A Screening of Antineoplastic Drugs for Acute Myeloid Leukemia Reveals Contrasting Immunogenic Effects of Etoposide and Fludarabine


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
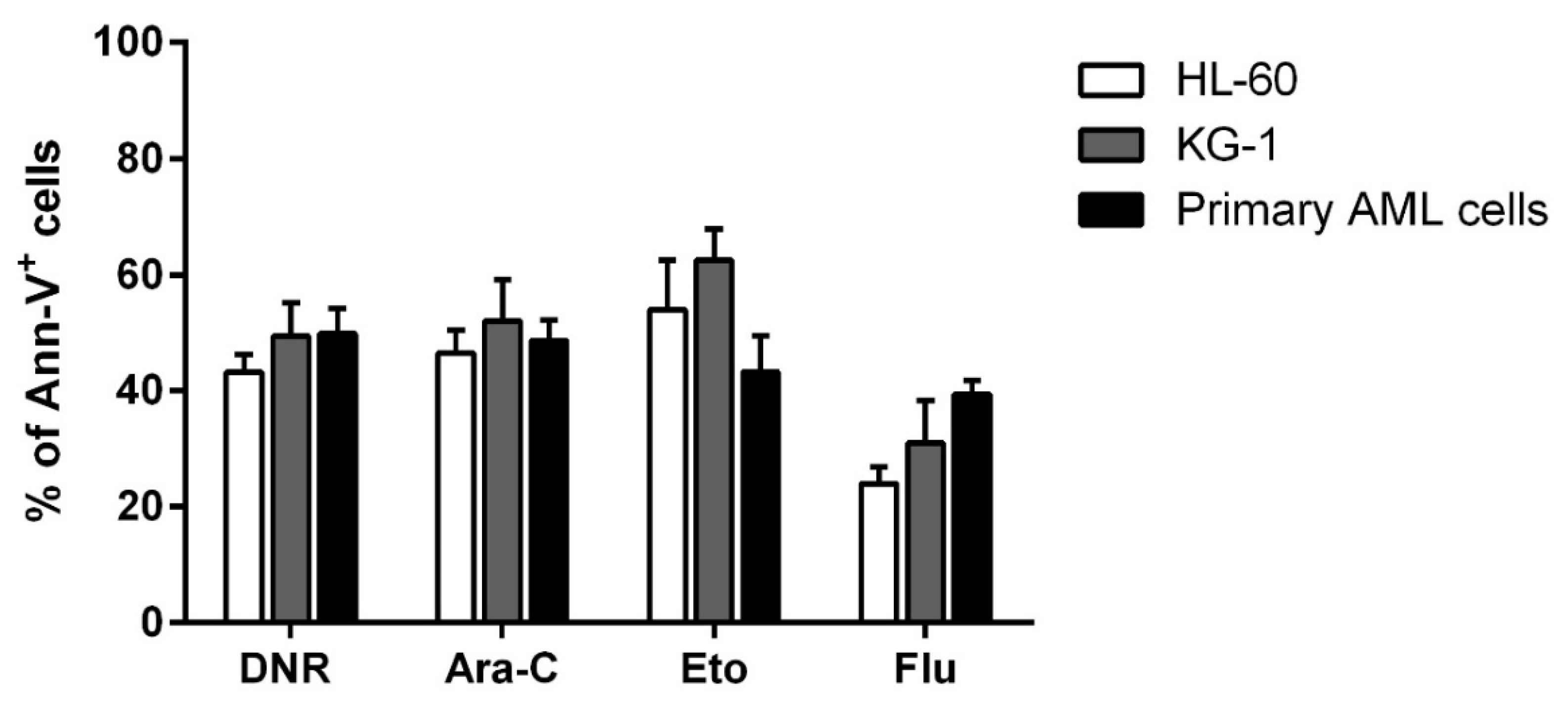
2.1. DNR, Eto, Ara-C and Flu Induced a Similar Apoptosis Level in HL-60, KG-1 and Primary AML Cells
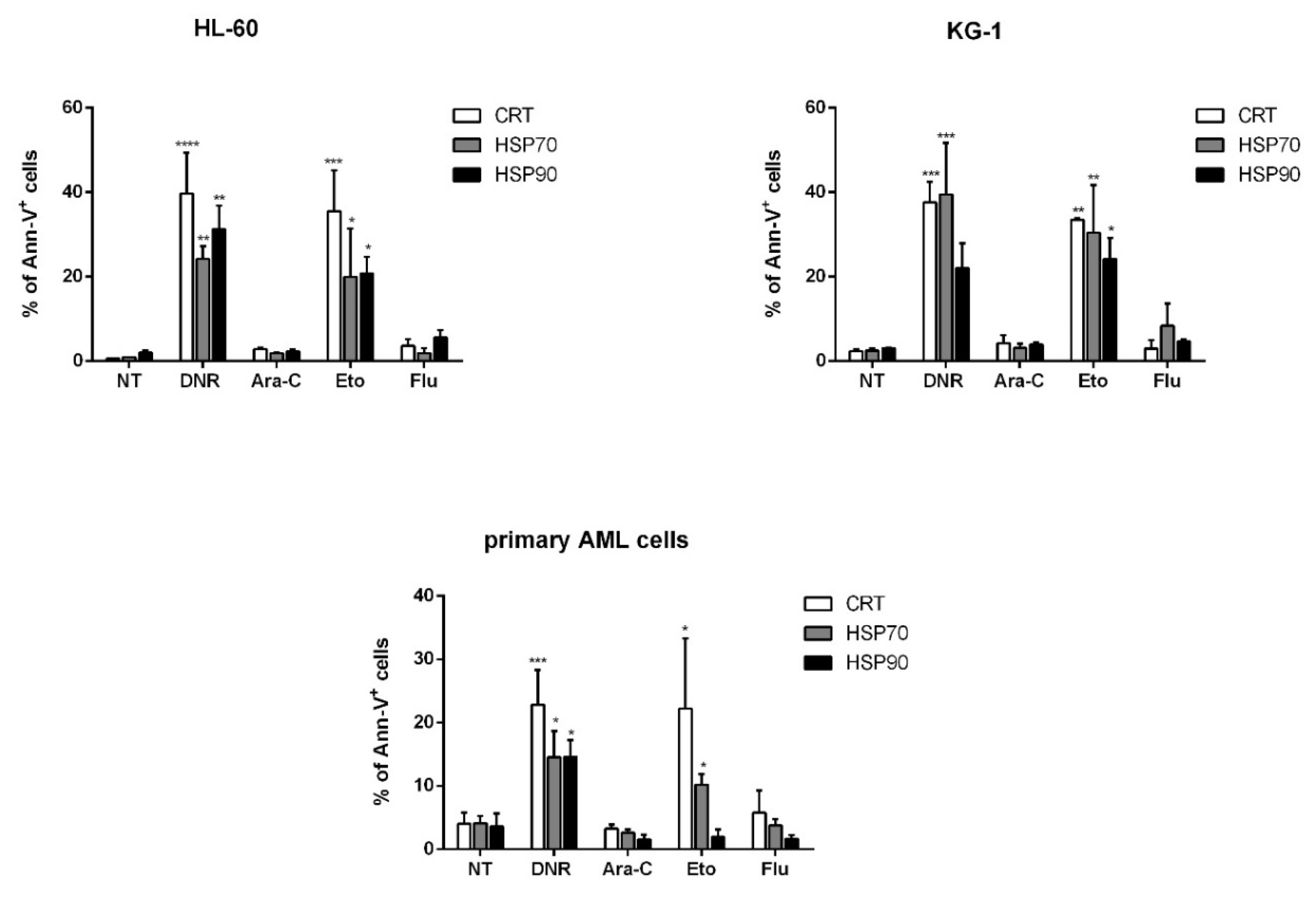
2.2. Only Treatment with DNR and Eto, but Not Flu and Ara-C, Induced Translocation of CRT and HSPs from ER to Plasma Cell Membrane in AML Cells
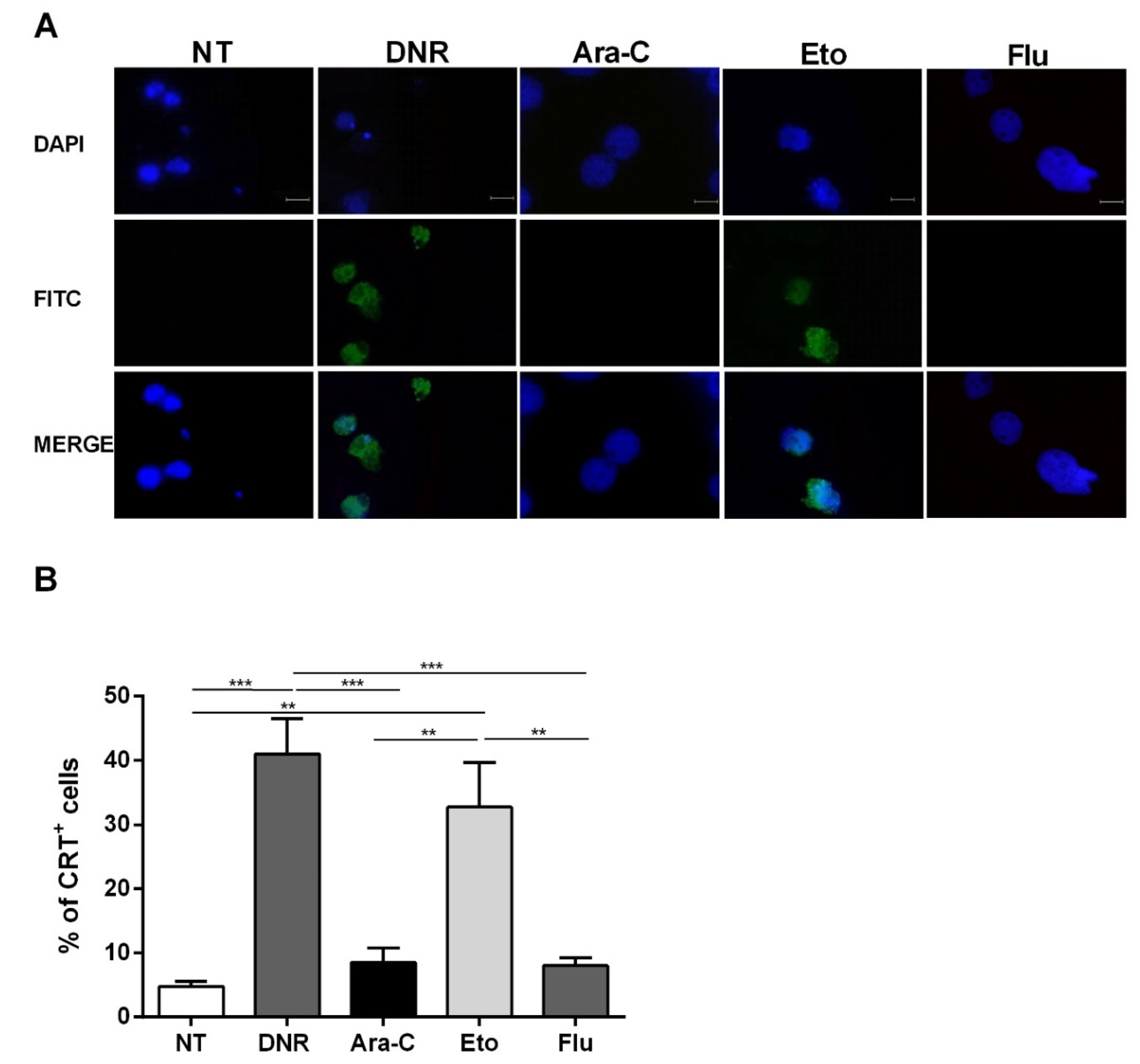
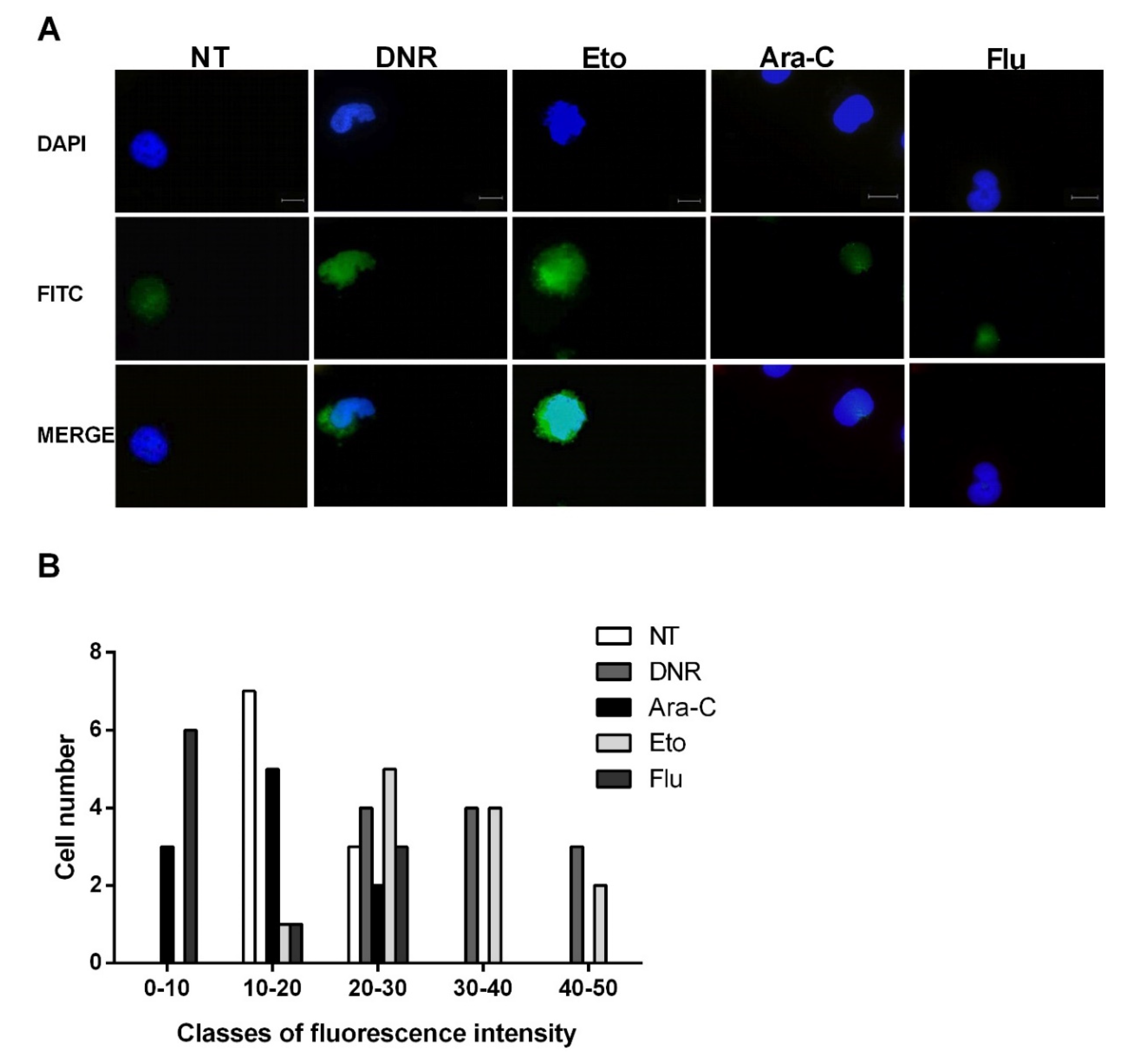
2.3. Only Treatment with DNR and Eto, but Not Flu and Ara-C, Induced the HMGB1 Release from the Nucleus to the Extracellular Space of AML Cells
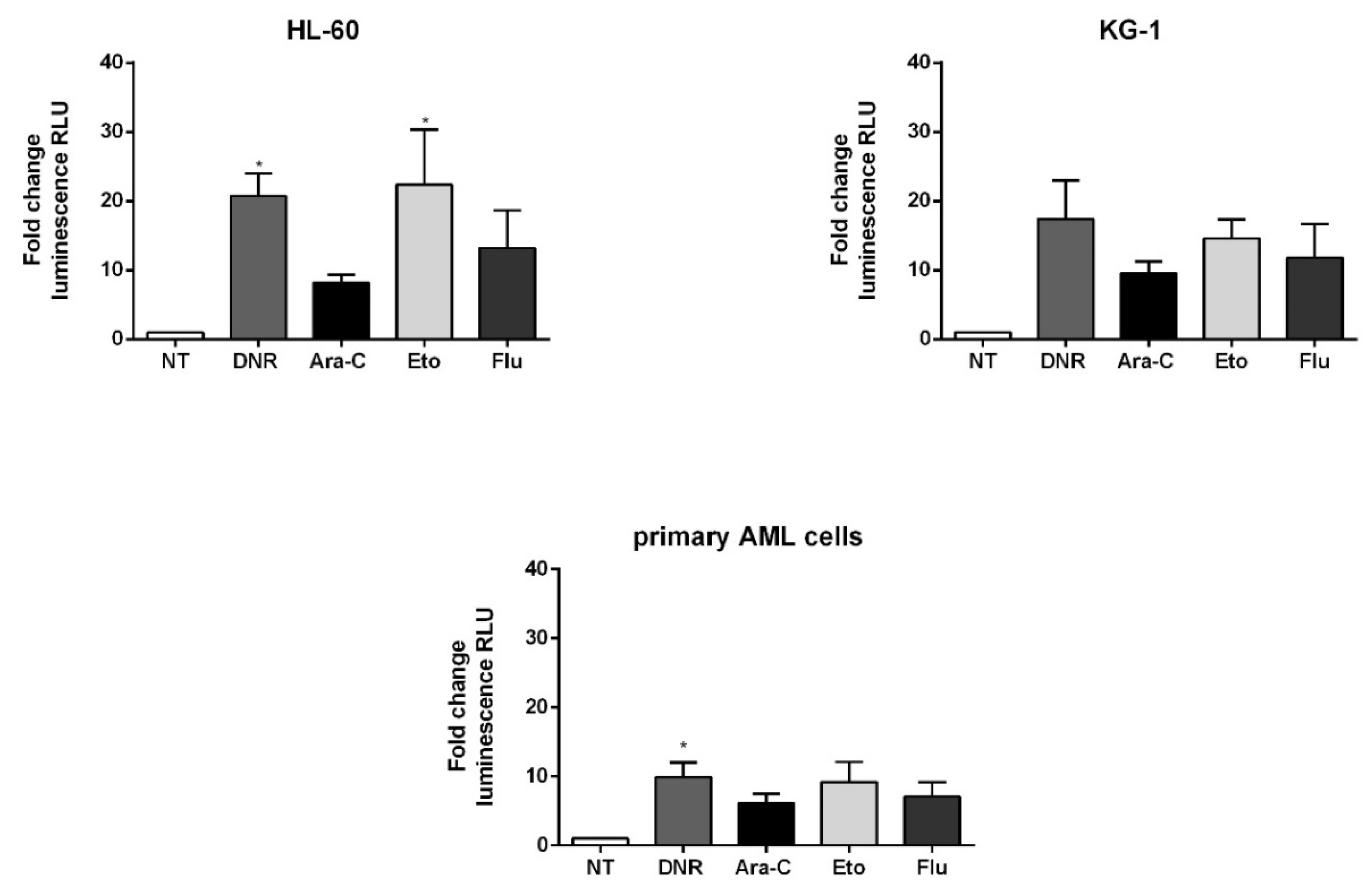
2.4. Only Treatment with DNR and Eto, but Not Flu and Ara-C, Induced ATP Release to the Extracellular Space of AML Cells
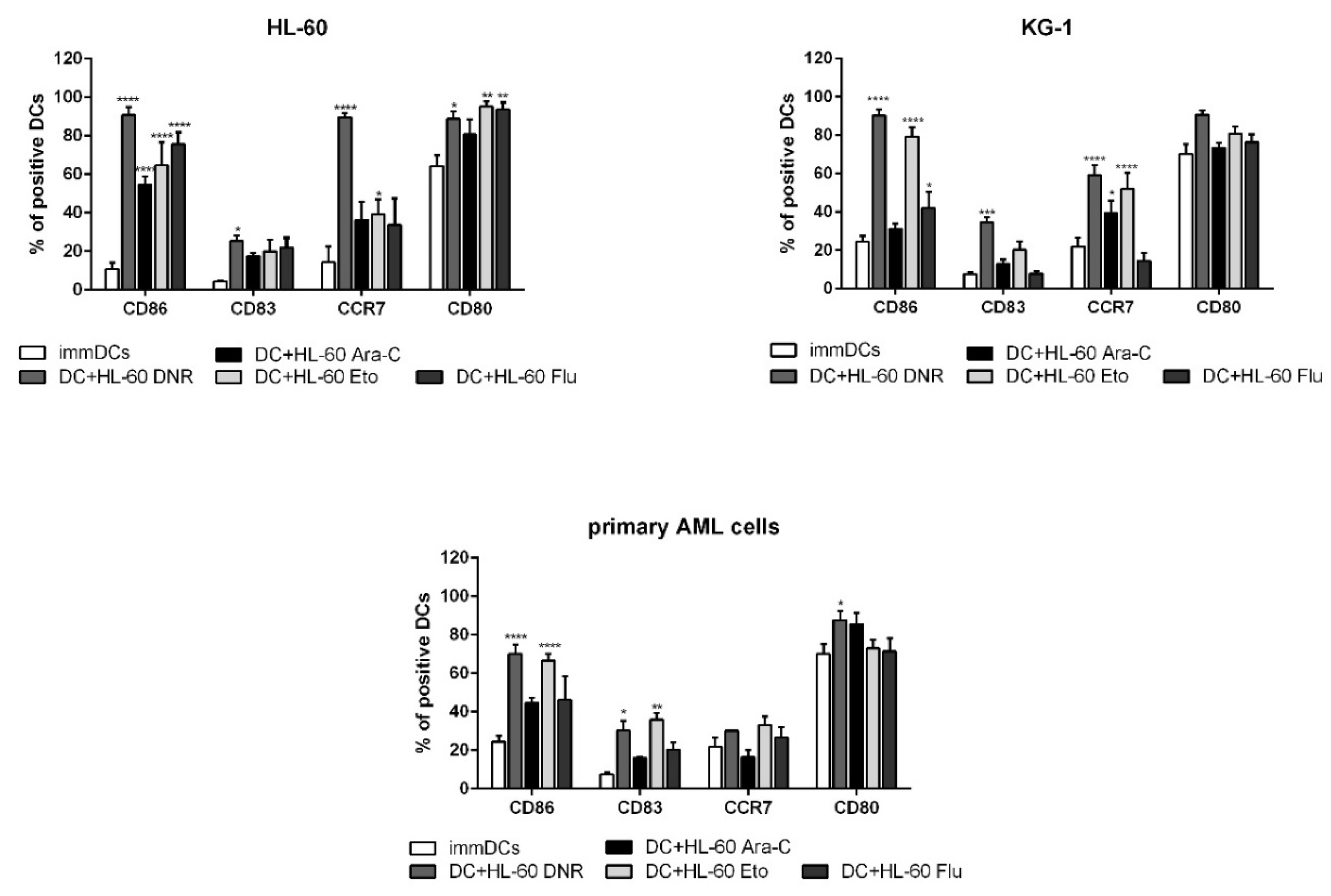
2.5. All Tested Drugs Induced DC Maturation Mediated by Chemotherapy-Treated HL-60, KG-1 and Primary AML Cells, but Only DNR and Eto Induced CD83 Up-Regulation, and Only DNR, Ara-C and Eto Induced CCR7 Expression
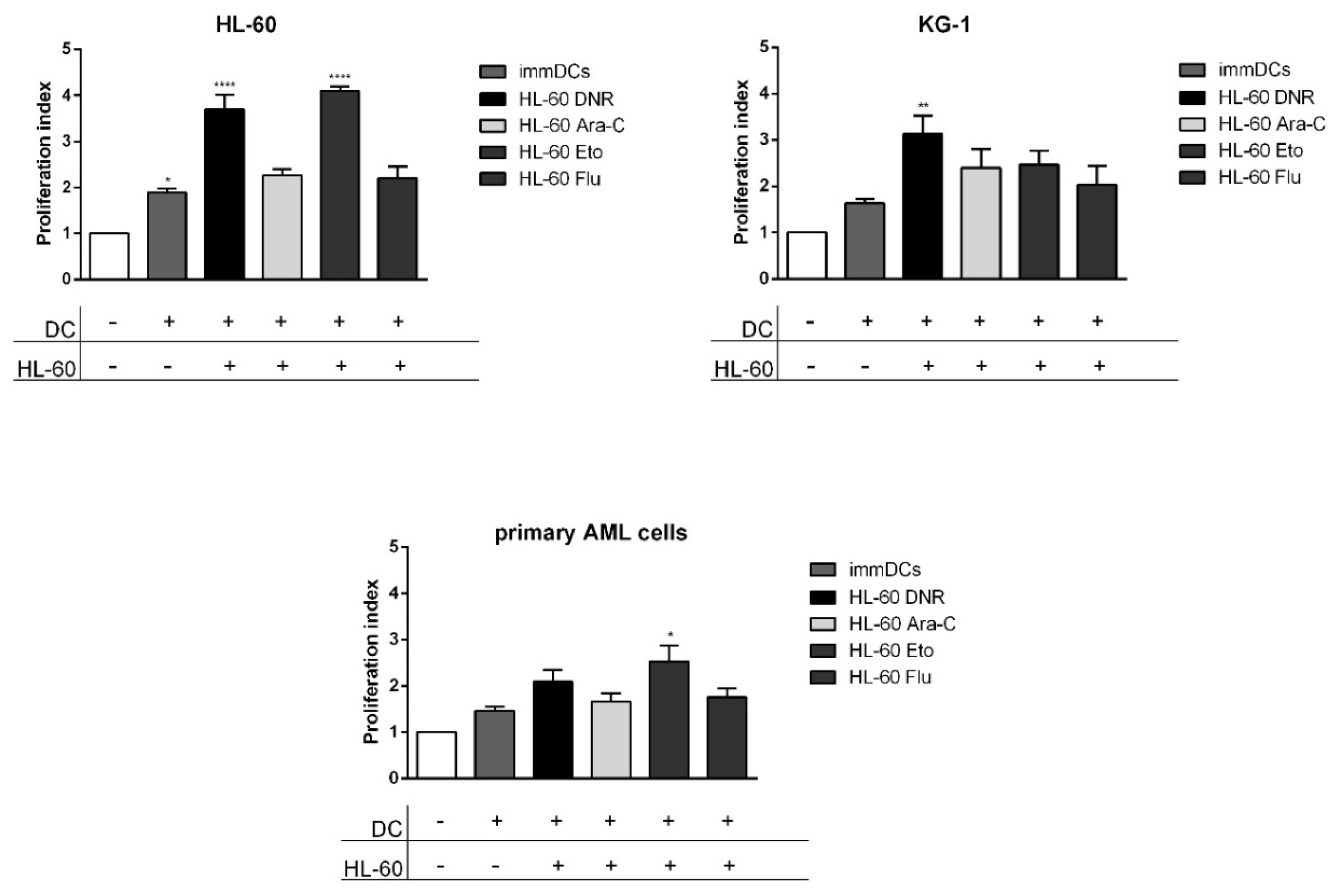
2.6. Only DNR and Eto, but Not Flu and Ara-C, Induced the Proliferation of Allogeneic T Cells by DCs Loaded with HL-60, KG-1 and Primary AML Cells Treated Cells
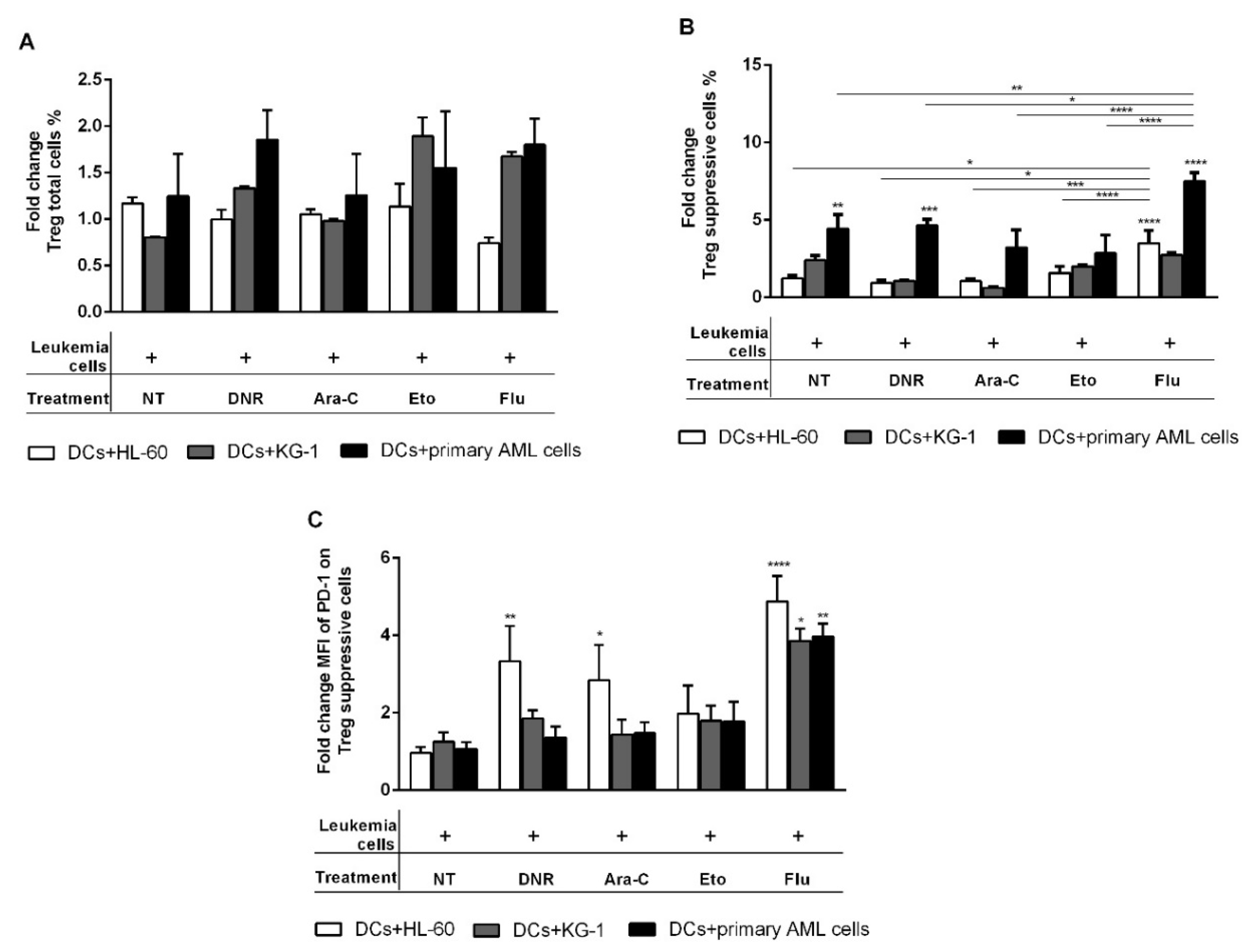
2.7. Flu-Treated Leukemic Cells Induced a Population of Suppressive T Regulatory Cells via DCs
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. HL-60, KG-1 and Primary AML Cell Treatment
4.3. CRT and HSP70 and 90 Staining by Flow Cytometry
4.4. Quantification of ATP Release
4.5. CRT and HMGB1 Staining by Immunofluorescence
4.5.1. CRT Exposure
4.5.2. HMGB1 Release
4.6. DC Generation, Pulsing and Maturation
4.7. DC Phenotype by Flow Cytometry
4.8. Proliferation Test
4.9. Treg Induction
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AML | Acute myeloid leukemia |
| DNR | Daunorubicin |
| Ara-C | Cytarabine |
| ICD | Immunogenic cell death |
| Eto | Etoposide |
| Flu | Fludarabine |
| DCs | Dendritic cells |
| DAMPs | Danger associated molecular patterns |
| ER | Endoplasmic reticulum |
| CRT | Calreticulin |
| HSPs | Heat shock proteins |
| ATP | Adenosine triphosphate |
| HMGB1 | Non-histone chromatin-binding protein high mobility group box 1 |
| TAAs | Tumor associated antigens |
| TLR4 | Toll like receptor 4 |
| CR | Complete remission |
| NT | No treatment |
| immDCs | Immature DCs |
| Tregs | T regulatory cells |
| PD-1 | Programmed cell death protein 1 |
| BM | Bone marrow |
| mAbs | Monoclonal antibodies |
| MFI | Mean of fluorescence intensity |
| 2-DG | 2-Deoxyglucose |
References
- Lecciso, M.; Ocadlikova, D.; Sangaletti, S.; Trabanelli, S.; De Marchi, E.; Orioli, E.; Pegoraro, A.; Portararo, P.; Jandus, C.; Bontadini, A.; et al. ATP Release from Chemotherapy-Treated Dying Leukemia Cells Elicits an Immune Suppressive Effect by Increasing Regulatory T Cells and Tolerogenic Dendritic Cells. Front. Immunol. 2017, 8, 1918. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Kralikova, P.; Fialova, A.; Brtnicky, T.; Rob, L.; Bartunkova, J.; Spísek, R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71, 4821–4833. [Google Scholar] [CrossRef]
- Fucikova, J.; Truxova, I.; Hensler, M.; Becht, E.; Kasikova, L.; Moserova, I.; Vosahlikova, S.; Klouckova, J.; Church, S.E.; Cremer, I.; et al. Calreticulin exposure by malignant blasts correlates with robust anticancer immunity and improved clinical outcome in AML patients. Blood 2016, 128, 3113–3124. [Google Scholar] [CrossRef] [PubMed]
- Wemeau, M.; Kepp, O.; Tesnière, A.; Panaretakis, T.; Flament, C.; De Botton, S.; Zitvogel, L.; Kroemer, G.; Chaput, N. Calreticulin exposure on malignant blasts predicts a cellular anticancer immune response in patients with acute myeloid leukemia. Cell Death Dis. 2010, 1, e104. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef]
- Fredly, H.; Ersvær, E.; Gjertsen, B.T.; Bruserud, O. Immunogenic apoptosis in human acute myeloid leukemia (AML): Primary human AML cells expose calreticulin and release heat shock protein (HSP) 70 and HSP90 during apoptosis. Oncol. Rep. 2011, 25, 1549–1556. [Google Scholar] [CrossRef]
- Spisek, R.; Charalambous, A.; Mazumder, A.; Vesole, D.H.; Jagannath, S.; Dhodapkar, M.V. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: Therapeutic implications. Blood 2007, 109, 4839–4845. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Martin, S.; Golab, J.; Agostinis, P. Danger signalling during cancer cell death: Origins, plasticity and regulation. Cell Death Differ. 2014, 21, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.D.; Dudek, A.M.; Agostinis, P. Cancer immunogenicity, danger signals, and DAMPs: What, when, and how? Biofactors 2013, 39, 355–367. [Google Scholar] [CrossRef]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef]
- Sistigu, A.; Yamazaki, T.; Vacchelli, E.; Chaba, K.; Enot, D.P.; Adam, J.; Vitale, I.; Goubar, A.; Baracco, E.E.; Remédios, C.; et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 2014, 20, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Panaretakis, T.; Tufi, R.; Joza, N.; van Endert, P.; Ghiringhelli, F.; Apetoh, L.; Chaput, N.; Flament, C.; et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 2007, 220, 22–34. [Google Scholar] [CrossRef]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Criollo, A.; Ortiz, C.; Lidereau, R.; Mariette, C.; Chaput, N.; Mira, J.P.; Delaloge, S.; et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 2007, 220, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Adjemian, S.; Yang, H.; Catani, J.P.; Hannani, D.; Martins, I.; Michaud, M.; Kepp, O.; Sukkurwala, A.Q.; Vacchelli, E.; et al. ATP-dependent recruitment, survival and differentiation of dendritic cell precursors in the tumor bed after anticancer chemotherapy. Oncoimmunology 2013, 2, e24568. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef]
- Cirone, M.; Di Renzo, L.; Lotti, L.V.; Conte, V.; Trivedi, P.; Santarelli, R.; Gonnella, R.; Frati, L.; Faggioni, A. Activation of dendritic cells by tumor cell death. Oncoimmunology 2012, 1, 1218–1219. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Chen, X.; Fosco, D.; Kline, D.E.; Kline, J. Calreticulin promotes immunity and type I interferon-dependent survival in mice with acute myeloid leukemia. Oncoimmunology 2017, 6, e1278332. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Kasikova, L.; Truxova, I.; Laco, J.; Skapa, P.; Ryska, A.; Spisek, R. Relevance of the chaperone-like protein calreticulin for the biological behavior and clinical outcome of cancer. Immunol. Lett. 2018, 193, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Alfayez, M.; Borthakur, G. Checkpoint inhibitors and acute myelogenous leukemia: Promises and challenges. Expert Rev. Hematol. 2018, 11, 373–389. [Google Scholar] [CrossRef]
- Hobo, W.; Hutten, T.J.A.; Schaap, N.P.M.; Dolstra, H. Immune checkpoint molecules in acute myeloid leukaemia: Managing the double-edged sword. Br. J. Haematol. 2018, 181, 38–53. [Google Scholar] [CrossRef]
- Bradstock, K.F.; Link, E.; Di Iulio, J.; Szer, J.; Marlton, P.; Wei, A.H.; Enno, A.; Schwarer, A.; Lewis, I.D.; D’Rozario, J.; et al. Idarubicin Dose Escalation during Consolidation Therapy for Adult Acute Myeloid Leukemia. J. Clin. Oncol. 2017, 35, 1678–1685. [Google Scholar] [CrossRef]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Kell, J.; Cavenagh, J.; Kjeldsen, L.; McMullin, M.F.; Cahalin, P.; Dennis, M.; Friis, L.; et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: Results from the UK NCRI AML17 trial in 1206 patients. Blood 2015, 125, 3878–3885. [Google Scholar] [CrossRef]
- Balakrishnan, V.S. Escalated anthracycline dose in adult AML. Lancet Oncol. 2017, 18, e253. [Google Scholar] [CrossRef]
- Magina, K.N.; Pregartner, G.; Zebisch, A.; Wölfler, A.; Neumeister, P.; Greinix, H.T.; Berghold, A.; Sill, H. Cytarabine dose in the consolidation treatment of AML: A systematic review and meta-analysis. Blood 2017, 130, 946–948. [Google Scholar] [CrossRef]
- Murphy, T.; Yee, K.W.L. Cytarabine and daunorubicin for the treatment of acute myeloid leukemia. Expert Opin. Pharmacother. 2017, 18, 1765–1780. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed]
- Estey, E. Acute myeloid leukemia: 2016 Update on risk-stratification and management. Am. J. Hematol. 2016, 91, 824–846. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.; Wetzler, M.; Löwenberg, B. Therapeutic advances in acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H.; Gardin, C. An update of current treatments for adult acute myeloid leukemia. Blood 2016, 127, 53–61. [Google Scholar] [CrossRef]
- Cerrano, M.; Candoni, A.; Crisà, E.; Dubbini, M.V.; D’Ardia, S.; Zannier, M.E.; Boccadoro, M.; Audisio, E.; Bruno, B.; Ferrero, D. FLAI induction regimen in elderly patients with acute myeloid leukemia. Leuk. Lymphoma 2019, 60, 1–2. [Google Scholar] [CrossRef]
- Kim, I.; Koh, Y.; Yoon, S.S.; Park, S.; Kim, B.K.; Kim, D.Y.; Lee, J.H.; Lee, K.H.; Cheong, J.W.; Lee, H.K.; et al. Fludarabine, cytarabine, and attenuated-dose idarubicin (m-FLAI) combination therapy for elderly acute myeloid leukemia patients. Am. J. Hematol. 2013, 88, 10–15. [Google Scholar] [CrossRef]
- Gebremeskel, S.; Johnston, B. Concepts and mechanisms underlying chemotherapy induced immunogenic cell death: Impact on clinical studies and considerations for combined therapies. Oncotarget 2015, 6, 41600–41619. [Google Scholar] [CrossRef]
- Bezu, L.; Gomes-da-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Corrigendum: “Combinatorial Strategies for the Induction of Immunogenic Cell Death”. Front. Immunol. 2015, 6, 275. [Google Scholar] [CrossRef]
- Jung, K.-H.; Paik, J.-Y.; Park, J.; Quach, C.; Lee, E.J.; Choe, Y.S.; Lee, K.-H. Targeting of calreticulin exposure for optical imaging of etoposide-induced immunogenic tumor cell death. In Proceedings of the Molecular Targets and Cancer Therapeutics, San Francisco, CA, USA, 12–16 November 2011. [Google Scholar]
- Garg, A.D.; Romano, E.; Rufo, N.; Agostinis, P. Immunogenic versus tolerogenic phagocytosis during anticancer therapy: Mechanisms and clinical translation. Cell Death Differ. 2016, 23, 938–951. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef]
- Knaus, H.A.; Berglund, S.; Hackl, H.; Blackford, A.L.; Zeidner, J.F.; Montiel-Esparza, R.; Mukhopadhyay, R.; Vanura, K.; Blazar, B.R.; Karp, J.E.; et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Kroemer, G.; Zitvogel, L. Immunogenic and Non-immunogenic Cell Death in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Ocadlikova, D.; Lecciso, M.; Isidori, A.; Loscocco, F.; Visani, G.; Amadori, S.; Cavo, M.; Curti, A. Chemotherapy-Induced Tumor Cell Death at the Crossroads Between Immunogenicity and Immunotolerance: Focus on Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1004. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, F.; Cao, Y.; Zhang, M.; Wang, A.; Xu, M.; Su, M.; Zhuge, Y. Etoposide Induces Apoptosis in Activated Human Hepatic Stellate Cells via ER Stress. Sci. Rep. 2016, 6, 34330. [Google Scholar] [CrossRef] [PubMed]
- Bezu, L.; Sauvat, A.; Humeau, J.; Gomes-da-Silva, L.C.; Iribarren, K.; Forveille, S.; Garcia, P.; Zhao, L.; Liu, P.; Zitvogel, L.; et al. eIF2α phosphorylation is pathognomonic for immunogenic cell death. Cell Death Differ. 2018, 25, 1375–1393. [Google Scholar] [CrossRef] [PubMed]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; van Endert, P.; et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Bénéteau, M.; Zunino, B.; Jacquin, M.A.; Meynet, O.; Chiche, J.; Pradelli, L.A.; Marchetti, S.; Cornille, A.; Carles, M.; Ricci, J.E. Combination of glycolysis inhibition with chemotherapy results in an antitumor immune response. Proc. Natl. Acad. Sci. USA 2012, 109, 20071–20076. [Google Scholar] [CrossRef]
- Giglio, P.; Gagliardi, M.; Bernardini, R.; Mattei, M.; Cotella, D.; Santoro, C.; Piacentini, M.; Corazzari, M. Ecto-Calreticulin is essential for an efficient immunogenic cell death stimulation in mouse melanoma. Genes Immun. 2019, 20, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Senovilla, L.; Vitale, I.; Vacchelli, E.; Adjemian, S.; Agostinis, P.; Apetoh, L.; Aranda, F.; Barnaba, V.; Bloy, N.; et al. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 2014, 3, e955691. [Google Scholar] [CrossRef]
- Davidson-Moncada, J.; Viboch, E.; Church, S.E.; Warren, S.E.; Rutella, S. Dissecting the Immune Landscape of Acute Myeloid Leukemia. Biomedicines 2018, 6, 110. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Vadakekolathu, J.; Minden, M.D.; Hood, T.; Church, S.E.; Reeder, S.; Altmann, H.; Sullivan, A.H.; Viboch, E.J.; Patel, T.; Ibrahimova, N.; et al. Immune landscapes predict chemotherapy resistance and immunotherapy response in acute myeloid leukemia. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Curti, A.; Trabanelli, S.; Onofri, C.; Aluigi, M.; Salvestrini, V.; Ocadlikova, D.; Evangelisti, C.; Rutella, S.; De Cristofaro, R.; Ottaviani, E.; et al. Indoleamine 2,3-dioxygenase-expressing leukemic dendritic cells impair a leukemia-specific immune response by inducing potent T regulatory cells. Haematologica 2010, 95, 2022–2030. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Xu, Y. Clinical significance of Treg cell frequency in acute myeloid leukemia. Int. J. Hematol. 2013, 98, 558–562. [Google Scholar] [CrossRef]
- Kanakry, C.G.; Hess, A.D.; Gocke, C.D.; Thoburn, C.; Kos, F.; Meyer, C.; Briel, J.; Luznik, L.; Smith, B.D.; Levitsky, H.; et al. Early lymphocyte recovery after intensive timed sequential chemotherapy for acute myelogenous leukemia: Peripheral oligoclonal expansion of regulatory T cells. Blood 2011, 117, 608–617. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Czystowska, M.; Mandapathil, M.; Strauss, L.; Welsh, A.; Foon, K.A.; Whiteside, T.L.; Boyiadzis, M. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin. Cancer Res. 2009, 15, 3325–3332. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, C.; Tian, T.; Zhang, T.; Wang, R.; Han, F.; Zhong, C.; Hua, M.; Ma, D. Increased Regulatory T Cells in Peripheral Blood of Acute Myeloid Leukemia Patients Rely on Tumor Necrosis Factor (TNF)-α-TNF Receptor-2 Pathway. Front. Immunol. 2018, 9, 1274. [Google Scholar] [CrossRef]
- Brück, O.; Dufva, O.; Hohtari, H.; Blom, S.; Turkki, R.; Ilander, M.; Kovanen, P.; Pallaud, C.; Ramos, P.M.; Lähteenmäki, H.; et al. Immune profiles in acute myeloid leukemia bone marrow associate with patient age, T-cell receptor clonality, and survival. Blood Adv. 2020, 4, 274–286. [Google Scholar] [CrossRef]
- Zhang, S.; Han, Y.; Wu, J.; Yu, K.; Bi, L.; Zhuang, Y.; Xu, X. Elevated frequencies of CD4+ CD25+ CD127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int. J. Cancer 2011, 129, 1373–1381. [Google Scholar] [CrossRef]
- Johnson, T.S.; Mcgaha, T.; Munn, D.H. Chemo-Immunotherapy: Role of Indoleamine 2,3-Dioxygenase in Defining Immunogenic Versus Tolerogenic Cell Death in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 91–104. [Google Scholar] [CrossRef]
- de Boer, J.P.; Raderer, M.; van Tinteren, H.; Aleman, B.M.; Boot, H.; de Jong, D. Treatment of extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue with fludarabine: Effect on tumor microenvironment. Leuk. Lymphoma 2011, 52, 2262–2269. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kusnadi, A.; Lee, E.J.; Kim, W.W.; Cho, B.C.; Lee, I.J.; Seong, J.; Ha, S.J. Tumor-infiltrating regulatory T cells delineated by upregulation of PD-1 and inhibitory receptors. Cell Immunol. 2012, 278, 76–83. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Barber, D.L.; Wherry, E.J.; Masopust, D.; Zhu, B.; Allison, J.P.; Sharpe, A.H.; Freeman, G.J.; Ahmed, R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 2006, 439, 682–687. [Google Scholar] [CrossRef]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor cell expression of programmed cell death-1 ligand 1 is a prognostic factor for malignant melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef]
- Vignali, D.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Ciciarello, M.; Roscioli, E.; Di Fiore, B.; Di Francesco, L.; Sobrero, F.; Bernard, D.; Mangiacasale, R.; Harel, A.; Schininà, M.E.; Lavia, P. Nuclear reformation after mitosis requires downregulation of the Ran GTPase effector RanBP1 in mammalian cells. Chromosoma 2010, 119, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Trabanelli, S.; Lecciso, M.; Salvestrini, V.; Cavo, M.; Očadlíková, D.; Lemoli, R.M.; Curti, A. PGE2-induced IDO1 inhibits the capacity of fully mature DCs to elicit an in vitro antileukemic immune response. J. Immunol. Res. 2015, 2015, 253191. [Google Scholar] [CrossRef]
- Curti, A.; Pandolfi, S.; Aluigi, M.; Isidori, A.; Alessandrini, I.; Chiodoni, C.; Testoni, N.; Colombo, M.P.; Baccarani, M.; Lemoli, R.M. Interleukin-12 production by leukemia-derived dendritic cells counteracts the inhibitory effect of leukemic microenvironment on T cells. Exp. Hematol. 2005, 33, 1521–1530. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ocadlikova, D.; Iannarone, C.; Redavid, A.R.; Cavo, M.; Curti, A. A Screening of Antineoplastic Drugs for Acute Myeloid Leukemia Reveals Contrasting Immunogenic Effects of Etoposide and Fludarabine. Int. J. Mol. Sci. 2020, 21, 6802. https://doi.org/10.3390/ijms21186802
Ocadlikova D, Iannarone C, Redavid AR, Cavo M, Curti A. A Screening of Antineoplastic Drugs for Acute Myeloid Leukemia Reveals Contrasting Immunogenic Effects of Etoposide and Fludarabine. International Journal of Molecular Sciences. 2020; 21(18):6802. https://doi.org/10.3390/ijms21186802
Chicago/Turabian StyleOcadlikova, Darina, Clara Iannarone, Anna Rita Redavid, Michele Cavo, and Antonio Curti. 2020. "A Screening of Antineoplastic Drugs for Acute Myeloid Leukemia Reveals Contrasting Immunogenic Effects of Etoposide and Fludarabine" International Journal of Molecular Sciences 21, no. 18: 6802. https://doi.org/10.3390/ijms21186802
APA StyleOcadlikova, D., Iannarone, C., Redavid, A. R., Cavo, M., & Curti, A. (2020). A Screening of Antineoplastic Drugs for Acute Myeloid Leukemia Reveals Contrasting Immunogenic Effects of Etoposide and Fludarabine. International Journal of Molecular Sciences, 21(18), 6802. https://doi.org/10.3390/ijms21186802