Relative and Absolute Quantification of Aberrant and Normal Splice Variants in HBBIVSI−110 (G > A) β-Thalassemia

 , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Results
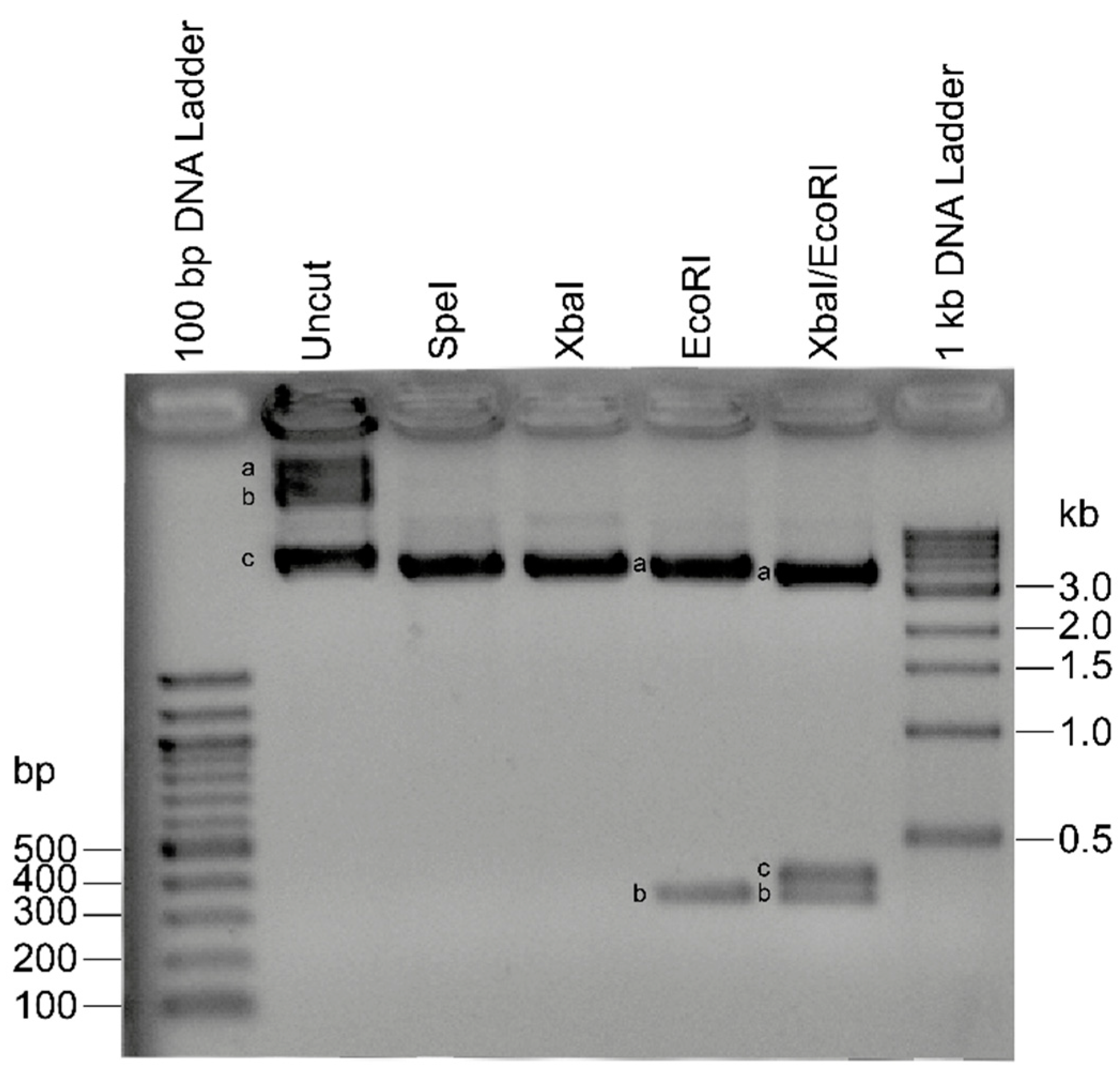
2.1. Recombinant Plasmids for Standard Curve Creation
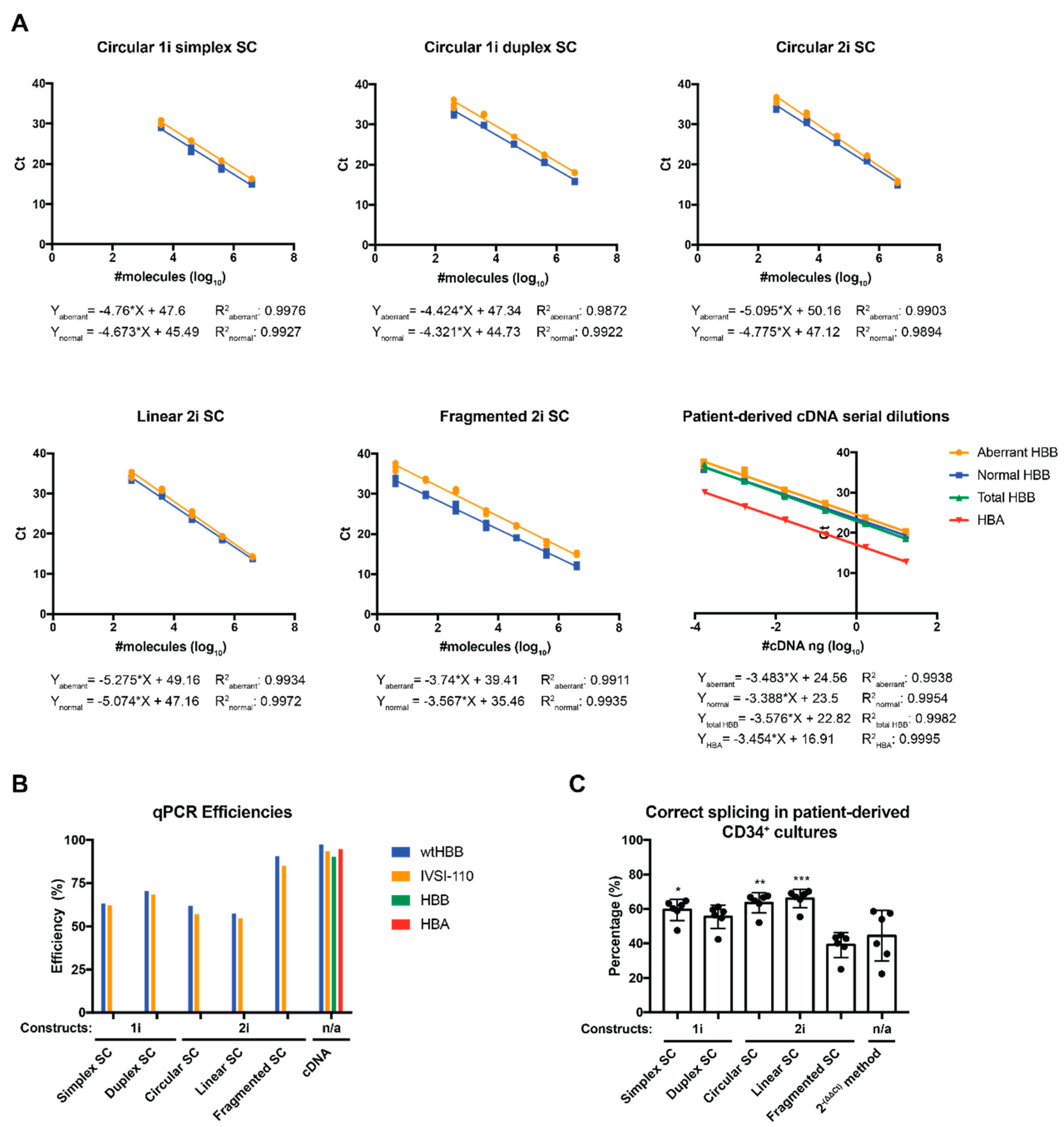
2.2. Evaluation of Alternative Standard Curve Designs
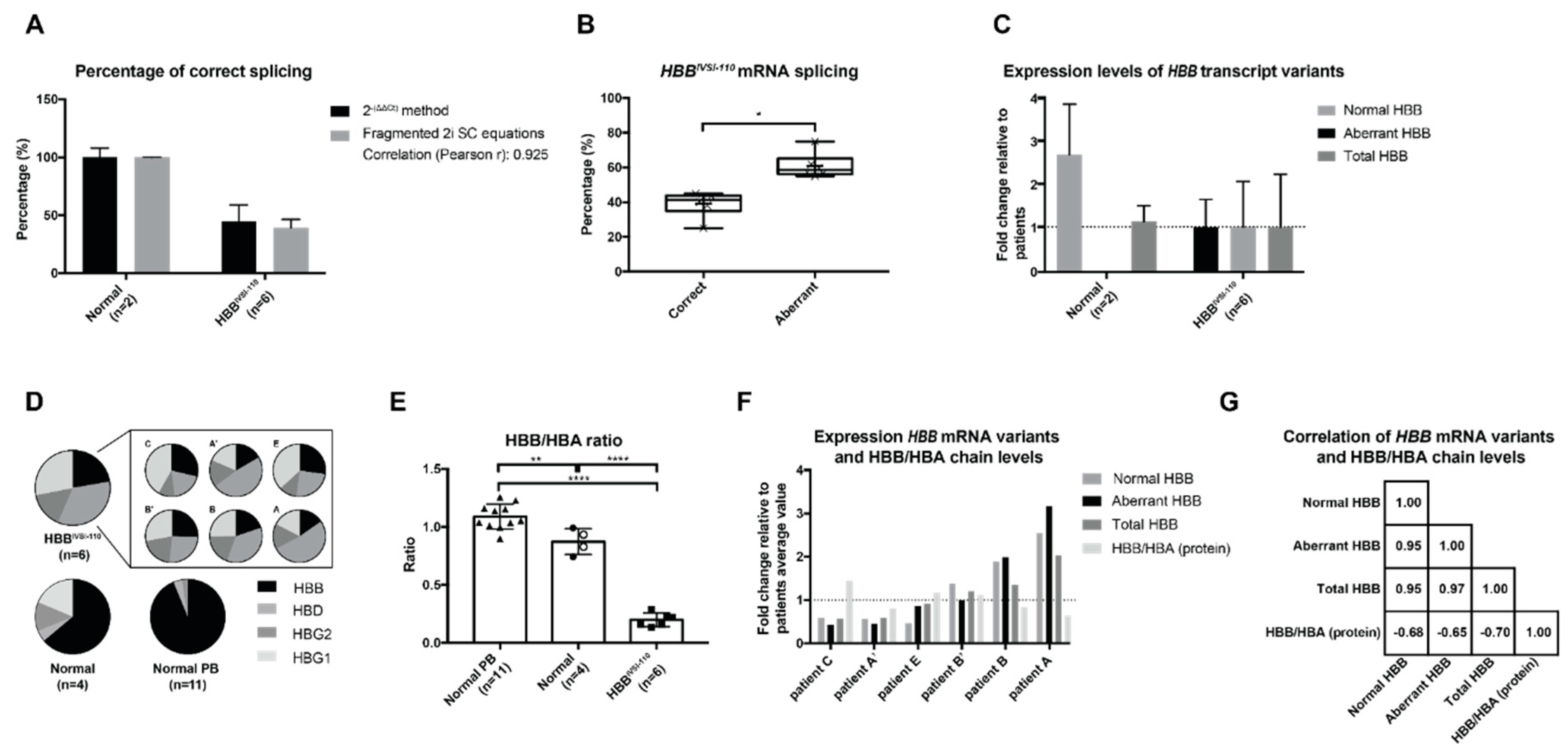
2.3. mRNA Levels in HBBIVSI−110-Homozygous Primary Cells
2.4. Correlation of mRNA and Protein Levels in HBBIVSI−110 Primary Erythroid Cultures
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Oligonucleotides and Probes
4.3. Plasmid Production
4.4. Plasmid-Based Standard Curve
4.5. Cell Culture
4.6. Quantification of mRNA Expression by RT-qPCR
4.6.1. Reverse Transcription PCR (1st Step)
4.6.2. Quantitative PCR (2nd Step A)
Transcript Percentage Contributions
4.6.3. Quantitative PCR (2nd Step B)
Relative Expression of Normal, Aberrant and Total HBB Variants (2 −(ΔΔCt) Method)
Percentage of Correct Splicing (2 −(ΔΔCt) Method)
4.7. Alternative Transcript Quantifications
4.8. High-Performance Liquid Chromatography
4.9. Bioinformatics and Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 1i | 1-insert |
| 2i | 2-insert |
| Ct | threshold cycle |
| FAM | 6-carboxyfluorescein amidite |
| HGVS | Human Genome Variation Society |
| HPLC | high-performance liquid chromatography |
| HSPC | hematopoietic stem/progenitor cell |
| IVS | intervening sequence |
| MGB | minor groove binder |
| RT-qPCR | reverse-transcription quantitative polymerase chain reaction |
| SC | standard curve |
| VIC | 2′-chloro-7′phenyl-1,4-dichloro-6-carboxy-fluorescein |
| ZNA | zip nucleic acid |
References
- Kountouris, P.; Lederer, C.W.; Fanis, P.; Feleki, X.; Old, J.; Kleanthous, M. IthaGenes: An interactive database for haemoglobin variations and epidemiology. PLoS ONE 2014, 9, e103020. [Google Scholar] [CrossRef] [PubMed]
- Peixeiro, I.; Silva, A.L.; Romao, L.; Romão, L. Control of human beta-globin mRNA stability and its impact on beta-thalassemia phenotype. Haematologica 2011, 96, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Danckwardt, S.; Neu-Yilik, G.; Thermann, R.; Frede, U.; Hentze, M.W.; Kulozik, A.E. Abnormally spliced β-globin mRNAs: A single point mutation generates transcripts sensitive and insensitive to nonsense-mediated mRNA decay. Blood 2002, 99, 1811–1816. [Google Scholar] [CrossRef] [PubMed]
- Vadolas, J.; Nefedov, M.; Wardan, H.; Mansooriderakshan, S.; Voullaire, L.; Jamsai, D.; Williamson, R.; Ioannou, P.A. Humanized β-thalassemia mouse model containing the common IVSI-110 splicing mutation. J. Biol. Chem. 2006, 281, 7399–7405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Beshlawy, A.; Mostafa, A.; Youssry, I.; Gabr, H.; Mansour, I.M.; El-Tablawy, M.; Aziz, M.; Hussein, I.R. Correction of Aberrant Pre-mRNA Splicing by Antisense Oligonucleotides in b-Thalassemia Egyptian Patients. J. Pediatr. Hematol. Oncol. 2008, 30, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Breda, L.; Casu, C.; Casula, L.; Kleinert, D.A.; Bianchi, N.; Prus, E.; Cartegni, L.; Fibach, E.; Gardner, L.B.; Giardina, P.J.; et al. Following Beta-Globin Gene Transfer, the Production of Hemoglobin Depends Upon the Beta-Thalassemia Genotype. Blood 2009, 114, 978. [Google Scholar] [CrossRef]
- Breda, L.; Casu, C.; Gardenghi, S.; Bianchi, N.; Cartegni, L.; Narla, M.; Yazdanbakhsh, K.; Musso, M.; Manwani, D.; Little, J.; et al. Therapeutic hemoglobin levels after gene transfer in β-thalassemia mice and in hematopoietic cells of β-thalassemia and sickle cells disease patients. PLoS ONE 2012, 7, e32345. [Google Scholar] [CrossRef] [Green Version]
- Breda, L.; Casu, C.; Kleinert, D.A.; Cartegni, L.; Bianchi, N.; Prus, E.; Cartegni, L.; Fibach, E.; Gardner, L.B.; Giardina, P.J.; et al. Following β-globin Gene Transfer, the Production of Hemoglobin Depends upon the β-Thalassemia Genotype: The “Traffic Jam Hypothesis”. In Proceedings of the 12th Annual Meeting of the American Society of Gene Therapy, San Diego, CA, USA, 27–30 May 2009. [Google Scholar]
- Busslinger, M.; Moschonas, N.; Flavell, R.A. Beta + thalassemia: Aberrant splicing results from a single point mutation in an intron. Cell 1981, 27, 289–298. [Google Scholar] [CrossRef]
- Fukumaki, Y.; Ghosh, P.K.; Benz, E.J.; Reddy, V.B.; Lebowitz, P.; Forget, B.G.; Weissman, S.M. Abnormally spliced messenger RNA in erythroid cells from patients with β+ thalassemia and monkey cells expressing a cloned β+-thalassemic gene. Cell 1982, 28, 585–593. [Google Scholar] [CrossRef]
- Gringras, P.; Wonke, B.; Old, J.; Fitches, A.; Valler, D.; Kuan, A.M.; Hoffbrand, V.; Old, J. Effect of alpha thalassaemia trait and enhanced gamma chain production on disease severity in beta thalassaemia major and intermedia. Arch. Dis. Child. 1994, 70, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Hall, G.W.; Luo, L.Y.; Weatherall, D.J.; Thein, S.L. Beta-thalassaemia intermedia: Is it possible consistently to predict phenotype from genotype? Br. J. Haematol. 1998, 100, 70–78. [Google Scholar]
- Patsali, P.; Papasavva, P.; Stephanou, C.; Christou, S.; Sitarou, M.; Antoniou, M.N.; Lederer, C.W.; Kleanthous, M. Short-hairpin RNA against aberrant HBBIVSI-110(G>A) mRNA restores β-globin levels in a novel cell model and acts as mono- and combination therapy for β-thalassemia in primary hematopoietic stem cells. Haematologica 2018, 103, e419–e423. [Google Scholar] [CrossRef] [PubMed]
- Patsali, P.; Turchiano, G.; Papasavva, P.; Romito, M.; Loucari, C.C.; Stephanou, C.; Christou, S.; Sitarou, M.; Mussolino, C.; Cornu, T.I.; et al. Correction of IVS I-110(G>A) β-thalassemia by CRISPR/Cas-and TALEN-mediated disruption of aberrant regulatory elements in human hematopoietic stem and progenitor cells. Haematologica 2019, 104, e497–e501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsali, P.; Mussolino, C.; Ladas, P.; Floga, A.; Kolnagou, A.; Christou, S.; Sitarou, M.; Antoniou, M.N.; Cathomen, T.; Lederer, C.W.; et al. The Scope for Thalassemia Gene Therapy by Disruption of Aberrant Regulatory Elements. J. Clin. Med. 2019, 8, 1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raso, A.; Mascelli, S.; Nozza, P.; Ugolotti, E.; Vanni, I.; Capra, V.; Biassoni, R. Troubleshooting fine-tuning procedures for qPCR system design. J. Clin. Lab. Anal. 2011, 25, 389–394. [Google Scholar] [CrossRef]
- Hou, Y.; Zhang, H.; Miranda, L.; Lin, S. Serious Overestimation in Quantitative PCR by Circular (Supercoiled) Plasmid Standard: Microalgal pcna as the Model Gene. PLoS ONE 2010, 5, e9545. [Google Scholar] [CrossRef] [Green Version]
- Stephanou, C.; Papasavva, P.; Zachariou, M.; Patsali, P.; Epitropou, M.; Ladas, P.; Al-Abdulla, R.; Christou, S.; Antoniou, M.N.; Lederer, C.W.; et al. Suitability of small diagnostic peripheral-blood samples for cell-therapy studies. Cytotherapy 2017, 19, 311–326. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Kousholt, A.N.; Harmsen, T.; Leemans, C.; Chen, T.; Jonkers, J.; van Steensel, B. Easy quantification of template-directed CRISPR/Cas9 editing. Nucleic Acids Res. 2018, 46, e58. [Google Scholar] [CrossRef]
- Hsiau, T.; Conant, D.; Rossi, N.; Maures, T.; Waite, K.; Yang, J.; Joshi, S.; Kelso, R.; Holden, K.; Enzmann, B.; et al. Inference of CRISPR Edits from Sanger Trace Data. bioRxiv 2018, 251082. [Google Scholar]
- Chen, J.; Kadlubar, F.F.; Chen, J.Z. DNA supercoiling suppresses real-time PCR: A new approach to the quantification of mitochondrial DNA damage and repair. Nucleic Acids Res. 2007, 35, 1377–1388. [Google Scholar] [CrossRef]
- Lin, C.-H.; Chen, Y.-C.; Pan, T.-M. Quantification Bias Caused by Plasmid DNA Conformation in Quantitative Real-Time PCR Assay. PLoS ONE 2011, 6, e29101. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.L.; Medrano, J.F. Real-time PCR for mRNA quantitation. Biotechniques 2005, 39, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Kalle, E.; Kubista, M.; Rensing, C. Multi-template polymerase chain reaction. Biomol. Detect. Quantif. 2014, 2, 11–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.; Wang, J.; Komiyama, M.; Liang, X. Effects of secondary structures of DNA templates on the quantification of qPCR. J. Biomol. Struct. Dyn. 2019, 37, 2867–2874. [Google Scholar] [CrossRef] [PubMed]
- Kühne, B.S. Quantitative real-time RT-PCR using imported plasmid standard curves. Biochemica 2003, 3, 9–11. [Google Scholar]
- Xu, P.; Tong, Y.; Liu, X.; Wang, T.; Cheng, L.; Wang, B.; Lv, X.; Huang, Y.; Liu, D. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2-654 (C > T) mutation in beta-thalassemia-derived iPSCs. Sci. Rep. 2015, 5, 12065. [Google Scholar] [CrossRef] [Green Version]
- Broseus, L.; Ritchie, W. Challenges in detecting and quantifying intron retention from next generation sequencing data. Comput. Struct. Biotechnol. J. 2020, 18, 501–508. [Google Scholar] [CrossRef]
- Marco-puche, G.; Lois, S.; Benítez, J.; Trivino, J.C. RNA-Seq Perspectives to Improve Clinical Diagnosis. Front. Genet. Genet. 2019, 10, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 2013, 3, 71–85. [Google Scholar] [PubMed]
- Breda, L.; Motta, I.; Lourenco, S.; Gemmo, C.; Deng, W.; Rupon, J.W.; Abdulmalik, O.Y.; Manwani, D.; Blobel, G.A.; Rivella, S. Forced chromatin looping raises fetal hemoglobin in adult sickle cells to higher levels than pharmacologic inducers. Blood 2016, 128, 1139–1143. [Google Scholar] [CrossRef] [Green Version]
- Lederer, C.W.; Pavlou, E.; Tanteles, G.A.; Evangelidou, P.; Sismani, C.; Kolnagou, A.; Sitarou, M.; Christou, S.; Hadjigavriel, M.; Kleanthous, M. Hb A 2 Episkopi—A novel δ-globin chain variant [HBD:c.428C>T] in a family of mixed Cypriot–Lebanese descent. Hematology 2016, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Borg, J.; Papadopoulos, P.; Georgitsi, M.; Gutiérrez, L.; Grech, G.; Fanis, P.; Phylactides, M.; Verkerk, A.J.M.H.; van der Spek, P.J.; Scerri, C.A.; et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat. Genet. 2010, 42, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ThermoFisher Scientific qPCR Efficiency Calculator|Thermo Fisher Scientific—CY. Available online: https://www.thermofisher.com/cy/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/thermo-scientific-web-tools/qpcr-efficiency-calculator.html (accessed on 11 June 2020).
- Kirkwood, T.B.L. Geometric Means and Measures of Dispersion. Biometrics 1979, 35, 908–909. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Standard Curve (SC) Composition | SC Equation | R2 | Efficiency |
|---|---|---|---|
| pCR2.1_HBB_N (Circular 1i simplex SC) | Y = −4.673 × X + 45.49 | 0.9927 | 63.28 |
| pCR2.1_HBB_A (Circular 1i simplex SC) | Y = −4.76 × X + 47.61 | 0.9976 | 62.21 |
| pCR2.1_HBB_N (Circular 1i duplex SC) | Y = −4.321 × X + 44.73 | 0.9922 | 70.38 |
| pCR2.1_HBB_A (Circular 1i duplex SC) | Y = −4.424 × X + 47.34 | 0.9872 | 68.28 |
| pCR2.1_HBB_N (Circular2i SC) | Y = −4.775 × X + 47.12 | 0.9894 | 61.97 |
| pCR2.1_HBB_A (Circular 2i SC) | Y = −5.095 × X + 50.16 | 0.9903 | 57.13 |
| pCR2.1_HBB_N (Linear 2i SC) | Y = −5.074 × X + 47.16 | 0.9972 | 57.43 |
| pCR2.1_HBB_A (Linear 2i SC) | Y = −5.275 × X + 49.16 | 0.9934 | 54.73 |
| pCR2.1_HBB_N (Fragmented 2i SC) | Y = −3.567 × X + 35.46 | 0.9935 | 90.70 |
| pCR2.1_HBB_A (Fragmented 2i SC) | Y = −3.74 × X + 39.41 | 0.9911 | 85.09 |
| Normal HBB cDNA sample dilution | Y = −3.388 × X + 23.5 | 0.9954 | 97.31 |
| Abnormal HBB cDNA sample dilution | Y = −3.483 × X + 24.56 | 0.9938 | 93.69 |
| Total HBB cDNA sample dilution | Y = −3.576 × X + 22.82 | 0.9982 | 90.39 |
| HBA cDNA sample dilution | Y = −3.454 × X + 16.91 | 0.9995 | 94.77 |
| Primer | Sequence (5′–3′) |
|---|---|
| IVSI-110_FW | TTCACTAGCAACCTCAAACAGACACC |
| IVSI-110_RV | CACAGTGCAGCTCACTCAG |
| HBB_Ex1_FW | GGGCAAGGTGAACGTG |
| HBB_Ex2_RV | GGACAGATCCCCAAAGGAC |
| A_MGB_VIC | VIC-TAAGGGTGGGAAAATAGA-MGB |
| N_ZNA_FAM | 6-FAM-TGG G(PDC)A GG(PDC) TG(PDC) TG-ZNA-3-BHQ-1 |
| HBA FW | GGTCAACTTCAAGCTCCTAAGC [35] |
| HBA RV | GCTCACAGAAGCCAGGAACTTG [35] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patsali, P.; Papasavva, P.; Christou, S.; Sitarou, M.; Antoniou, M.N.; Lederer, C.W.; Kleanthous, M. Relative and Absolute Quantification of Aberrant and Normal Splice Variants in HBBIVSI−110 (G > A) β-Thalassemia. Int. J. Mol. Sci. 2020, 21, 6671. https://doi.org/10.3390/ijms21186671
Patsali P, Papasavva P, Christou S, Sitarou M, Antoniou MN, Lederer CW, Kleanthous M. Relative and Absolute Quantification of Aberrant and Normal Splice Variants in HBBIVSI−110 (G > A) β-Thalassemia. International Journal of Molecular Sciences. 2020; 21(18):6671. https://doi.org/10.3390/ijms21186671
Chicago/Turabian StylePatsali, Petros, Panayiota Papasavva, Soteroulla Christou, Maria Sitarou, Michael N. Antoniou, Carsten W. Lederer, and Marina Kleanthous. 2020. "Relative and Absolute Quantification of Aberrant and Normal Splice Variants in HBBIVSI−110 (G > A) β-Thalassemia" International Journal of Molecular Sciences 21, no. 18: 6671. https://doi.org/10.3390/ijms21186671
APA StylePatsali, P., Papasavva, P., Christou, S., Sitarou, M., Antoniou, M. N., Lederer, C. W., & Kleanthous, M. (2020). Relative and Absolute Quantification of Aberrant and Normal Splice Variants in HBBIVSI−110 (G > A) β-Thalassemia. International Journal of Molecular Sciences, 21(18), 6671. https://doi.org/10.3390/ijms21186671