Psoriasis and Antimicrobial Peptides


Abstract
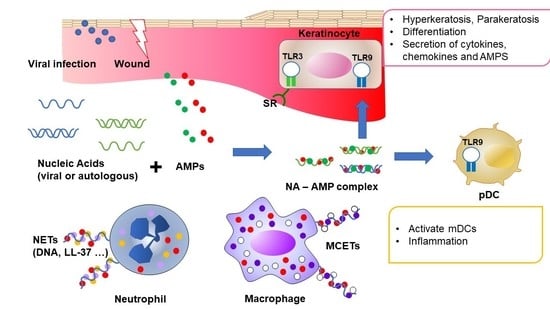
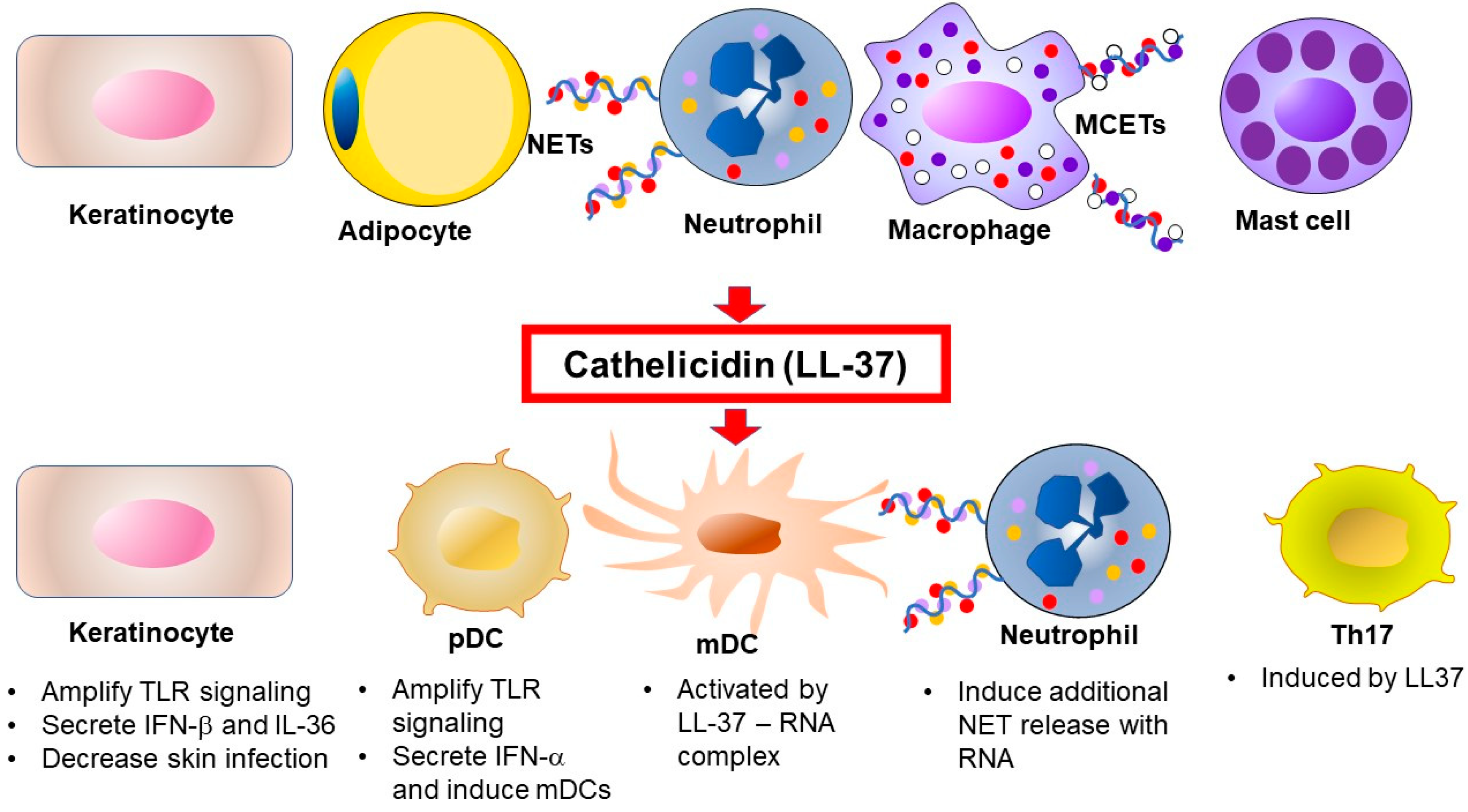
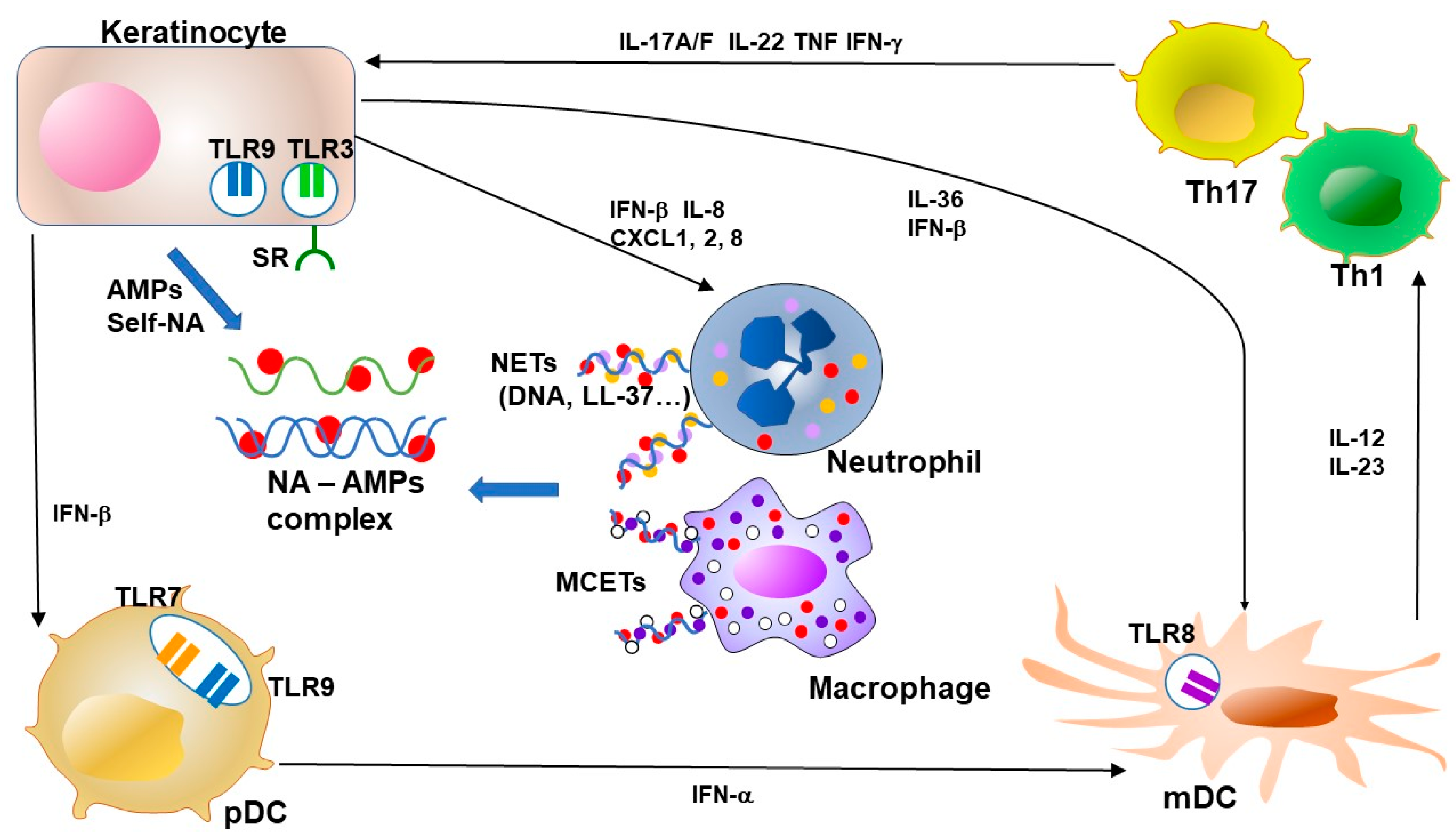
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. AMPs Expressed in Skin and Dermatoses
2.1. Cathelicidin Antimicrobial Peptides (CAMPs)
2.2. Defensins
2.3. S100 Proteins
2.4. Other AMPs
2.5. AMPs from Skin-Commensal Staphylococci Serve as a Skin Barrier to Control Microbiota
3. Cell-Specific Regulation of AMPs in Psoriasis
3.1. Keratinocytes and AMPs
3.2. Neutrophils, Neutrophil Extracellular Traps (NETs) and AMPs
3.3. Dendritic Cells and AMPs
3.4. T Cells and AMPs
4. Cytokine and Intracellular Signaling Regulation by AMPs in Psoriasis
4.1. Interferon and AMPs
4.2. TLRs and AMPs
4.3. Scavenger Receptors and AMPs
5. Involvement of AMPs in Clinical Aspects of Psoriasis
5.1. Psoriasis Phenotypes and AMPs
5.2. Psoriasis Treatments and AMPs
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPs | antimicrobial peptides and proteins |
| CLA | cutaneous lymphocyte antigen |
| DAMPs | damage-associated molecular patterns |
| hBD | human β-defensin |
| HLA | human leukocyte antigen |
| HNP | human neutrophil peptide |
| IFN | interferon |
| IL | interleukin |
| KLKs | kallikreins |
| LPS | lipopolysaccharide |
| MCETs | mast cell extracellular traps |
| mDC | myeloid / conventional DC |
| NETs | neutrophil extracellular traps |
| pDC | plasmacytoid dendritic cells |
| PRRs | pattern recognition receptors |
| PsA | psoriatic arthritis |
| PSM | phenol-soluble modulin |
| SF | synovial fluid |
| TCR | T cell receptor |
| Th | T-helper cells |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TRAF | TNF receptor-associated factor |
| VDRE | vitamin D response element |
References
- Griffiths, C.E.; Barker, J.N. Pathogenesis and clinical features of psoriasis. Lancet 2007, 370, 263–271. [Google Scholar]
- Yin, X.; Low, H.Q.; Wang, L.; Li, Y.; Ellinghaus, E.; Han, J.; Estivill, X.; Sun, L.; Zuo, X.; Shen, C.; et al. Genome-wide meta-analysis identifies multiple novel associations and ethnic heterogeneity of psoriasis susceptibility. Nat. Commun. 2015, 6, 6916. [Google Scholar]
- Krueger, G.G.; Duvic, M. Epidemiology of psoriasis: Clinical issues. J. Investig. Dermatol. 1994, 102. [Google Scholar] [CrossRef]
- Balak, D.M.; Hajdarbegovic, E. Drug-induced psoriasis: Clinical perspectives. Psoriasis 2017, 7, 87–94. [Google Scholar]
- Ritchlin, C.T.; Colbert, R.A.; Gladman, D.D. Psoriatic Arthritis. N. Engl. J. Med. 2017, 376, 957–970. [Google Scholar] [CrossRef] [Green Version]
- Conrad, C.; Gilliet, M. Psoriasis: From Pathogenesis to Targeted Therapies. Clin. Rev. Allergy Immunol. 2018, 54, 102–113. [Google Scholar]
- Armstrong, A.W.; Harskamp, C.T.; Armstrong, E.J. Psoriasis and metabolic syndrome: A systematic review and meta-analysis of observational studies. J. Am. Acad. Dermatol. 2013, 68, 654–662. [Google Scholar] [CrossRef]
- Takeshita, J.; Grewal, S.; Langan, S.M.; Mehta, N.N.; Ogdie, A.; Van Voorhees, A.S.; Gelfand, J.M. Psoriasis and comorbid diseases. J. Am. Acad. Dermatol. 2017, 76, 377–390. [Google Scholar]
- Nestle, F.O.; Kaplan, D.H.; Barker, J. Psoriasis. N. Engl. J. Med. 2009, 361, 496–509. [Google Scholar]
- Di Cesare, A.; Di Meglio, P.; Nestle, F.O. The IL-23/Th17 Axis in the Immunopathogenesis of Psoriasis. J. Investig. Dermatol. 2009, 129, 1339–1350. [Google Scholar] [CrossRef] [Green Version]
- Papp, K.A.; Reich, K.; Paul, C.; Blauvelt, A.; Baran, W.; Bolduc, C.; Toth, D.; Langley, R.G.; Cather, J.; Gottlieb, A.B.; et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis. Br. J. Dermatol. 2016, 175, 273–286. [Google Scholar] [CrossRef]
- Chiu, H.-Y.; Hui, R.C.-Y.; Tsai, T.-F.; Chen, Y.-C.; Chang Liao, N.-F.; Chen, P.-H.; Lai, P.-J.; Wang, T.-S.; Huang, Y.-H. Predictors of time to relapse following ustekinumab withdrawal in patients with psoriasis who had responded to therapy: An eight-year multicenter study. J. Am. Acad. Dermatol. 2019, in press. [Google Scholar] [CrossRef]
- Papp, K.; Crowley, J.; Ortonne, J.P.; Leu, J.; Okun, M.; Gupta, S.R.; Gu, Y.; Langley, R.G. Adalimumab for moderate to severe chronic plaque psoriasis: Efficacy and safety of retreatment and disease recurrence following withdrawal from therapy. Br. J. Dermatol. 2011, 164, 434–441. [Google Scholar] [CrossRef]
- Gallo, R.L.; Hooper, L.V. Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol. 2012, 12, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Lai, Y.; Gallo, R.L. AMPed up immunity: How antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009, 30, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Glaser, R.; Harder, J.; Lange, H.; Bartels, J.; Christophers, E.; Schroder, J.M. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat. Immunol. 2005, 6, 57–64. [Google Scholar] [CrossRef]
- Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [Green Version]
- Buchau, A.S.; Gallo, R.L. Innate immunity and antimicrobial defense systems in psoriasis. Clin. Dermatol. 2007, 25, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Harder, J.; Schroder, J.M. Psoriatic scales: A promising source for the isolation of human skin-derived antimicrobial proteins. J. Leukoc. Biol. 2005, 77, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Vaishnava, S.; Hooper, L.V. Multi-layered regulation of intestinal antimicrobial defense. Cell Mol. Life Sci. 2008, 65, 3019–3027. [Google Scholar]
- Yamasaki, K.; Di Nardo, A.; Bardan, A.; Murakami, M.; Ohtake, T.; Coda, A.; Dorschner, R.A.; Bonnart, C.; Descargues, P.; Hovnanian, A.; et al. Increased serine protease activity and cathelicidin promotes skin inflammation in rosacea. Nat. Med. 2007, 13, 975–980. [Google Scholar]
- Yamasaki, K.; Kanada, K.; Macleod, D.T.; Borkowski, A.W.; Morizane, S.; Nakatsuji, T.; Cogen, A.L.; Gallo, R.L. TLR2 expression is increased in rosacea and stimulates enhanced serine protease production by keratinocytes. J. Investig. Dermatol. 2011, 131, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, K.; Gallo, R.L. Rosacea as a disease of cathelicidins and skin innate immunity. J. Investig. Dermatol. Symp. Proc. 2011, 15, 12–15. [Google Scholar]
- Ong, P.Y.; Ohtake, T.; Brandt, C.; Strickland, I.; Boguniewicz, M.; Ganz, T.; Gallo, R.L.; Leung, D.Y. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med. 2002, 347, 1151–1160. [Google Scholar]
- Morizane, S.; Yamasaki, K.; Kabigting, F.D.; Gallo, R.L. Kallikrein expression and cathelicidin processing are independently controlled in keratinocytes by calcium, vitamin D(3), and retinoic acid. J. Investig. Dermatol. 2010, 130, 1297–1306. [Google Scholar] [CrossRef] [Green Version]
- Gallo, R.L.; Ono, M.; Povsic, T.; Page, C.; Eriksson, E.; Klagsbrun, M.; Bernfield, M. Syndecans, cell surface heparan sulfate proteoglycans, are induced by a proline-rich antimicrobial peptide from wounds. Proc. Natl. Acad. Sci. USA 1994, 91, 11035–11039. [Google Scholar]
- Larrick, J.W.; Lee, J.; Ma, S.; Li, X.; Francke, U.; Wright, S.C.; Balint, R.F. Structural, functional analysis and localization of the human CAP18 gene. FEBS Lett. 1996, 398, 74–80. [Google Scholar]
- Gudmundsson, G.H.; Agerberth, B.; Odeberg, J.; Bergman, T.; Olsson, B.; Salcedo, R. The human gene FALL39 and processing of the cathelin precursor to the antibacterial peptide LL-37 in granulocytes. FEBS J. 1996, 238, 325–332. [Google Scholar]
- Méndez-Samperio, P. The human cathelicidin hCAP18/LL-37: A multifunctional peptide involved in mycobacterial infections. Peptides 2010, 31, 1791–1798. [Google Scholar] [PubMed]
- Yamasaki, K.; Gallo, R.L. Antimicrobial peptides in human skin disease. Eur. J. Dermatol. 2008, 18, 11–21. [Google Scholar]
- Morizane, S.; Gallo, R.L. Antimicrobial peptides in the pathogenesis of psoriasis. J. Dermatol. 2012, 39, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Ohtake, T.; Dorschner, R.A.; Schittek, B.; Garbe, C.; Gallo, R.L. Cathelicidin anti-microbial peptide expression in sweat, an innate defense system for the skin. J. Investig. Dermatol. 2002, 119, 1090–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Nardo, A.; Vitiello, A.; Gallo, R.L. Cutting edge: Mast cell antimicrobial activity is mediated by expression of cathelicidin antimicrobial peptide. J. Immunol. 2003, 170, 2274–2278. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lai, Y.; Bernard, J.J.; Macleod, D.T.; Cogen, A.L.; Moss, B.; Di Nardo, A. Skin mast cells protect mice against vaccinia virus by triggering mast cell receptor S1PR2 and releasing antimicrobial peptides. J. Immunol. 2012, 188, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, N.N.; Takahashi, T.; Sanford, J.A.; Tong, Y.; Gombart, A.F.; Hinds, B.; Cheng, J.Y.; Gallo, R.L. Innate Immune Dysfunction in Rosacea Promotes Photosensitivity and Vascular Adhesion Molecule Expression. J. Investig. Dermatol. 2020, 140, 645.e6–655.e6. [Google Scholar] [CrossRef]
- Zhang, L.-j.; Guerrero-Juarez, C.F.; Hata, T.; Bapat, S.P.; Ramos, R.; Plikus, M.V.; Gallo, R.L. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 2015, 347, 67–71. [Google Scholar] [CrossRef] [Green Version]
- Frohm, M.; Agerberth, B.; Ahangari, G.; Stahle-Backdahl, M.; Liden, S.; Wigzell, H.; Gudmundsson, G.H. The expression of the gene coding for the antibacterial peptide LL-37 is induced in human keratinocytes during inflammatory disorders. J. Biol. Chem. 1997, 272, 15258–15263. [Google Scholar] [CrossRef] [Green Version]
- Dorschner, R.A.; Pestonjamasp, V.K.; Tamakuwala, S.; Ohtake, T.; Rudisill, J.; Nizet, V.; Agerberth, B.; Gudmundsson, G.H.; Gallo, R.L. Cutaneous injury induces the release of cathelicidin anti-microbial peptides active against group A Streptococcus. J. Investig. Dermatol. 2001, 117, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Nizet, V.; Ohtake, T.; Lauth, X.; Trowbridge, J.; Rudisill, J.; Dorschner, R.A.; Pestonjamasp, V.; Piraino, J.; Huttner, K.; Gallo, R.L. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 2001, 414, 454–457. [Google Scholar] [CrossRef]
- Grether-Beck, S.; Felsner, I.; Brenden, H.; Kohne, Z.; Majora, M.; Marini, A.; Jaenicke, T.; Rodriguez-Martin, M.; Trullas, C.; Hupe, M. Urea uptake enhances barrier function and antimicrobial defense in humans by regulating epidermal gene expression. J. Investig. Dermatol. 2012, 132, 1561–1572. [Google Scholar] [CrossRef] [Green Version]
- Bernard, J.J.; Cowing-Zitron, C.; Nakatsuji, T.; Muehleisen, B.; Muto, J.; Borkowski, A.W.; Martinez, L.; Greidinger, E.L.; Benjamin, D.Y.; Gallo, R.L. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat. Med. 2012, 18, 1286–1290. [Google Scholar] [CrossRef]
- Yamasaki, K.; Schauber, J.; Coda, A.; Lin, H.; Dorschner, R.A.; Schechter, N.M.; Bonnart, C.; Descargues, P.; Hovnanian, A.; Gallo, R.L. Kallikrein-mediated proteolysis regulates the antimicrobial effects of cathelicidins in skin. FASEB J. 2006, 20, 2068–2080. [Google Scholar] [CrossRef] [Green Version]
- Sorensen, O.; Arnljots, K.; Cowland, J.B.; Bainton, D.F.; Borregaard, N. The human antibacterial cathelicidin, hCAP-18, is synthesized in myelocytes and metamyelocytes and localized to specific granules in neutrophils. Blood 1997, 90, 2796–2803. [Google Scholar] [CrossRef]
- Braff, M.H.; Hawkins, M.A.; Di Nardo, A.; Lopez-Garcia, B.; Howell, M.D.; Wong, C.; Lin, K.; Streib, J.E.; Dorschner, R.; Leung, D.Y.; et al. Structure-function relationships among human cathelicidin peptides: Dissociation of antimicrobial properties from host immunostimulatory activities. J. Immunol. 2005, 174, 4271–4278. [Google Scholar] [CrossRef] [Green Version]
- Lichtenstein, A.; Ganz, T.; Selsted, M.E.; Lehrer, R.I. In vitro tumor cell cytolysis mediated by peptide defensins of human and rabbit granulocytes. Blood 1986, 68, 1407–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasca, L.; Palazzo, R.; Chimenti, M.S.; Alivernini, S.; Tolusso, B.; Bui, L.; Botti, E.; Giunta, A.; Bianchi, L.; Petricca, L.; et al. Anti-LL37 Antibodies Are Present in Psoriatic Arthritis (PsA) Patients: New Biomarkers in PsA. Front. Immunol. 2018, 9, 1936. [Google Scholar] [CrossRef] [Green Version]
- Schutte, B.C.; McCray, P.B., Jr. [beta]-defensins in lung host defense. Annu. Rev. Physiol. 2002, 64, 709–748. [Google Scholar] [CrossRef]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.M. A peptide antibiotic from human skin. Nature 1997, 387, 861. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Guttman-Yassky, E.; Suárez-Fariñas, M.; Nograles, K.E.; Tian, S.; Cardinale, I.; Chimenti, S.; Krueger, J.G. Integrative Responses to IL-17 and TNF-α in Human Keratinocytes Account for Key Inflammatory Pathogenic Circuits in Psoriasis. J. Investig. Dermatol. 2011, 131, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Johansen, C.; Bertelsen, T.; Ljungberg, C.; Mose, M.; Iversen, L. Characterization of TNF-α– and IL-17A–Mediated Synergistic Induction of DEFB4 Gene Expression in Human Keratinocytes through IκBζ. J. Investig. Dermatol. 2016, 136, 1608–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohrl, J.; Yang, D.; Oppenheim, J.J.; Hehlgans, T. Specific Binding and Chemotactic Activity of mBD4 and Its Functional Orthologue hBD2 to CCR6-expressing Cells. J. Biol. Chem. 2010, 285, 7028–7034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabuchi, T.; Singh, T.P.; Takekoshi, T.; Jia, G.-F.; Wu, X.; Kao, M.C.; Weiss, I.; Farber, J.M.; Hwang, S.T. CCR6 Is Required for Epidermal Trafficking of γδ-T Cells in an IL-23-Induced Model of Psoriasiform Dermatitis. J. Investig. Deramtol. 2013, 133, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Kolbinger, F.; Loesche, C.; Valentin, M.-A.; Jiang, X.; Cheng, Y.; Jarvis, P.; Peters, T.; Calonder, C.; Bruin, G.; Polus, F.; et al. β-Defensin 2 is a responsive biomarker of IL-17A–driven skin pathology in patients with psoriasis. J. Allergy Clin. Immunol. 2017, 139, 923.e8–932.e8. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, C.M.; Shane, R.; Malara, A.; Kelly, G.; Hughes, R.; Tobin, A.-M.; Adamzik, K.; Walsh, P.T.; Kirby, B. Human β-Defensin 3 and Its Mouse Ortholog Murine β-Defensin 14 Activate Langerhans Cells and Exacerbate Psoriasis-Like Skin Inflammation in Mice. J. Investig. Dermatol. 2016, 136, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Eckert, R.L.; Broome, A.M.; Ruse, M.; Robinson, N.; Ryan, D.; Lee, K. S100 proteins in the epidermis. J. Investig. Dermatol. 2004, 123, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Madsen, P.; Rasmussen, H.H.; Leffers, H.; Honore, B.; Dejgaard, K.; Olsen, E.; Kiil, J.; Walbum, E.; Andersen, A.H.; Basse, B.; et al. Molecular cloning, occurrence, and expression of a novel partially secreted protein “psoriasin” that is highly up-regulated in psoriatic skin. J. Investig. Dermatol. 1991, 97, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Jinquan, T.; Vorum, H.; Larsen, C.G.; Madsen, P.; Rasmussen, H.H.; Gesser, B.; Etzerodt, M.; Honore, B.; Celis, J.E.; Thestrup-Pedersen, K. Psoriasin: A novel chemotactic protein. J. Investig. Dermatol. 1996, 107, 5–10. [Google Scholar] [CrossRef] [Green Version]
- Hegyi, Z.; Zwicker, S.; Bureik, D.; Peric, M.; Koglin, S.; Batycka-Baran, A.; Prinz, J.C.; Ruzicka, T.; Schauber, J.; Wolf, R. Vitamin D analog calcipotriol suppresses the Th17 cytokine–induced proinflammatory S100 “alarmins” psoriasin (S100A7) and koebnerisin (S100A15) in psoriasis. J. Investig. Dermatol. 2012, 132, 1416–1424. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Cao, T.; Jin, L.; Li, B.; Fang, H.; Zhang, J.; Zhang, Y.; Hu, J.; Wang, G. Increased Lipocalin-2 Contributes to the Pathogenesis of Psoriasis by Modulating Neutrophil Chemotaxis and Cytokine Secretion. J. Investig. Dermatol. 2016, 136, 1418–1428. [Google Scholar] [CrossRef] [Green Version]
- Elgharib, I.; Khashaba, S.A.; Elsaid, H.H.; Sharaf, M.M. Serum elafin as a potential inflammatory marker in psoriasis. Int. J. Dermatol. 2018, 58, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Cogen, A.L.; Yamasaki, K.; Sanchez, K.M.; Dorschner, R.A.; Lai, Y.; MacLeod, D.T.; Torpey, J.W.; Otto, M.; Nizet, V.; Kim, J.E.; et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J. Investig. Dermatol. 2010, 130, 192–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwase, T.; Uehara, Y.; Shinji, H.; Tajima, A.; Seo, H.; Takada, K.; Agata, T.; Mizunoe, Y. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature 2010, 465, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Otto, M.; Sussmuth, R.; Vuong, C.; Jung, G.; Gotz, F. Inhibition of virulence factor expression in Staphylococcus aureus by the Staphylococcus epidermidis agr pheromone and derivatives. FEBS Lett. 1999, 450, 257–262. [Google Scholar] [CrossRef] [Green Version]
- Albanesi, C.; Madonna, S.; Gisondi, P.; Girolomoni, G. The Interplay Between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front. Immunol. 2018, 9, 1549. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yao, Q.; Mariscal, A.G.; Wu, X.; Hülse, J.; Pedersen, E.; Helin, K.; Waisman, A.; Vinkel, C.; Thomsen, S.F.; et al. Epigenetic control of IL-23 expression in keratinocytes is important for chronic skin inflammation. Nat. Commun. 2018, 9, 1549. [Google Scholar] [CrossRef]
- Xu, M.; Lu, H.; Lee, Y.-H.; Wu, Y.; Liu, K.; Shi, Y.; An, H.; Zhang, J.; Wang, X.; Lai, Y.; et al. An Interleukin-25-Mediated Autoregulatory Circuit in Keratinocytes Plays a Pivotal Role in Psoriatic Skin Inflammation. Immunity 2018, 48, 787.e4–798.e4. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat. Med. 2009, 15, 1377–1382. [Google Scholar] [CrossRef]
- Zhang, L.-j.; Sen, G.L.; Ward, N.L.; Johnston, A.; Chun, K.; Chen, Y.; Adase, C.; Sanford, J.A.; Gao, N.; Chensee, M. Antimicrobial Peptide LL37 and MAVS Signaling Drive Interferon-β Production by Epidermal Keratinocytes during Skin Injury. Immunity 2016, 45, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, R.; Dainichi, T.; Tsuchiya, S.; Nomura, T.; Kitoh, A.; Hayden, M.S.; Ishii, K.J.; Tanaka, M.; Honda, T.; Egawa, G.; et al. Epithelial TRAF6 drives IL-17–mediated psoriatic inflammation. JCI Insight 2018, 3, e121175. [Google Scholar] [CrossRef]
- Li, N.; Yamasaki, K.; Saito, R.; Fukushi-Takahashi, S.; Shimada-Omori, R.; Asano, M.; Aiba, S. Alarmin function of cathelicidin antimicrobial peptide LL37 through IL-36gamma induction in human epidermal keratinocytes. J. Immunol. 2014, 193, 5140–5148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.M. Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic. J. Biol. Chem. 2001, 276, 5707–5713. [Google Scholar] [PubMed] [Green Version]
- Hollox, E.J.; Huffmeier, U.; Zeeuwen, P.L.; Palla, R.; Lascorz, J.; Rodijk-Olthuis, D.; van de Kerkhof, P.C.; Traupe, H.; de Jongh, G.; den Heijer, M.; et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat. Genet. 2008, 40, 23–25. [Google Scholar] [CrossRef] [Green Version]
- Qiao, P.; Guo, W.; Ke, Y.; Fang, H.; Zhuang, Y.; Jiang, M.; Zhang, J.; Shen, S.; Qiao, H.; Dang, E.; et al. Mechanical Stretch Exacerbates Psoriasis by Stimulating Keratinocyte Proliferation and Cytokine Production. J. Dermatol. 2019, 139, 1470–1479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, T.; Bhosale, A.; Bajantri, B.; Mathapathi, M.S.; Rizvi, A.; Scita, G.; Majumdar, A.; Jamora, C. Sustained Secretion of the Antimicrobial Peptide S100A7 Is Dependent on the Downregulation of Caspase-8. Cell Rep. 2019, 29, 2546.e4–2555.e4. [Google Scholar] [CrossRef]
- Brinkmann, V. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Martinelli, S.; Urosevic, M.; Daryadel, A.; Oberholzer, P.A.; Baumann, C.; Fey, M.F.; Dummer, R.; Simon, H.-U.; Yousefi, S. Induction of Genes Mediating Interferon-dependent Extracellular Trap Formation during Neutrophil Differentiation. J. Biol. Chem. 2004, 279, 44123–44132. [Google Scholar] [CrossRef] [Green Version]
- Lambert, S.; Hambro, C.A.; Johnston, A.; Stuart, P.E.; Tsoi, L.C.; Nair, R.P.; Elder, J.T. Neutrophil Extracellular Traps Induce Human Th17 Cells: Effect of Psoriasis-Associated TRAF3IP2 Genotype. J. Investig. Dermatol. 2019, 139, 1245–1253. [Google Scholar]
- Herster, F.; Bittner, Z.; Archer, N.K.; Dickhöfer, S.; Eisel, D.; Eigenbrod, T.; Knorpp, T.; Schneiderhan-Marra, N.; Löffler, M.W.; Kalbacher, H.; et al. Neutrophil extracellular trap-associated RNA and LL37 enable self-amplifying inflammation in psoriasis. Nat. Commun. 2020, 11, 105. [Google Scholar]
- von Köckritz-Blickwede, M.; Goldmann, O.; Thulin, P.; Heinemann, K.; Norrby-Teglund, A.; Rohde, M.; Medina, E. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 2008, 111, 3070–3080. [Google Scholar]
- Nestle, F.O.; Conrad, C.; Tun-Kyi, A.; Homey, B.; Gombert, M.; Boyman, O.; Burg, G.; Liu, Y.J.; Gilliet, M. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J. Exp. Med. 2005, 202, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B.; Bunin, A.; Ghosh, H.S.; Lewis, K.L.; Sisirak, V. Plasmacytoid Dendritic Cells: Recent Progress and Open Questions. Annu. Rev Immunol. 2011, 29, 163–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregorio, J.; Meller, S.; Conrad, C.; Di Nardo, A.; Homey, B.; Lauerma, A.; Arai, N.; Gallo, R.L.; DiGiovanni, J.; Gilliet, M. Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J. Exp. Med. 2010, 207, 2921–2930. [Google Scholar] [CrossRef]
- Lande, R.; Gregorio, J.; Facchinetti, V.; Chatterjee, B.; Wang, Y.-H.; Homey, B.; Cao, W.; Wang, Y.-H.; Su, B.; Nestle, F.O.; et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 2007, 449, 564–569. [Google Scholar] [CrossRef]
- Gilliet, M.; Cao, W.; Liu, Y.-J. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 2008, 8, 594–606. [Google Scholar] [CrossRef]
- Lowes, M.A.; Suárez-Fariñas, M.; Krueger, J.G. Immunology of Psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef] [Green Version]
- Chiricozzi, A.; Romanelli, P.; Volpe, E.; Borsellino, G.; Romanelli, M. Scanning the Immunopathogenesis of Psoriasis. Int. J. Mol. Sci. 2018, 19, 179. [Google Scholar] [CrossRef] [Green Version]
- Lowes, M.A.; Chamian, F.; Abello, M.V.; Fuentes-Duculan, J.; Lin, S.-L.; Nussbaum, R.; Novitskaya, I.; Carbonaro, H.; Cardinale, I.; Kikuchi, T. Increase in TNF-α and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc. Natl. Acad. Sci. USA 2005, 102, 19057–19062. [Google Scholar] [CrossRef] [Green Version]
- Fuentes-Duculan, J.; Suárez-Fariñas, M.; Zaba, L.C.; Nograles, K.E.; Pierson, K.C.; Mitsui, H.; Pensabene, C.A.; Kzhyshkowska, J.; Krueger, J.G.; Lowes, M.A. A Subpopulation of CD163-Positive Macrophages Is Classically Activated in Psoriasis. J. Investig. Dermatol. 2010, 130, 2412–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Lande, R.; Botti, E.; Jandus, C.; Dojcinovic, D.; Fanelli, G.; Conrad, C.; Chamilos, G.; Feldmeyer, L.; Marinari, B.; Chon, S.; et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat. Commun. 2014, 5, 5621. [Google Scholar] [PubMed]
- Peric, M.; Koglin, S.; Kim, S.M.; Morizane, S.; Besch, R.; Prinz, J.C.; Ruzicka, T.; Gallo, R.L.; Schauber, J. IL-17A enhances vitamin D3-induced expression of cathelicidin antimicrobial peptide in human keratinocytes. J. Immunol. 2008, 181, 8504–8512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banchereau, J.; Pascual, V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 2006, 25, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.-J. Type1 Interferons Potential Initiating Factors Linking Skin Wounds with Psoriasis Pathogenesis. Front. Immunol. 2019, 10, 1440. [Google Scholar]
- Banchereau, J.; Pascual, V.; Palucka, A.K. Autoimmunity through cytokine-induced dendritic cell activation. Immunity 2004, 20, 539–550. [Google Scholar] [CrossRef] [Green Version]
- Baccala, R.; Hoebe, K.; Kono, D.H.; Beutler, B.; Theofilopoulos, A.N. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat. Med. 2007, 13, 543–551. [Google Scholar]
- Ganguly, D.; Chamilos, G.; Lande, R.; Gregorio, J.; Meller, S.; Facchinetti, V.; Homey, B.; Barrat, F.J.; Zal, T.; Gilliet, M. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J. Exp. Med. 2009, 206, 1983–1994. [Google Scholar]
- Takahashi, T.; Kulkarni, N.N.; Lee, E.Y.; Zhang, L.-j.; Wong, G.C.L.; Gallo, R.L. Cathelicidin promotes inflammation by enabling binding of self-RNA to cell surface scavenger receptors. Sci. Rep. 2018, 8, 4032. [Google Scholar]
- Lande, R.; Chamilos, G.; Ganguly, D.; Demaria, O.; Frasca, L.; Durr, S.; Conrad, C.; Schröder, J.; Gilliet, M. Cationic antimicrobial peptides in psoriatic skin cooperate to break innate tolerance to self-DNA. Eur. J. Immunol. 2015, 45, 203–213. [Google Scholar]
- Morizane, S.; Yamasaki, K.; Muhleisen, B.; Kotol, P.F.; Murakami, M.; Aoyama, Y.; Iwatsuki, K.; Hata, T.; Gallo, R.L. Cathelicidin antimicrobial peptide LL-37 in psoriasis enables keratinocyte reactivity against TLR9 ligands. J. Investig. Dermatol. 2012, 132, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Y.; Takahashi, T.; Curk, T.; Dobnikar, J.; Gallo, R.L.; Wong, G.C.L. Crystallinity of Double-Stranded RNA-Antimicrobial Peptide Complexes Modulates Toll-Like Receptor 3-Mediated Inflammation. ACS Nano 2017, 11, 12145–12155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabuchi, T.; Hirayama, N. Binding Affinity and Interaction of LL-37 with HLA-C*06:02 in Psoriasis. J. Investig. Dermatol. 2016, 136, 1901–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Qiu, J.; Lin, Z.T.; Li, W.; Haley, C.; Mui, U.N.; Ning, J.; Tyring, S.K.; Wu, T. Identification of Novel Autoantibodies Associated With Psoriatic Arthritis. Arthritis Rheumatol. 2019, 71, 941–951. [Google Scholar] [CrossRef]
- Vahavihu, K.; Ala-Houhala, M.; Peric, M.; Karisola, P.; Kautiainen, H.; Hasan, T.; Snellman, E.; Alenius, H.; Schauber, J.; Reunala, T. Narrowband ultraviolet B treatment improves vitamin D balance and alters antimicrobial peptide expression in skin lesions of psoriasis and atopic dermatitis. Br. J. Dermatol. 2010, 163, 321–328. [Google Scholar] [CrossRef]
- Kanda, N.; Ishikawa, T.; Kamata, M.; Tada, Y.; Watanabe, S. Increased serum leucine, leucine-37 levels in psoriasis: Positive and negative feedback loops of leucine, leucine-37 and pro- or anti-inflammatory cytokines. Hum. Immunol. 2010, 71, 1161–1171. [Google Scholar] [CrossRef]
- Gambichler, T.; Kobus, S.; Kobus, A.; Tigges, C.; Scola, N.; Altmeyer, P.; Kreuter, A.; Bechara, F.G.; Skrygan, M. Expression of antimicrobial peptides and proteins in etanercept-treated psoriasis patients. Regul. Pept. 2011, 167, 163–166. [Google Scholar] [CrossRef]
- Chamorro, C.I.; Weber, G.; Gronberg, A.; Pivarcsi, A.; Stahle, M. The human antimicrobial peptide LL-37 suppresses apoptosis in keratinocytes. J. Investig. Dermatol. 2009, 129, 937–944. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.T.; Nestel, F.P.; Bourdeau, V.; Nagai, Y.; Wang, Q.; Liao, J.; Tavera-Mendoza, L.; Lin, R.; Hanrahan, J.W.; Mader, S.; et al. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J. Immunol. 2004, 173, 2909–2912. [Google Scholar] [CrossRef] [Green Version]
- Gombart, A.F.; Borregaard, N.; Koeffler, H.P. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1.25-dihydroxyvitamin D3. FASEB J. 2005, 19, 1067–1077. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef]
- Schauber, J.; Dorschner, R.A.; Coda, A.B.; Buchau, A.S.; Liu, P.T.; Kiken, D.; Helfrich, Y.R.; Kang, S.; Elalieh, H.Z.; Steinmeyer, A.; et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J. Clin. Investig. 2007, 117, 803–811. [Google Scholar] [PubMed] [Green Version]
- Dixon, B.M.; Barker, T.; McKinnon, T.; Cuomo, J.; Frei, B.; Borregaard, N.; Gombart, A.F. Positive correlation between circulating cathelicidin antimicrobial peptide (hCAP18/LL-37) and 25-hydroxyvitamin D levels in healthy adults. BMC Res. Notes 2012, 5, 575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, T.R.; Kotol, P.; Jackson, M.; Nguyen, M.; Paik, A.; Udall, D.; Kanada, K.; Yamasaki, K.; Alexandrescu, D.; Gallo, R.L. Administration of oral vitamin D induces cathelicidin production in atopic individuals. J. Allergy Clin. Immunol. 2008, 122, 829–831. [Google Scholar] [PubMed] [Green Version]
- Hong, S.P.; Kim, M.J.; Jung, M.Y.; Jeon, H.; Goo, J.; Ahn, S.K.; Lee, S.H.; Elias, P.M.; Choi, E.H. Biopositive effects of low-dose UVB on epidermis: Coordinate upregulation of antimicrobial peptides and permeability barrier reinforcement. J. Investig. Dermatol. 2008, 128, 2880–2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjabringa, G.S.; Aarbiou, J.; Ninaber, D.K.; Drijfhout, J.W.; Sorensen, O.E.; Borregaard, N.; Rabe, K.F.; Hiemstra, P.S. The antimicrobial peptide LL-37 activates innate immunity at the airway epithelial surface by transactivation of the epidermal growth factor receptor. J. Immunol. 2003, 171, 6690–6696. [Google Scholar]
- Tokumaru, S.; Sayama, K.; Shirakata, Y.; Komatsuzawa, H.; Ouhara, K.; Hanakawa, Y.; Yahata, Y.; Dai, X.; Tohyama, M.; Nagai, H.; et al. Induction of keratinocyte migration via transactivation of the epidermal growth factor receptor by the antimicrobial peptide LL-37. J. Immunol. 2005, 175, 4662–4668. [Google Scholar]
- Peric, M.; Koglin, S.; Dombrowski, Y.; Gross, K.; Bradac, E.; Buchau, A.; Steinmeyer, A.; Zugel, U.; Ruzicka, T.; Schauber, J. Vitamin D analogs differentially control antimicrobial peptide/”alarmin” expression in psoriasis. PLoS ONE 2009, 4, e6340. [Google Scholar]
- Gorman, S.; Kuritzky, L.A.; Judge, M.A.; Dixon, K.M.; McGlade, J.P.; Mason, R.S.; Finlay-Jones, J.J.; Hart, P.H. Topically applied 1,25-dihydroxyvitamin D3 enhances the suppressive activity of CD4+CD25+ cells in the draining lymph nodes. J. Immunol. 2007, 179, 6273–6283. [Google Scholar] [CrossRef] [Green Version]
- Dombrowski, Y.; Peric, M.; Koglin, S.; Kammerbauer, C.; Goss, C.; Anz, D.; Simanski, M.; Glaser, R.; Harder, J.; Hornung, V.; et al. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011, 3, 82ra38. [Google Scholar]
- Brown, G.; Wang, E.; Leon, A.; Huynh, M.; Wehner, M.; Matro, R.; Linos, E.; Liao, W.; Haemel, A. Tumor necrosis factor-α inhibitor-induced psoriasis: Systematic review of clinical features, histopathological findings, and management experience. J. Acad. Dermatol. 2017, 76, 334–341. [Google Scholar]
- Conrad, C.; Di Domizio, J.; Mylonas, A.; Belkhodja, C.; Demaria, O.; Navarini, A.A.; Lapointe, A.-K.; French, L.E.; Vernez, M.; Gilliet, M. TNF blockade induces a dysregulated type I interferon response without autoimmunity in paradoxical psoriasis. Nat. Commun. 2018, 9, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takahashi, T.; Yamasaki, K. Psoriasis and Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 6791. https://doi.org/10.3390/ijms21186791
Takahashi T, Yamasaki K. Psoriasis and Antimicrobial Peptides. International Journal of Molecular Sciences. 2020; 21(18):6791. https://doi.org/10.3390/ijms21186791
Chicago/Turabian StyleTakahashi, Toshiya, and Kenshi Yamasaki. 2020. "Psoriasis and Antimicrobial Peptides" International Journal of Molecular Sciences 21, no. 18: 6791. https://doi.org/10.3390/ijms21186791
APA StyleTakahashi, T., & Yamasaki, K. (2020). Psoriasis and Antimicrobial Peptides. International Journal of Molecular Sciences, 21(18), 6791. https://doi.org/10.3390/ijms21186791