiment in which the calibrator dilution series had been prepared in a slightly deviant way (to allow for determination of the ULOQ in this particular assay run). Accordingly, we conclude that a strict SOP covering all pre-analytical and analytical steps in detail has to be implemented to minimize inter-assay variability.
The primary intended purpose of the novel assay is the assessment of Aβ
−3–40 and the Aβ
42/Aβ
−3–40 ratio (or reverse) in human blood plasma as a candidate biomarker of AD. In view of published observations from IP mass spectrometry studies [
22,
23,
24], the blood concentration of Aβ
−3–40 was expected to be small. Thus, we decided to pre-concentrate Aβs from plasma by magnetic bead immunoprecipitation and to analyze the Aβ
−3–40 plasma level by a two-step immunoassay procedure, essentially as described previously for the measurement of Aβ
38, Aβ
40, and Aβ
42 [
21]. A control experiment with a pooled blood plasma sample confirmed high yield of Aβ
−3–40 after a single round of 1E8 magnetic bead IP. Pre-analytical freezing and thawing of diluted IP eluates did not seem to substantially and systematically affect the measurement of Aβ
−3–40. Consequently, freezing of the diluted IP eluates at −80 °C was implemented in our two-step immunoassay protocol to completely separate pre-analytical Aβ-enrichment from the actual measurement, and thus facilitate the execution of the two-step immunoassay under routine laboratory conditions. Furthermore, this allowed for testing a semi-automated prototypic IP procedure executed at a different site (see below). We have shown that the two-step immunoassay for Aβ
−3–40 is highly selective. We did not observe appreciable signals in plasma IP eluates from magnetic bead IP with an unrelated antibody or from a simulated IP with 1E8 magnetic beads. Thus, unspecific binding of other plasma components to functionalized anti mouse IgG magnetic beads or background signal directly stemming from the 1E8 coupled beads could be excluded. Depleting Aβ by two rounds of IP with anti Aβ mAbs 6E10 plus 4G8 prior to the 1E8 IP reduced the measured Aβ
−3–40 concentration by approximately 95%.
Analytical spike recoveries in the two-step immunoassay varied substantially between the tested plasma samples and different spike levels indicating matrix interferences. Thus, the measured Aβ−3–40 levels in the diluted IP eluates do not allow for calculating the true, absolute concentrations of this Aβ peptide in plasma samples, but have to be considered relative.
The intermediate imprecision (between-run variance) of the complete manual two-step immunoassay reached an acceptable CV of 16.2% only after normalization. Notably, in this case, the normalization addressed the technical error contributed by the Aβ−3–40 assay only, not the scatter of the manual IP procedure. Without normalization, the mean between-run CV of the measured Aβ−3–40 concentrations in four QC-samples was 29.6%, and thus clearly above the predefined acceptance criterion of <20% CV. In conclusion, appropriate QC samples allowing for normalization should be included in future experiments, whenever possible, and further technical improvement is desirable.
To finally test the novel Aβ
−3–40 immunoassay on biological samples in a pilot study and to demonstrate the possibility of automation of the magnetic bead IP, we studied the same small clinical sample we have previously described in detail [
21]. In our current study, Aβ was pre-concentrated from 1:2 dilutions of blood plasma with water by a prototypic semi-automated 1E8 magnetic bead IP protocol. The IP eluates were diluted 4.8-fold with Diluent-35 and frozen in aliquots for single use at −80 °C. For measuring Aβ
−3–40, Aβ
40, and Aβ
42 at a different site, the samples were transported on dry ice. We did not observe a statistically significant difference in the mean (log2 transformed) Aβ
−3–40 levels between patients with AD-D (
n = 23) and OD (
n = 17). In contrast, Aβ
42 and the Aβ ratios Aβ
42/Aβ
−3–40 and Aβ
42/Aβ
40 were statistically significantly lower in patients with AD-D compared with those in the OD group. These findings are in agreement with the IP mass spectrometry data collected in two independent cohorts by Nakamura et al. [
23]. The areas under the ROC curves in our small data set were 0.76 (Aβ
42), 0.79 (Aβ
42/Aβ
−3–40 ratio), and 0.85 (Aβ
42/Aβ
40 ratio). These observations support the idea that a suitable reference may serve to accentuate the pathological AD-associated decrease in soluble Aβ
1–42 [
22] (or Aβ
42, respectively). Furthermore, our findings show that a prototypic semi-automated pre-analytical IP protocol works and that the adaptions and modifications to the original manual two-step immunoassay that were implemented did not lead to masking or loss of the reduced Aβ
42/Aβ
40 plasma ratio as a peripheral diagnostic biomarker of AD. Further studies employing independent and larger clinical cohorts will be required to assess the diagnostic potential of the two-step immunoassay for the Aβ
42/Aβ
−3–40 ratio in detail. Preferably, healthy controls and patients in different stages of AD should be included
In summary, we have developed a novel, highly selective sandwich immunoassay for measuring Aβ−3–40 in biological samples. In combination with pre-analytical Aβ enrichment by magnetic bead IP, the assay can serve to measure the relative levels of Aβ−3–40 in human blood plasma. Thus, the methodological groundwork has been set for future studies addressing the diagnostic potential of the Aβ42/Aβ−3–40 ratio (or reverse) as a novel surrogate biomarker candidate of cerebral amyloid deposition.
4. Materials and Methods
Antibodies: The mouse monoclonal antibody (mAb) 101-1-1 was developed, produced, and purified by Biogenes GmbH (Berlin, Germany) on a fee for service basis. The synthetic peptide VKMDAEFRC-amide (Biosyntan, Berlin, Germany) was conjugated to bovine thyroglobulin as carrier protein and served for immunization of mice. Hybridoma cells were screened by ELISA using Aβ1–40 and Aβ2–40 for negative screening and VKMDAEFRC-BSA-conjugate for positive screening. Clone 101-1-1 was obtained after two rounds of cloning. We received 88 mg of purified monoclonal antibody mAb 101-1-1 for antibody characterization, assay development, assay validation, and further use. Monoclonal anti Aβ−3–x, clone 14-2-4, was obtained from IBL International/Tecan, (Hamburg, Germany). MAb 1E8 (anti Aβ N-terminus) and mAb 5C3 (anti Aβ40) were purchased from nanoTools GmbH (Teningen, Germany). MAb 280F2 (anti Aβ40) was obtained from Synaptic Systems (Goettingen, Germany), mAb 6E10 from Biolegend (San Diego, CA, USA), anti APP mAb 22C11 from Merck-Millipore (Darmstadt, Germany), and anti-Human Tau mAb HT7 from Thermo Scientific (Waltham, MA USA). The rabbit monoclonal anti Aβ42 antibody mAb D3E10 was purchased from Cell Signaling Technology (Danvers, MA, USA), while the rabbit polyclonal anti C-terminus APP A8717 was purchased from Sigma-Aldrich (Munich, Germany). SULFO-TAG Aβ40 detection antibody was obtained from Mesoscale Discovery, Rockville, MD, USA. Biotinylated horse anti-mouse IgG was purchased from Vector Laboratories (Burlingame, CA, USA), streptavidin-biotinylated peroxidase complex from GE Healthcare, Chicago, IL, USA), and goat anti mouse IgG peroxidase conjugate from Calbiochem/Millipore/Merck (Darmstadt, Germany). Biotinylated secondary anti-mouse and anti-rabbit antibodies, used in DAB-immunohistochemical staining, were obtained from DAKO/Agilent (Waldbronn, Germany). Fluorescent goat anti-Mouse IgG (DyLight 594) donkey anti-Rabbit IgG (DyLight 488) secondary antibodies were purchased from Thermo Scientific (Waltham, MA, USA).
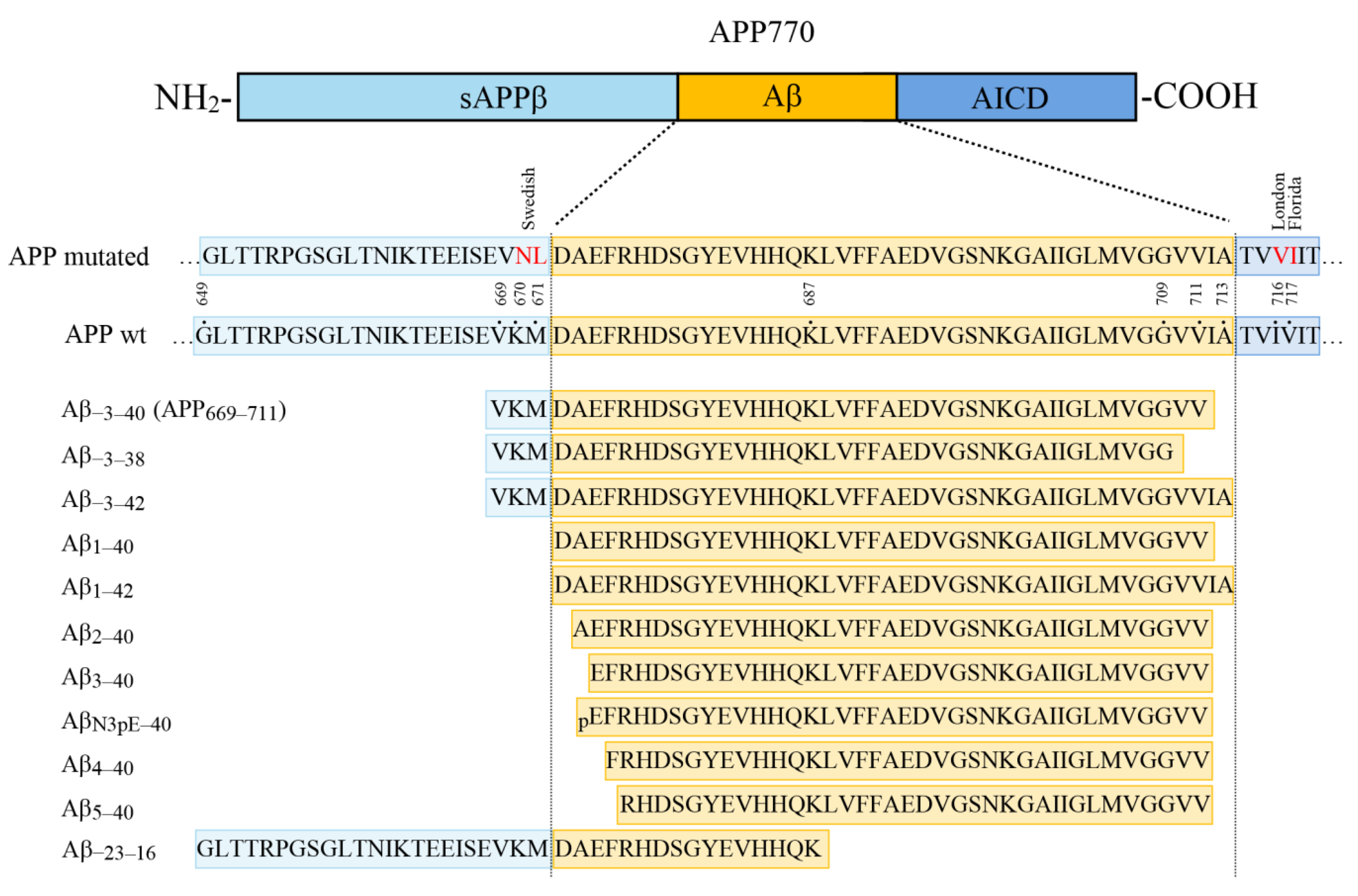
Recombinant sAPPα was obtained from Sigma-Aldrich (Munich, Germany). Aβ
−3–40, Aβ
−3–38, and Aβ
−3–42 were synthesized as described in detail previously [
25]. The synthetic peptides Aβ
1–40, Aβ
2–40, Aβ
3–40, Aβ
N3pE–40, Aβ
4–40, and Aβ
5–40 were obtained from AnaSpec (Fremont, CA, USA). The synthetic model peptide Aβ
−23–16 (APP
649–687) (H
2N-GLTTRPGSGLTNIKTEEISEVKMDAEFRHDSGYEVHHQK-CONH
2) was synthesized using standard solid-phase fluorenylmethoxycarbonyl (Fmoc) chemistry. Quality control of the purified product was performed by reversed-phase high-performance liquid chromatography coupled to a single-quadrupole mass spectrometer (Alliance/QDa, Waters Corporation, Milford, MA, USA) and by high-resolution mass spectrometry using a MALDI-TOF/TOF mass spectrometer (UltrafleXtreme, Bruker, Billerica, MA, USA).
The CIEF immunoassay was performed as described before [
8,
26]. For chemiluminescence detection, goat anti-mouse IgG, streptavidin-peroxidase conjugate, and Luminol/peroxide (all 3 reagents obtained from ProteinSimple, San Jose, CA, USA) were employed.
Preparation of functionalized anti-Aβ magnetic beads: Dynabeads M280 sheep anti mouse IgG (ThermoFisher Scientific, Waltham, MA, USA) were functionalized by covalent coupling to mAb 1E8 according to the manufacturer’s instructions and as described before [
21]. Dynabeads M-270 Epoxy (Invitrogen/ThermoFisher Scientific, Waltham, MA, USA) were covalently coupled to mAb 101-1-1 following the manufacturer’s instructions.
Antibody labeling: For small-scale biotinylation and SULFO-Tag labeling, 80 µL of a 1 mg/mL stock solution of mAb 101-1-1 or 280F2 was buffer-exchanged into MSD conjugation buffer (PBS, pH 7.9, preservative-free) on a Zeba Spin Desalting column 40 K MWCO, 0.5 mL (Thermo Scientific, Waltham, MA, USA). The buffer-exchanged antibody solution was divided into two halves, each containing approximately 40 µg of IgG. For biotinylation, 3 µL of a 0.9 nmol/µL Sulfo-NHS-LC biotin solution (EZ-Link Micro Sulfo-NHS-LC Biotinylation kit, Thermo Scientific, Waltham, MA, USA) was added to approximately 40 µg of IgG (challenge ratio: 10:1) and mixed immediately on a vortex mixer. After 2 h incubation at 23 °C, excess biotin was removed by buffer exchange into MSD conjugate storage buffer (PBS, pH 7.4 containing 0.05% sodium azide) on a Zeba Spin Desalting column (40 K MWCO, 0.5 mL). For storage at −20 °C, glycerole was added to a final concentration of 50% (v/v). For SULFO-TAG labeling, 1.8 µL of a 3 nmol/µL solution of MSD Gold SULFO-TAG NHS-Ester (Mesoscale Discovery, Rockville, MD, USA) was added dropwise to approximately 40 µg of buffer exchanged IgG (see above, challenge ratio: 20:1). After mixing by pipetting up and down, the reaction was incubated for 2 h at room temperature in the dark. The remaining reagent was removed by buffer exchange into MSD conjugate storage buffer on a Zeba Spin desalting column (40 K MWCO, 0.5 mL). Storage: at 4 °C in the dark.
For the biotinylation of mAb 101-1-1 on a larger scale, 159 µL of 101-1-1 [6.3 mg/mL] was mixed with 841 µL of PBS (0.1 M sodium phosphate/0.15 M NaCl, pH 7.2, Thermo Scientific, Waltham, MA, USA) and buffer exchanged into PBS (0.1 M sodium phosphate/0.15 M NaCl, pH 7.2, Thermo Scientific) on a Zeba Spin desalting column (5 mL, 7000 MWCO, Thermo Scientific, Waltham, MA, USA). Then, 7.4 µL of a 9 nmol/µL Sulfo-NHS-LC biotin stock solution was added (challenge ratio 10:1) and mixed on a vortex immediately. After incubation for 2 h at 23 °C, excess biotin was removed by buffer exchange into PBS. The protein concentration was measured by microplate BCA protein assay (PIERCE/ThermoFisher Scientific, Waltham, MA, USA) and the level of biotin incorporation was determined by HABA (4-hydroxyazobenzene-2-carboxylic acid) assay in a microplate according to the instructions to the EZ-Link Sulfo-NHS-Biotinylation Kit (Themo Scientific, Waltham, MA, USA). BSA and sodium azide were added to the biotinylated antibody to final concentrations of 1% and 0.05%, respectively. For long term storage, half of the material was aliquoted and frozen at −80 °C. The remaining biotinylated antibody stock was supplemented with 40% (v/v) ethylene glycol and stored in aliquots at −20 °C.
Urea-SDS-PAGE and semi dry blotting: Aβ-peptides were separated by urea-bicine/bis-tris/tris/sulfate SDS-polyacrylamide gel electrophoresis [
31] on 12%T/5% C gels and subsequently transferred on PVDF membranes by semi dry blotting, essentially as described previously [
32,
33]. The protein transfer occurred at a constant current of 0.8 mA/cm
2 for 30 min with a voltage limit of maximum 30 V.
Bis-Tris Gradient Gel electrophoresis and Western blotting: Proteins and peptides were separated on a 4–12% Bis-Tris gradient gel (Anamed Elektrophorese GmbH, Groß-Bieberau, Germany) at 200 V for 30 min with MES (2-(N-morpholino)ethanesulfonic acid) running buffer (50 mM MES, 50 mM Tris, 0.1% w/v SDS, 1 mM EDTA). NuPAGE antioxidant (Thermo Fisher Sientific, Waltham, MA, USA) was added to the upper buffer chamber. The proteins were subsequently blotted onto PVDF membranes using 25 mM Bicine, 25 mM Bis-Tris pH 7.2, 1 mM EDTA, and 10% (v/v) methanol transfer buffer with NuPAGE antioxidant at 20 V for 60 min (MiniGel Tank and Blot Module obtained from Invitrogen/Thermoscientific, Waltham, MA, USA).
Immunohistochemistry: Experiments involving animal tissues were approved by the local animal care and use committee (Landesamt für Verbraucherschutz und Lebensmittelsicherheit (LAVES), Lower Saxony). Brain tissue slides from 12-month-old 5XFAD [
34] and 15-month-old APP/
Ld transgenic mice [
35] were stained as published elsewhere [
36]. In brief, 4 µm paraffin sections were deparaffinized in xylene and rehydrated in a series of ethanol, followed by incubation in PBS containing 1% H
2O
2 to block endogenous peroxidases. Antigen-retrieval was carried using microwave treatment in 0.01 M citrate buffer pH 6.0 and incubation in 88% formic acid. Non-specific binding sites were blocked with 4% skim milk in PBS containing 10% fetal calf serum. This was followed by overnight incubation with 101-1-1 (1:10,000) or APP C-term (A8717, Sigma Aldrich, Munich, Germany, 1:3000) in a humid chamber. Staining was visualized using the ABC method with a Vectastain Kit (Vector Laboratories, Burlingame, CA, USA) and diaminobenzidine. In the case of fluorescent staining, 101-1-1 (1:6000) and the Aβ
42-specific antibody D3E10 (1:1000) (Cell Signaling Danvers, MA, USA) were used and detected using anti-mouse-DyLight-594 or anti-rabbit-Dylight-488 antibodies (both Thermo Fisher, Waltham, MA, USA).
Manual and automated immunoprecipitations: Magnetic bead immunoprecipitations (IPs) by hand starting from 400 µL of EDTA-blood plasma were performed as described previously [
21]. An adapted protocol for a semi-automated IP procedure was executed on a CyBio FeliX liquid handling Instrument (Analytik Jena, Jena, Germany) equipped with BioShake 3000-Telm (Q Instruments, Jena, Germany) and MAGNUM FLX Enhanced Universal Magnet Plate, Alpaqua Engineering, LLC, Beverly, MA, USA.
Aliquots of EDTA-blood plasma samples (approximately 500 µL per sample), which had been stored at −80 °C in Matrix vials, were thawed and transferred into 1.5 mL reaction vials. The vials were mixed vigorously on a vortex mixer (5 × 10 s), and insoluble material was pelleted by centrifugation for 10 min at 10,000× g at room temperature in a fixed angle rotor. Then, 220 µL of each supernatant was transferred into a 96-well sample plate (Deep Well MegaBlock®, 96 wells, 2.2 mL, PP (Sarstedt, Nümbrecht, Germany)) and manually mixed with 220 µL of H2O.
The sample plate was mounted into the CyBio FeliX instrument and preparation of immunoprecipitation mixes was carried out automatically by adding 100 µL of 5× IP buffer concentrate [
21] and 25 µL of 1E8 magnetic beads to 400 µL of the diluted samples. Finally, the instrument transferred the preparations into a process plate (Deep Well MegaBlock®, 96 wells, 2.2 mL, PP (Sarstedt, Nümbrecht, Germany)).
The process plate was then manually placed in the “process” position of the CyBio FeliX instrument and the immunoprecipitation process was executed automatically. The process included an 18-h incubation at room temperature, where the samples were regularly mixed. Then, the process plate was moved into a magnet position for 120 s for immobilization of the magnetic beads. The supernatant was aspirated and discarded, and the process plate was moved back to the process position. Per well, 1 mL of wash buffer (PBS containing 0.1% w/v BSA) was added and the beads were washed for 5 min at room temperature with agitation. The plate was moved to the magnet position for bead immobilization and removal of the wash fluid as described. Then, the plate was moved back to the process position for a second wash. In total, the magnetic bead immune complexes were washed 3× for 5 min with PBS/0.1% w/v BSA and once for 3 min with 1 mL of 10 mM Tris-HCl, pH 7.5. After removal of the wash buffer, 2 × 25 µL of elution buffer (20 mM Bicine, pH 7.6, 0.6% w/v CHAPS) were added, and the beads were resuspended by aspirating and dispensing. The resuspended beads were then transferred into a preheated elution plate (Deep Well MegaBlock®, 96 wells, 1.2 mL PP (Sarstedt, Nümbrecht, Germany)) positioned on the BioShake and incubated for 5 min at 95 °C and 800 rpm to allow elution of the bound Aβ peptides. Then, the suspension was quantitatively moved to a 96-well plate (Deepwell plate 96/500 µL Protein LoBind (Eppendorf, Hamburg, Germany)) located on the magnet and incubated for 120 s for cooling and bead immobilization. Subsequently, 190 µL of Diluent-35 was added to each well (4.8-fold dilution of the IP eluates) and the samples were mixed. Finally, 200 µL of each diluted IP eluate was transferred into a separate plate (Deepwell plate 96/500 µL Protein LoBind (Eppendorf, Hamburg, Germany)). Aliquots of 60 µL each were manually pipetted into 1.5 mL reaction vials and frozen at −80 °C.
Measurements of Aβ
40 and Aβ
42: Aβ
40 and Aβ
42 levels in 4.8-fold diluted IP eluates were measured with the V-Plex Aβ panel 1 (6E10) multiplex assay kit (Mesoscale Discovery, MSD, Rockville, MD, USA) according to the manufacturer’s instructions and as described previously [
21]. In the current version of the two-step immunoassay protocol, the 4.8-fold diluted IP eluates were measured after storage at −80 °C and singular thawing.
Development of the Aβ−3–40 immunoassay: The novel sandwich immunoassay for the measurement of Aβ−3–40 was developed on the MSD technology platform (Mesoscale Discovery, MSD, Rockville, MD, USA) using MSD Gold 96-well Small Spot streptavidin plates and various MSD reagents, including 20× wash buffer, Diluent-100, Diluent-35, Blocker A, 4× Read buffer, and SULFO-TAG Aβ40 detection antibody. In the final assay protocol (SOP), 150 µL of 3% (w/v) Blocker-A in wash buffer was added to the wells of a 96-well Small Spot streptavidin plate. The plate was sealed with an adhesive foil and incubated for 1 h at room temperature with shaking (500 rpm) on an Eppendorf Thermomix with a lightproof cover. The blocking solution was discarded and the plate was washed three times with 200 µL of wash buffer per well. For plate coating, 25 µL of a 2 µg/mL dilution of the biotinylated capture antibody in Diluent-100 was added per well. The plate was sealed with an adhesive foil and incubated for 1 h at room temperature with shaking (as described above). After three washes with 200 µL of wash buffer per well, 25 µL of sample or calibrator dilution per well was pipetted into the assay plate. The plate was sealed with an adhesive foil and incubated for 1 h at room temperature with shaking (see above). After three washes with 200 µL of wash buffer per well, 25 µL of detection antibody diluted in Diluent-100 was added to each well. Again, the plate was sealed with an adhesive foil and incubated for 1 h at room temperature with shaking (500 rpm) on an Eppendorf Thermomix with a lightproof cover. After three washes with 200 µL of wash buffer per well, 150 µL of 2× Read buffer was added to each well by reverse pipetting. Formation of bubbles must be avoided at this stage. The electro-chemiluminescent signals were recorded on an MSD Quickplex SQ120 reader and analyzed with the MSD Discovery Workbench Software version 4.0.12.
The synthetic calibrator peptide Aβ
−3–40 was synthesized as described before [
25]. The lyophilized peptide was initially solubilized in DMSO (dimethyl sulfoxide) at a concentration of 1 mg/mL and stored in aliquots at −80 °C. Starting from this master stock solution in DMSO, a 600 ng/mL calibrator stock solution for use in the novel Aβ
−3–40 immunoassay was prepared in Diluent-35 and stored in aliquots for single use at −80 °C. In most experiments, seven serial calibrator dilutions in Diluent-35 plus a zero calibrator (blank) were measured for calculating the standard curve by four-parameter logistic curve fitting. The seven calibrator solutions (standards) were prepared by serial fourfold dilution in Diluent-35 starting with the highest standard to be measured. During the assay validation campaign, in most cases, the highest standard had a concentration of 1875 pg/mL. For the study of the clinical sample, the highest standard contained 7.5 ng/mL of Aβ
−3–40. In one experiment, an extended fourfold calibrator dilution series including ten fourfold serial dilutions starting at 120 ng/mL plus zero calibrator (blank) was analyzed to determine the ULOQ. The alternative synthetic D7-Calibrator (VKMDAEFRH followed by 5× PEG2 (8-amino-3,6-dioxaoctanoic acid) and the Aβ
40 C-terminus GLMVGGVV) (Mr: 2571.4)) was synthesized by Biosyntan (Berlin, Germany).
Cell culture: H4 cells stably overexpressing Swedish mutant APP751SWE [
37] were cultured in D-MEM/F12, supplemented with 10% fetal calf serum, 2 mM L-Alanyl-L-Glutamine, non-essential amino acids, and 500 µg/mL Hygromycin. To generate H4 cells stably overexpressing wild type APP751, the human amyloid beta A4 protein isoform b precursor cDNA (also known as PreA4 751, APP 751) was subcloned into a pCI-neo mammalian expression vector (Promega, Walldorf, Germany). H4 neuroglioma cells from the exponential growth phase were seeded at 5 × 10
5 cells/cm² in H4 growth medium (DMEM supplemented with 4.5 g/L glucose, 4 mM L-glutamine, 10% (
v/
v) fetal bovine serum, 100 IU/mL penicillin, and 100 μg/mL streptomycin) 24 h before the transfection. Medium changes with serum free DMEM/Ham´s F12 with G5 supplement were performed 3 h before and directly before the transfection. Plasmid DNA and CaCl
2 were mixed with N,N-Bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES) buffer with pH 6.98, yielding final concentrations of 25 mM BES, 140 mM NaCl, 0.75 mM Na
2HPO
4, 125 mM CaCl
2, and 37.5 ng/µL plasmid DNA. For the transfection of a growth area of 9.5 cm², 200 µL of the mixture was incubated for 20 min at room temperature in the dark and subsequently added to the cells. The culture medium was changed to growth medium 24 h after transfection. After 48 h, 500 µg/mL G418 (Thermo Fisher Scientific, Waltham, MA, USA) was added and resistant clones were selected by limiting dilution at <0.2 cells/well in H4 growth medium with 500 µg/mL G418. The cells were maintained in the presence of 500 µg/mL G418. Six clones were isolated and assessed for the level of APP expression. The cells were cultured in DMEM GlutaMAX (Thermo Fisher, Waltham, MA, USA) supplemented with 10% superior fetal bovine serum (Biochrom, Berlin, Germany) and 500 µg/mL G418.
Study approval and study cohort. The study was conducted in accordance with the Declaration of Helsinki. The pseudonomized collection of biological samples and clinical data in a local biobank and their use in biomarker studies was approved by the ethics committee of the University of Göttingen (9/2/16). All subjects or their legal representatives gave their informed consent prior to inclusion. The clinical sample comprising
n = 23 patients with dementia of the Alzheimer’s type (AD-D) and
n = 17 patients with dementia due to other reasons (OD) has been described in detail in a previous study [
21]. The preparation and storage of blood plasma samples according to standardized protocol were described before (ibid.).
Statistical analysis: For statistical analysis and graphical representation of data, we employed GraphPad Prism 8.4 and IBM SPSS Statistics 26.
Normalization between assay plates. On each assay plate in the core validation campaign, we included the measurement of an aliquot of a quality control (QC) IP eluate in four technical replicates. It served to assess the between-run variance of the Aβ
−3–40 assay and to allow for testing a method for normalization between assay plates. The QC-IP eluate was prepared by running several IPs in parallel from pooled human blood plasma and combining the obtained IP eluates after 4.8-fold dilution to a single QC-IP eluate. Aliquots for single use were stored frozen at −80 °C until the analysis. For normalization, the average concentration of Aβ
−3–40 in the QC-IP eluate (mean of four independent assay runs) was calculated as follows:
where c
m = measured concentration, mean of the replicate measurements on the respective assay plate, and c
avg = mean concentration (average of
n = 4 plates considered).
Subsequently, plate correction factors (k) for each assay plate were calculated as follows:
Finally, the normalized Aβ
−3–40 concentrations (c
norm) were calculated with the following formula:
The normalization by the QC IP eluate was applied to the data obtained with aliquots of a set of three individual control plasma samples and a control plasma pool that were analyzed by the complete two-step immunoassay procedure (i.e., IP followed by Aβ−3–40 measurement) in four independent experiments. It should be noted that the described normalization procedure considered only the experimental error contributed by the 96-well plate immunoassay, but not the error contributed by the manual immunoprecipitation.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}