Tofacitinib and Baricitinib Are Taken up by Different Uptake Mechanisms Determining the Efficacy of Both Drugs in RA

 ,
,  and
and 
Abstract
1. Introduction
2. Results
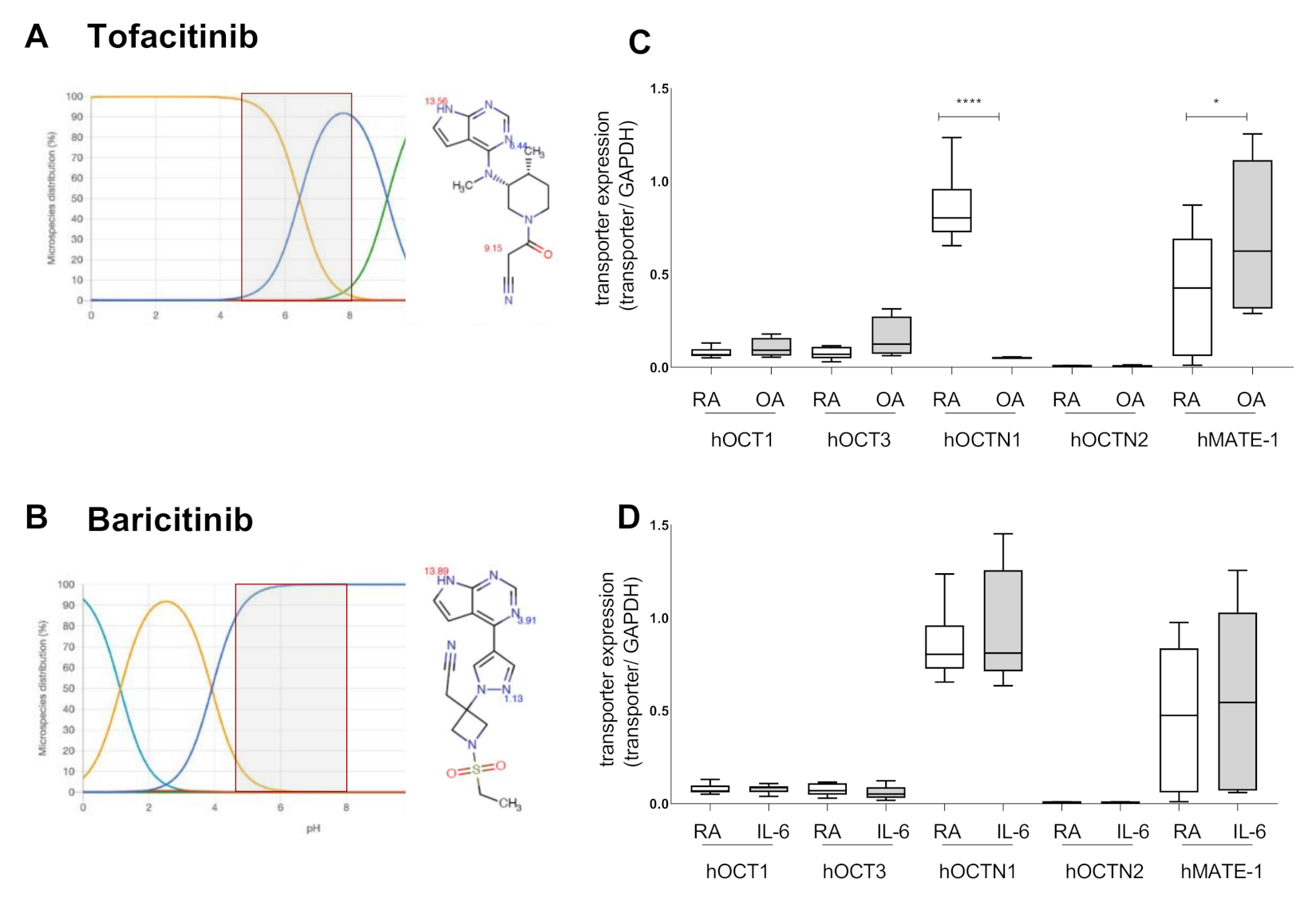
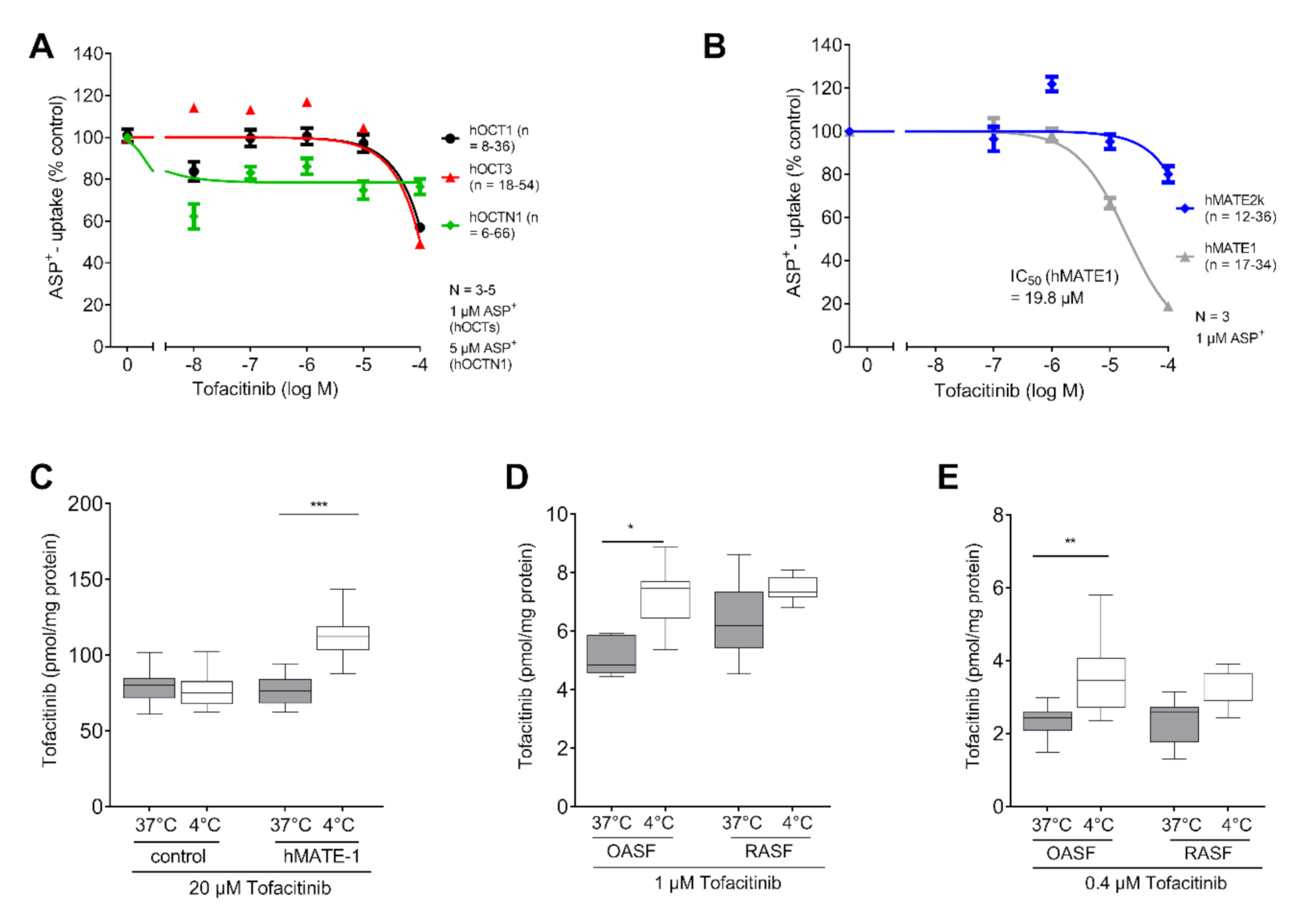
2.1. Tofacitinib Could Be a Target for OCT Mediated Cellular Uptake
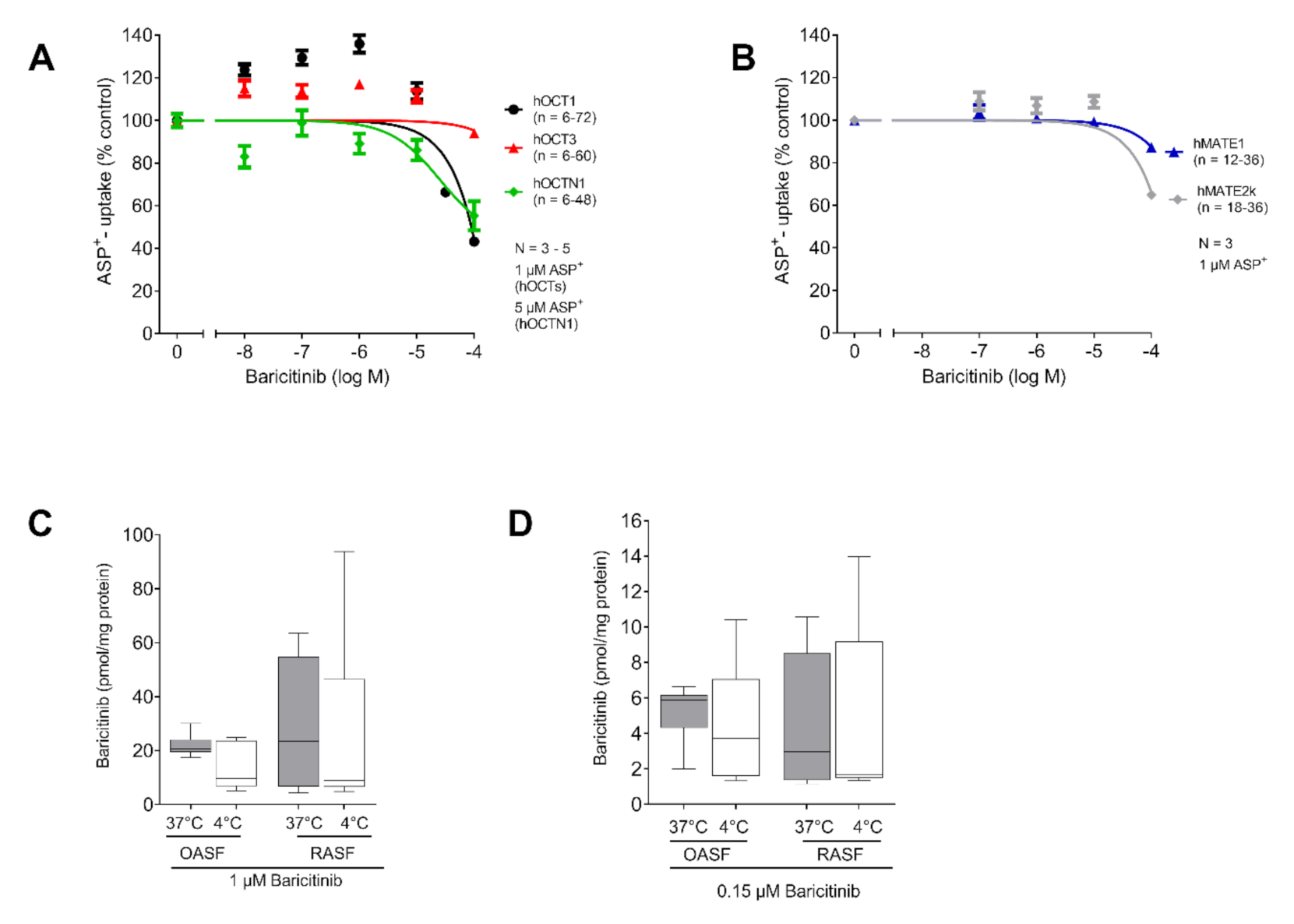
2.2. Baricitinib Uptake Is Not Transporter Dependent
2.3. MATE-1 Mediates Tofacitinib Transport
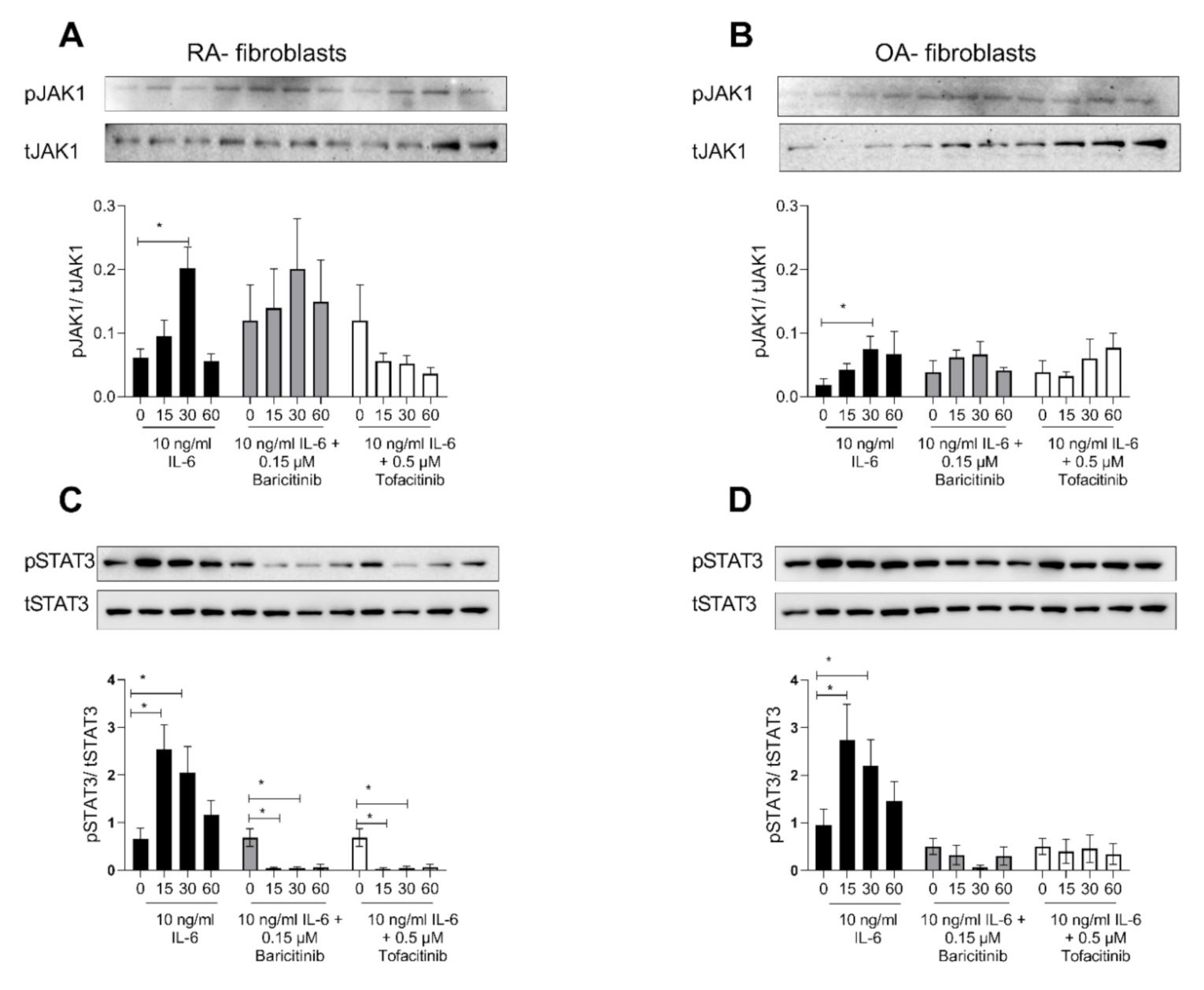
2.4. Tofacitinib and Baricitinib Showed Comparable Inhibition of IL-6-Induced STAT3-Phosphorylation
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Lines
5.2. Synovial Fibroblasts (SF) Culture and Isolation
5.3. Peripheral Blood Mononuclear Cell (PBMC) Isolation and Cultivation
5.4. Apparent Affinities of Baricitinib and Tofacitinib for OCTs With 4-(4-(Dimethylamino)styryl)-N-Methylpyridinium (ASP+)
5.5. Quantification of Baricitinib and Tofacitinib Uptake by Liquid Chromatography Mass Spectrometry (LC/MS)
5.6. Quantitative Real-Time PCR (qRT-PCR)
5.7. Western Blot Analysis
5.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| ASP | 4-(4-(dimethylamino)styryl)-N-methylpyridinium |
| AU | Arbitrary units |
| BSA | Bovine serum albumin |
| cDNA | complementary Deoxyribonucleic Acid |
| csDMARD-IR | conventional synthetic disease-modifying antirheumatic drug inadequate response |
| DMEM | Dulbecco’s modified eagle medium |
| DMSO | Dimethyl sulfoxide |
| ECL | Enhanced chemiluminescence |
| EMA | European Medicines Agency |
| ESI | electrospray ionization |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| gp130 | Glycoprotein 130 |
| HEK 293 cells | Human embryonic kidney 293 cells |
| HPLC | High Performance Liquid Chromatography |
| IL-6 | Interleukin-6 |
| IL-6Ra | Interleukin 6 Receptor Alpha |
| IS | Internal standrad |
| JAK | Janus Kinase |
| LC/MS | Liquid chromatography/mass spectrometry |
| MATE | multidrug and toxin extrusion |
| MRM | multiple reaction mode |
| MTX | methotrexate |
| NP-40 | Nonidet P-40 |
| OA | osteoarthrits |
| OC | Organic cation |
| OCT | Organic cation transporter |
| OCTN | Novel organic cation transporter |
| PBMCs | peripheral blood mononuclear cell |
| pKa | Ionization Constant |
| RA | Rheumatoid arthritis |
| RLS | ringer-like solution |
| RM-ANOVA | Repeated Measures Analysis of variance |
| RNA | Ribonucleic acid |
| RT-PCR | Reverse transcription polymerase chain reaction |
| SF | Synovial fibroblasts |
| STAT | Signal transducer and activator of transcription |
| TKI | Tyrosine kinase inhibitor |
| TNF-alpha | tumour necrosis factor alpha |
| TYK | Tyrosine kinase |
References
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Gay, S.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology 2006, 45, 669–675. [Google Scholar] [CrossRef]
- Ospelt, C. Synovial fibroblasts in 2017. RMD Open 2017, 3, e000471. [Google Scholar] [CrossRef] [PubMed]
- Brentano, F.; Schorr, O.; Gay, R.E.; Gay, S.; Kyburz, D. RNA released from necrotic synovial fluid cells activates rheumatoid arthritis synovial fibroblasts via Toll-like receptor 3. Arthritis Rheum. 2005, 52, 2656–2665. [Google Scholar] [CrossRef] [PubMed]
- Ospelt, C.; Brentano, F.; Rengel, Y.; Stanczyk, J.; Kolling, C.; Tak, P.P.; Gay, R.E.; Gay, S.; Kyburz, D. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: Toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum. 2008, 58, 3684–3692. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, H.G. Histomorphological basis of articular cartilage destruction in rheumatoid arthritis. Coll. Relat. Res. 1983, 3, 141–155. [Google Scholar] [CrossRef]
- Wei, S.T.; Sun, Y.H.; Zong, S.H.; Xiang, Y.B. Serum Levels of IL-6 and TNF-alpha May Correlate with Activity and Severity of Rheumatoid Arthritis. Med. Sci. Monit. 2015, 21, 4030–4038. [Google Scholar] [CrossRef]
- Malemud, C.J. The role of the JAK/STAT signal pathway in rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2018, 10, 117–127. [Google Scholar] [CrossRef]
- Harrison, D.A. The Jak/STAT Pathway. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Smolen, J.S.; Breedveld, F.C.; Burmester, G.R.; Bykerk, V.; Dougados, M.; Emery, P.; Kvien, T.K.; Navarro-Compan, M.V.; Oliver, S.; Schoels, M.; et al. Treating rheumatoid arthritis to target: 2014 update of the recommendations of an international task force. Ann. Rheum. Dis. 2016, 75, 3–15. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewe, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann. Rheum. Dis. 2020, 79, 685–699. [Google Scholar] [CrossRef]
- Al-Salama, Z.T.; Scott, L.J. Baricitinib: A Review in Rheumatoid Arthritis. Drugs 2018, 78, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Tofacitinib: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1987–2001. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Lauber, C.; Harrach, S.; Pap, T.; Fischer, M.; Victor, M.; Heitzmann, M.; Hansen, U.; Fobker, M.; Brand, S.M.; Sindic, A.; et al. Transport mechanisms and their pathology-induced regulation govern tyrosine kinase inhibitor delivery in rheumatoid arthritis. PLoS ONE 2012, 7, e52247. [Google Scholar] [CrossRef] [PubMed]
- Harrach, S.; Barz, V.; Pap, T.; Pavenstadt, H.; Schlatter, E.; Edemir, B.; Distler, J.; Ciarimboli, G.; Bertrand, J. Notch Signaling Activity Determines Uptake and Biological Effect of Imatinib in Systemic Sclerosis Dermal Fibroblasts. J. Investig. Dermatol. 2019, 139, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Harrach, S.; Edemir, B.; Schmidt-Lauber, C.; Pap, T.; Bertrand, J.; Ciarimboli, G. Importance of the novel organic cation transporter 1 for tyrosine kinase inhibition by saracatinib in rheumatoid arthritis synovial fibroblasts. Sci. Rep. 2017, 7, 1258. [Google Scholar] [CrossRef] [PubMed]
- Harrach, S.; Schmidt-Lauber, C.; Pap, T.; Pavenstadt, H.; Schlatter, E.; Schmidt, E.; Berdel, W.E.; Schulze, U.; Edemir, B.; Jeromin, S.; et al. MATE1 regulates cellular uptake and sensitivity to imatinib in CML patients. Blood Cancer J. 2016, 6, e470. [Google Scholar] [CrossRef]
- Hediger, M.A.; Romero, M.F.; Peng, J.B.; Rolfs, A.; Takanaga, H.; Bruford, E.A. The ABCs of solute carriers: Physiological, pathological and therapeutic implications of human membrane transport proteinsIntroduction. Pflügers Arch. 2004, 447, 465–468. [Google Scholar] [CrossRef]
- Koepsell, H.; Endou, H. The SLC22 drug transporter family. Pflugers Arch. 2004, 447, 666–676. [Google Scholar] [CrossRef]
- Takeda, M.; Khamdang, S.; Narikawa, S.; Kimura, H.; Kobayashi, Y.; Yamamoto, T.; Cha, S.H.; Sekine, T.; Endou, H. Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. J. Pharmacol. Exp. Ther. 2002, 300, 918–924. [Google Scholar] [CrossRef]
- Tanihara, Y.; Masuda, S.; Sato, T.; Katsura, T.; Ogawa, O.; Inui, K. Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H(+)-organic cation antiporters. Biochem. Pharmacol. 2007, 74, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Westhovens, R. Clinical efficacy of new JAK inhibitors under development. Just more of the same? Rheumatology 2019, 58, i27–i33. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Kontzias, A.; Yamaoka, K.; Tanaka, Y.; Laurence, A. Janus kinase inhibitors in autoimmune diseases. Ann. Rheum. Dis. 2013, 72, 111–115. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat. Rev. Immunol. 2007, 7, 429–442. [Google Scholar] [CrossRef]
- Farr, M.; Garvey, K.; Bold, A.M.; Kendall, M.J.; Bacon, P.A. Significance of the hydrogen ion concentration in synovial fluid in rheumatoid arthritis. Clin. Exp. Rheumatol. 1985, 3, 99–104. [Google Scholar]
- Dowty, M.E.; Lin, J.; Ryder, T.F.; Wang, W.; Walker, G.S.; Vaz, A.; Chan, G.L.; Krishnaswami, S.; Prakash, C. The pharmacokinetics, metabolism, and clearance mechanisms of tofacitinib, a janus kinase inhibitor, in humans. Drug Metab. Dispos. 2014, 42, 759–773. [Google Scholar] [CrossRef]
- Shi, J.G.; Chen, X.; Lee, F.; Emm, T.; Scherle, P.A.; Lo, Y.; Punwani, N.; Williams, W.V.; Yeleswaram, S. The pharmacokinetics, pharmacodynamics, and safety of baricitinib, an oral JAK 1/2 inhibitor, in healthy volunteers. J. Clin. Pharmacol. 2014, 54, 1354–1361. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Transition [m/z > m/z] | Collision Energy |
|---|---|---|
| Tofacitinib | 313 > 149 | 28 |
| 313 > 98 | 31 | |
| 313 > 173 | 37 | |
| Tofacitinib-13C3 | 316 > 149 | 28 |
| 316 > 98 | 31 | |
| 316 > 173 | 37 | |
| Baricitinib | 372 > 251 | 26 |
| 372 > 186 | 31 | |
| 372 > 159 | 43 | |
| Baricitinib-d5 | 377 > 251 | 26 |
| 377 > 186 | 31 | |
| 377 > 159 | 43 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amrhein, J.; Drynda, S.; Schlatt, L.; Karst, U.; Lohmann, C.H.; Ciarimboli, G.; Bertrand, J. Tofacitinib and Baricitinib Are Taken up by Different Uptake Mechanisms Determining the Efficacy of Both Drugs in RA. Int. J. Mol. Sci. 2020, 21, 6632. https://doi.org/10.3390/ijms21186632
Amrhein J, Drynda S, Schlatt L, Karst U, Lohmann CH, Ciarimboli G, Bertrand J. Tofacitinib and Baricitinib Are Taken up by Different Uptake Mechanisms Determining the Efficacy of Both Drugs in RA. International Journal of Molecular Sciences. 2020; 21(18):6632. https://doi.org/10.3390/ijms21186632
Chicago/Turabian StyleAmrhein, Jan, Susanne Drynda, Lukas Schlatt, Uwe Karst, Christoph H. Lohmann, Giuliano Ciarimboli, and Jessica Bertrand. 2020. "Tofacitinib and Baricitinib Are Taken up by Different Uptake Mechanisms Determining the Efficacy of Both Drugs in RA" International Journal of Molecular Sciences 21, no. 18: 6632. https://doi.org/10.3390/ijms21186632
APA StyleAmrhein, J., Drynda, S., Schlatt, L., Karst, U., Lohmann, C. H., Ciarimboli, G., & Bertrand, J. (2020). Tofacitinib and Baricitinib Are Taken up by Different Uptake Mechanisms Determining the Efficacy of Both Drugs in RA. International Journal of Molecular Sciences, 21(18), 6632. https://doi.org/10.3390/ijms21186632