The Emerging Role of ATP-Dependent Chromatin Remodeling in Memory and Substance Use Disorders



Abstract
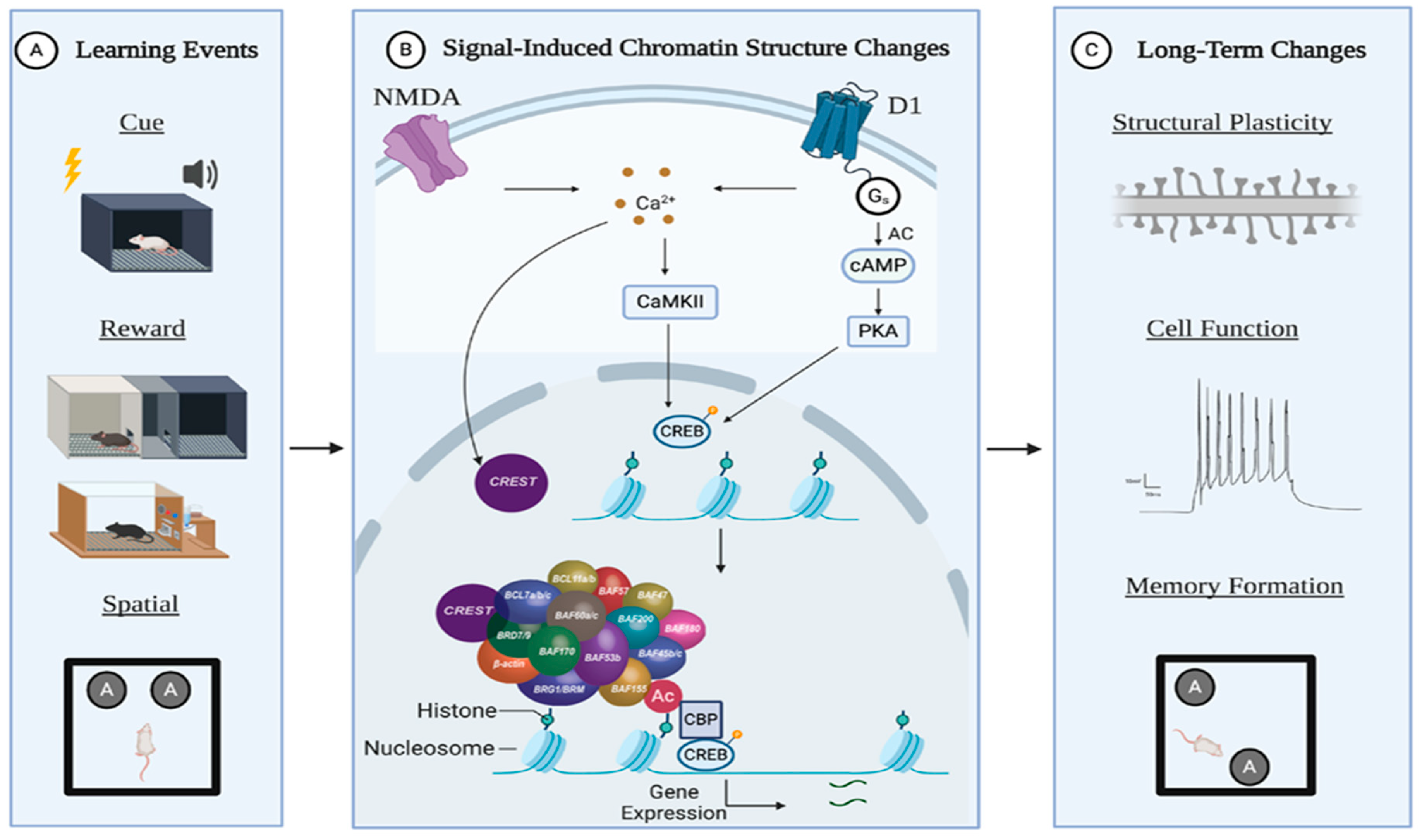
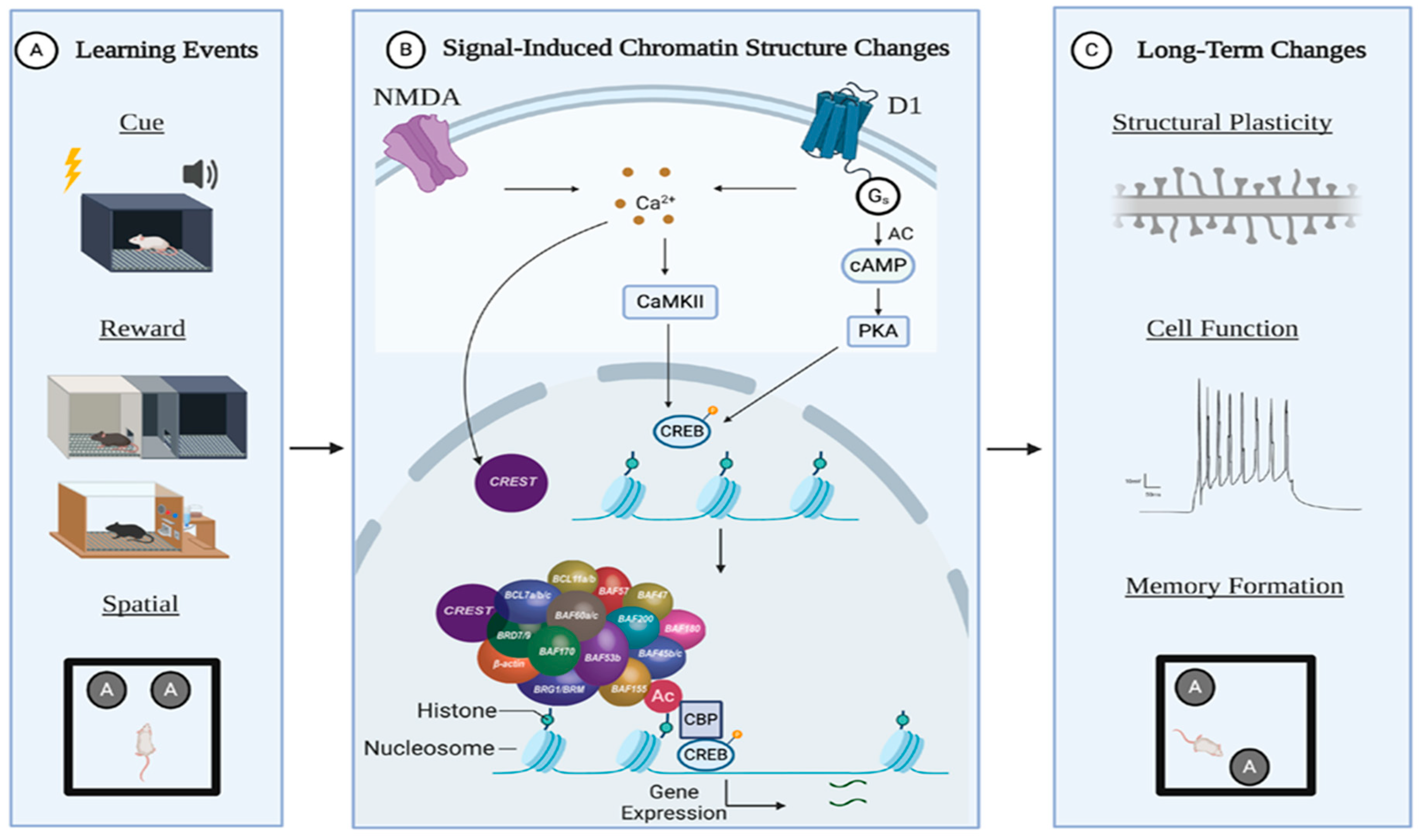
:1. Introduction
2. Epigenetics in Learning and Memory
2.1. Overview
2.2. Histone Modifications in Learning and Memory
3. Introduction to Chromatin Remodeling Complexes
4. Chromatin Remodeling Complexes in Neurodevelopment
4.1. ISWI
4.2. nBAF

{kind=link}
| Remodeling Complex | Neuron-Relevant Subunit | Target Residues | Transcriptional Effect | References |
|---|---|---|---|---|
| nBAF | BAF53B/ACTL6B CREST BAF45B BAF45C | Acetylated Histones (via SMARCA bromodomains) | Permissive (Mor1, Bdnf, Mef2d, Cap2, Dbn1) Repressive (Fos, Fosl2, Fosb, Junb) | [78,79,80,81,82,83] |
| ISWI | SMARCA1 SMARCA5 BAZ1A BAZ1B BAZ2B CECR2 RSF1 | Acetylated Histones (via CECR2 and SMARCA SANT-domains; BAZ bromodomains) Methylated Histones (via chromodomains and PHDs) | Permissive | [84,85,86,87,88,89,90,91] |
| NuRD | Mi-2a/b MBD3 MTA1-3 RbAp46 RbAp48 HDAC1/2 | Histone Lysine Residue (via HDAC1/2 bromodomains) Methylated Histones (via Mi-2a/b chromodomains) | Repressive | [92,93,94,95,96,97,98,99,100,101,102,103,104] |
5. Chromatin Remodeling Complexes in Adult Cognitive Function
5.1. ISWI Subunits Regulate Depressive-Like Phenotypes
5.2. nBAF is Essential for Synaptic Plasticity and Memory Processes
6. Epigenetics in Substance Use Disorders
6.1. Histone Modifications
6.2. ISWI and nBAF Chromatin Remodeling Complexes
7. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alberini, C.M. Transcription Factors in Long-Term Memory and Synaptic Plasticity. Physiol. Rev. 2009, 89, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gräff, J.; Tsai, L.-H. Histone acetylation: Molecular mnemonics on the chromatin. Nat. Rev. Neurosci. 2013, 14, 97. [Google Scholar] [PubMed]
- Campbell, R.R.; Wood, M.A. How the epigenome integrates information and reshapes the synapse. Nat. Rev. Neurosci. 2019, 20, 133–147. [Google Scholar] [PubMed]
- Hyman, S.E. Addiction: A disease of learning and memory. Am. J. Psychiatry 2005, 162, 1414–1422. [Google Scholar]
- Hyman, S.E.; Malenka, R.C.; Nestler, E.J. NEURAL MECHANISMS OF ADDICTION: The Role of Reward-Related Learning and Memory. Annu. Rev. Neurosci. 2006, 29, 565–598. [Google Scholar] [CrossRef] [Green Version]
- Robison, A.J.; Nestler, E.J. Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 2011, 12, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Jarome, T.J.; Lubin, F.D. Epigenetic mechanisms of memory formation and reconsolidation. Neurobiol. Learn. Mem. 2014, 115, 116–127. [Google Scholar] [CrossRef] [Green Version]
- López, A.J.; Siciliano, C.A.; Calipari, E.S. Activity-dependent epigenetic remodeling in cocaine use disorder. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 258, pp. 231–263. [Google Scholar]
- Barrett, R.M.; Wood, M.A. Beyond transcription factors: The role of chromatin modifying enzymes in regulating transcription required for memory. Learn. Mem. 2008, 15, 460–467. [Google Scholar] [CrossRef] [Green Version]
- Nelson, E.D.; Monteggia, L.M. Epigenetics in the mature mammalian brain: Effects on behavior and synaptic transmission. Neurobiol. Learn. Mem. 2011, 96, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Kwapis, J.L.; Wood, M.A. Epigenetic mechanisms in fear conditioning: Implications for treating post-traumatic stress disorder. Trends Neurosci. 2014, 37, 706–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Z.; Giustetto, M.; Lomvardas, S.; Kim, J.H.; Miniaci, M.C.; Schwartz, J.H.; Thanos, D.; Kandel, E.R. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell 2002, 111, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Latham, J.A.; Dent, S.Y.R. Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 2007, 14, 1017–1024. [Google Scholar] [CrossRef]
- Matzke, M.A.; Birchler, J.A. RNAi-mediated pathways in the nucleus. Nat. Rev. Genet. 2005, 6, 24–35. [Google Scholar] [CrossRef]
- Jin, C.; Zang, C.; Wei, G.; Cui, K.; Peng, W.; Zhao, K.; Felsenfeld, G. H3.3/H2A.Z double variant-containing nucleosomes mark ‘nucleosome-free regions’ of active promoters and other regulatory regions. Nat. Genet. 2009, 41, 941–945. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, M.; Matthies, H. Biochemical studies on histones of the central nervous system. III. Incorporation of [14C]-acetate into the histones of different rat brain regions during a learning experiment. Acta Biol. Med. Ger. 1979, 38, 683–689. [Google Scholar]
- McQuown, S.C.; Wood, M.A. HDAC3 and the Molecular Brake Pad Hypothesis. Neurobiol. Learn. Mem. 2012, 96, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Vecsey, C.G.; Hawk, J.D.; Lattal, K.M.; Stein, J.M.; Fabian, S.A.; Attner, M.A.; Cabrera, S.M.; McDonough, C.B.; Brindle, P.K.; Abel, T.; et al. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP-dependent transcriptional activation. J. Neurosci. 2007, 27, 6128–6140. [Google Scholar] [PubMed]
- McQuown, S.C.; Barrett, R.M.; Matheos, D.P.; Post, R.J.; Rogge, G.A.; Alenghat, T.; Mullican, S.E.; Jones, S.; Rusche, J.R.; Lazar, M.A.; et al. HDAC3 is a critical negative regulator of long-term memory formation. J. Neurosci. 2011, 31, 764–774. [Google Scholar] [CrossRef] [Green Version]
- Kwapis, J.L.; Alaghband, Y.; Kramár, E.A.; López, A.J.; Vogel Ciernia, A.; White, A.O.; Shu, G.; Rhee, D.; Michael, C.M.; Montellier, E.; et al. Epigenetic regulation of the circadian gene Per1 contributes to age-related changes in hippocampal memory. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kwapis, J.L.; Alaghband, Y.; López, A.J.; White, A.O.; Campbell, R.R.; Dang, R.T.; Rhee, D.; Tran, A.V.; Carl, A.E.; Matheos, D.P.; et al. Context and Auditory Fear are Differentially Regulated by HDAC3 Activity in the Lateral and Basal Subnuclei of the Amygdala. Neuropsychopharmacology 2017, 42, 1284–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, M.A.; Kaplan, M.P.; Park, A.; Blanchard, E.J.; Oliveira, A.M.M.; Lombardi, T.L.; Abel, T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn. Mem. 2005, 12, 111–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gräff, J.; Kaplan, M.P.; Park, A.; Blanchard, E.J.; Oliveira, A.M.M.; Lombardi, T.L.; Abel, T. Attenuate Remote Fear Memories. Cell 2015, 156, 261–276. [Google Scholar] [CrossRef] [Green Version]
- Levenson, J.M.; O'Riordan, K.J.; Brown, K.D.; Trinh, M.A.; Molfese, D.L.; Sweatt, J.D. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004, 279, 40545–40559. [Google Scholar] [CrossRef] [Green Version]
- Stefanko, D.P.; Barrett, R.M.; Ly, A.R.; Reolon, G.K.; Wood, M.A. Modulation of long-term memory for object recognition via HDAC inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 9447–9452. [Google Scholar] [CrossRef] [Green Version]
- Haettig, J.; Stefanko, D.P.; Multani, M.L.; Figueroa, D.X.; McQuown, S.C.; Wood, M.A. HDAC inhibition modulates hippocampus-dependent long-term memory for object location in a CBP-dependent manner. Learn. Mem. 2011, 18, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.-S.; Haggarty, S.J.; Giacometti, E.; Dannenberg, J.-H.; Joseph, N.; Gao, J.; Nieland, T.J.F.; Zhou, Y.; Wang, X.; Mazitschek, R.; et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 2009, 459, 55–60. [Google Scholar] [CrossRef]
- Malvaez, M.; Mhillaj, E.; Matheos, D.P.; Palmery, M.; Wood, M.A. CBP in the Nucleus Accumbens Regulates Cocaine-Induced Histone Acetylation and Is Critical for Cocaine-Associated Behaviors. J. Neurosci. 2011, 31, 16941–16948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieszczad, K.M.; Bechay, K.; Rusche, J.R.; Jacques, V.; Kudugunti, S.; Miao, W.; Weinberger, N.M.; McGaugh, J.L.; Wood, M.A. Histone Deacetylase Inhibition via RGFP966 Releases the Brakes on Sensory Cortical Plasticity and the Specificity of Memory Formation. J. Neurosci. 2015, 35, 13124–13132. [Google Scholar] [CrossRef] [Green Version]
- Barrett, R.M.; Malvaez, M.; Kramar, E.; Matheos, D.P.; Arrizon, A.; Cabrera, S.M.; Lynch, G.; Greene, R.W.; Wood, M.A. Hippocampal focal knockout of CBP affects specific histone modifications, long-term potentiation, and long-term memory. Neuropsychopharmacology 2011, 36, 1545–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maddox, S.A.; Watts, C.S.; Schafe, G.E. p300/CBP histone acetyltransferase activity is required for newly acquired and reactivated fear memories in the lateral amygdala. Learn. Mem. 2013, 20, 109–119. [Google Scholar] [CrossRef] [Green Version]
- Kerimoglu, C.; Agis-Balboa, R.C.; Kranz, A.; Stilling, R.; Bahari-Javan, S.; Benito-Garagorri, E.; Halder, R.; Burkhardt, S.; Stewart, A.F.; Fischer, A. Histone-methyltransferase mll2 (kmt2B) is required for memory formation in mice. J. Neurosci. 2013, 33, 3452–3464. [Google Scholar] [CrossRef] [Green Version]
- Gupta-Agarwal, S.; Franklin, A.V.; DeRamus, T.; Wheelock, M.; Davis, R.L.; McMahon, L.L.; Lubin, F.D. G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J. Neurosci. 2012, 32, 5440–5453. [Google Scholar] [CrossRef] [Green Version]
- Gupta-Agarwal, S.; Jarome, T.J.; Fernandez, J.; Lubin, F.D. NMDA receptor- and ERK-dependent histone methylation changes in the lateral amygdala bidirectionally regulate fear memory formation. Learn. Mem. 2014, 21, 351–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.S.; Nam, H.J.; Lee, J.; Kim, D.; Choi, J.E.; Kang, S.J.J.; Kim, S.; Kim, H.; Kwak, C.; Shim, K.W. PKCα-mediated phosphorylation of LSD1 is required for presynaptic plasticity and hippocampal learning and memory. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Telese, F.; Tan, Y.; Li, W.; Jin, C.; He, X.; Basnet, H.; Ma, Q.; Merkurjev, D.; Zhu, X.; et al. LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat. Neurosci. 2015, 18, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, S.; Zhou, Y.; Han, Y.; Li, S.; Xu, Q.; Xu, L.; Zhu, Z.; Deng, Y.; Yu, L.; et al. Phf8 histone demethylase deficiency causes cognitive impairments through the mTOR pathway. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rakhimova, A.; Wang, S.; Zhou, Y.; Han, Y.; Li, S.; Xu, Q.; Xu, L.; Zhu, Z.; Deng, Y.; Yu, L. Site-specific ADP-ribosylation of histone H2B in response to DNA double strand breaks. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Azad, G.K.; Ito, K.; Sailaja, B.S.; Biran, A.; Nissim-Rafinia, M.; Yamada, Y.; Brown, D.T.; Takizawa, T.; Meshorer, E. PARP1-dependent eviction of the linker histone H1 mediates immediate early gene expression during neuronal activation. J. Cell Biol. 2018, 217, 473–481. [Google Scholar] [CrossRef]
- Goldberg, S.; Visochek, L.; Giladi, E.; Gozes, I.; Cohen-Armon, M. PolyADP-ribosylation is required for long-term memory formation in mammals. J. Neurochem. 2009, 111, 72–79. [Google Scholar] [CrossRef]
- Brami-Cherrier, K. Parsing Molecular and Behavioral Effects of Cocaine in Mitogen- and Stress-Activated Protein Kinase-1-Deficient Mice. J. Neurosci. 2005, 25, 11444–11454. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, M.; Queisser, G.; Eder, A.; Wiegert, J.S.; Bengtson, C.P.; Hellwig, A.; Wittum, G.; Bading, H. Synaptic activity induces dramatic changes in the geometry of the cell nucleus: Interplay between nuclear structure, histone H3 phosphorylation, and nuclear calcium signaling. J. Neurosci. 2009, 29, 14687–14700. [Google Scholar] [CrossRef] [PubMed]
- Koshibu, K.; Graff, J.; Beullens, M.; Heitz, F.D.; Berchtold, D.; Russig, H.; Farinelli, M.; Bollen, M.; Mansuy, I.M. Protein Phosphatase 1 Regulates the Histone Code for Long-Term Memory. J. Neurosci. 2009, 29, 13079–13089. [Google Scholar] [CrossRef] [Green Version]
- Lo, W.S.; Duggan, L.; Emre, N.C.T.; Belotserkovskya, R.; Lane, W.S.; Shiekhattar, R.; Berger, S.L. Snf1—A histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science 2001, 293, 1142–1146. [Google Scholar] [CrossRef]
- Stilling, R.M.; Rönicke, R.; Benito, E.; Urbanke, H.; Capece, V.; Burkhardt, S.; BahariJavan, S.; Barth, J.; Sananbenesi, F.; Schütz, A.L.; et al. KLysine acetyltransferase 2a regulates a hippocampal gene expression network linked to memory formation. EMBO J. 2014, 33, 19121927. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Song, J.; Milne, T.A.; Wang, G.G.; Li, H.; Allis, C.D.; Patel, D.J. Pro isomerization in MLL1 PHD3-Bromo cassette connects H3K4me readout to CyP33 and HDAC-mediated repression. Cell 2010, 141, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Kuo, Y.M.; Andrews, A.J. Quantitating the Specificity and Selectivity of Gcn5-Mediated Acetylation of Histone H3. PLoS ONE 2013, 8, e54896. [Google Scholar]
- Voss, A.K.; Thomas, T. Histone Lysine and Genomic Targets of Histone Acetyltransferases in Mammals. BioEssays 2018, 40, 1–16. [Google Scholar] [CrossRef]
- Fischer, A. The Role of Dynamic Histone Modifications in Learning Behavior. In Behavioral Neurogenomics; Binder, E.B., Klengel, T., Eds.; Springer International Publishing: New York, NY, USA, 2019; pp. 127–157. [Google Scholar] [CrossRef]
- Herre, M.; Korb, E. The chromatin landscape of neuronal plasticity. Curr. Opin. Neurobiol. 2019, 59, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Tanner, K.G.; Cheung, W.L.; Sassone-Corsi, P.; Denu, J.M.; Allis, C.D. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell 2000, 5, 905–915. [Google Scholar] [CrossRef]
- Jin, Q.; Yu, L.R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.E.; Wang, C.; Brindle, P.K.; Dent, S.Y.R.; Ge, K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011, 30, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.-H.; Brownell, J.E.; Sobel, R.E.; Ranalli, R.A.; Cook, R.G.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Lett. Nat. 1996, 383, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, L.; Abel, T. The Role of Histone Acetylation in Memory Formation and Cognitive Impairments. Neuropsychopharmacology 2012, 38, 62–76. [Google Scholar] [CrossRef] [Green Version]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Johnson, C.A.; White, D.A.; Lavender, J.S.; O’Neill, L.P.; Turner, B.M. Human class I histone deacetylase complexes show enhanced catalytic activity in the presence of ATP and co-immunoprecipitate with the ATP-dependent chaperone protein Hsp70. J. Biol. Chem. 2002, 277, 9590–9597. [Google Scholar] [CrossRef] [Green Version]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [Green Version]
- Agger, K.; Christensen, J.; Cloos, P.A.; Helin, K. The emerging functions of histone demethylases. Curr. Opin. Genet. Dev. 2008, 18, 159–168. [Google Scholar] [CrossRef]
- Kerimoglu, C.; Sakib, M.S.; Jain, G.; Benito, E.; Burkhardt, S.; Capece, V.; Kaurani, L.; Halder, R.; Agís-Balboa, R.C.; Stilling, R.; et al. KMT2A and KMT2B Mediate Memory Function by Affecting Distinct Genomic Regions. Cell Rep. 2017, 20, 538–548. [Google Scholar] [CrossRef] [Green Version]
- Jakovcevski, M.; Ruan, H.; Shen, E.Y.; Dincer, A.; Javidfar, B.; Ma, Q.; Peter, C.J.; Cheung, I.; Mitchell, A.C.; Jiang, Y.; et al. Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and working memory. J. Neurosci. 2015, 35, 5097–5108. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, M.; Matsumura, Y.; Fukuda, M.; Kimura, H.; Shinkai, Y. G9a/GLP complexes independently mediate H3K9 and DNA methylation to silence transcription. EMBO J. 2008, 27, 2681–2690. [Google Scholar] [CrossRef]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [PubMed]
- Hargreaves, D.C.; Crabtree, G.R. ATP-dependent chromatin remodeling: Genetics, genomics and mechanisms. Cell Res. 2011, 21, 396–420. [Google Scholar] [PubMed]
- Côté, J.; Quinn, J.; Workman, J.L.; Peterson, C.L.; Cote, J.; Quinn, J.; Workman, J.L.; Peterson, C.L. Stimulation of GAL4 Derivative Binding to Nucleosomal DNA by the Yeast SWI SNF Complex. Science 2018, 265, 53–60. [Google Scholar]
- Goodwin, L.R.; Picketts, D.J. The role of ISWI chromatin remodeling complexes in brain development and neurodevelopmental disorders. Mol. Cell. Neurosci. 2018, 87, 55–64. [Google Scholar] [CrossRef]
- Sansom, S.N.; Griffiths, D.S.; Faedo, A.; Kleinjan, D.J.; Ruan, Y.; Smith, J.; Van Heyningen, V.; Rubenstein, J.L.; Livesey, F.J. The level of the transcription factor Pax6 is essential for controlling the balance between neural stem cell self-renewal and neurogenesis. PLoS Genet. 2009, 5, 20–23. [Google Scholar]
- Belgacem, Y.H.; Hamilton, A.M.; Shim, S.; Spencer, K.A.; Borodinsky, L.N. The many hats of Sonic hedgehog signaling in nervous system development and disease. J. Dev. Biol. 2016, 4, 35. [Google Scholar]
- Alvarez-Saavedra, M.; Lagali, P.; Yan, K.; Hashem, E.; Mears, A.; De Repentigny, Y.; Wallace, V.A.; Kothary, R.; Stopka, T.; Skoultchi, A.I.; et al. Coordinated epigenetic regulation of Engrailed-1 by the chromatin remodelers Smarca1 and Smarca5 mediates cerebellar morphogenesis. Epigenet. Chromatin 2013, 6, P105. [Google Scholar]
- Alvarez-Saavedra, M.; De Repentigny, Y.; Lagali, P.S.; Raghu Ram, E.V.S.; Yan, K.; Hashem, E.; Ivanochko, D.; Huh, M.S.; Yang, D.; Mears, A.J.; et al. Snf2h-mediated chromatin organization and histone H1 dynamics govern cerebellar morphogenesis and neural maturation. Nat. Commun. 2014, 5, 1–18. [Google Scholar]
- Karaca, E.; Harel, T.; Pehlivan, D.; Jhangiani, S.N.; Gambin, T.; Coban Akdemir, Z.; Gonzaga-Jauregui, C.; Erdin, S.; Bayram, Y.; Campbell, I.M.; et al. Genes that Affect Brain Structure and Function Identified by Rare Variant Analyses of Mendelian Neurologic Disease. Neuron 2015, 88, 499–513. [Google Scholar]
- Homann, O.R.; Misura, K.; Lamas, E.; Sandrock, R.W.; Nelson, P.; McDonough, S.I.; DeLisi, L.E. Whole-genome sequencing in multiplex families with psychoses reveals mutations in the SHANK2 and SMARCA1 genes segregating with illness. Mol. Psychiatry 2016, 21, 1690–1695. [Google Scholar]
- Wu, J.I.; Lessard, J.; Olave, I.A.; Qiu, Z.; Ghosh, A.; Graef, I.A.; Crabtree, G.R. Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 2007, 56, 94–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lessard, J.; Wu, J.I.; Ranish, J.A.; Wan, M.; Winslow, M.M.; Staahl, B.T.; Wu, H.; Aebersold, R.; Graef, I.A.; Crabtree, G.R. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 2007, 55, 201–215. [Google Scholar] [PubMed] [Green Version]
- Aizawa, H. Dendrite Development Regulated by CREST, a Calcium-Regulated Transcriptional Activator. Science 2004, 303, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesi, A.; Staahl, B.T.; Jovičić, A.; Couthouis, J.; Fasolino, M.; Raphael, A.R.; Yamazaki, T.; Elias, L.; Polak, M.; Kelly, C.; et al. Exome sequencing to identify de novo mutations in sporadic ALS trios. Nat. Neurosci. 2013, 16, 851–855. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.; Ghosh, A. A Calcium-Dependent Switch in a CREST-BRG1 Complex Regulates Activity-Dependent Gene Expression. Neuron 2008, 60, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, A.J.; Wood, M.A. Role of nucleosome remodeling in neurodevelopmental and intellectual disability disorders. Front. Behav. Neurosci. 2015, 9, 1–10. [Google Scholar]
- Wenderski, W.; Wang, L.; Krokhotin, A.; Walsh, J.J.; Li, H.; Shoji, H.; Ghosh, S.; George, R.D.; Miller, E.L.; Elias, L.; et al. Loss of the neural-specific BAF subunit ACTL6B relieves repression of early response genes and causes recessive autism. Proc. Natl. Acad. Sci. USA 2020, 117, 10055–10066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel-Ciernia, A.; Wood, M.A. Neuron-specific chromatin remodeling: A missing link in epigenetic mechanisms underlying synaptic plasticity, memory, and intellectual disability disorders. Neuropharmacology 2014, 80, 18–27. [Google Scholar] [CrossRef] [Green Version]
- White, A.O.; Kramár, E.A.; López, A.J.; Kwapis, J.L.; Doan, J.; Saldana, D.; Davatolhagh, M.F.; Alaghband, Y.; Blurton-Jones, M.; Matheos, D.P.; et al. BDNF rescues BAF53b-dependent synaptic plasticity and cocaine-associated memory in the nucleus accumbens. Nat. Commun. 2016, 7, 11725. [Google Scholar] [CrossRef] [Green Version]
- Vogel-Ciernia, A.; Matheos, D.P.; Barrett, R.M.; Kramár, E.A.; Azzawi, S.; Chen, Y.; Magnan, C.N.; Zeller, M.; Sylvain, A.; Haettig, J.; et al. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat. Neurosci. 2013, 16, 552–561. [Google Scholar]
- Ciernia, A.V.; Kramár, E.A.; Matheos, D.P.; Havekes, R.; Hemstedt, T.J.; Magnan, C.N.; Sakata, K.; Tran, A.; Azzawi, S.; Lopez, A.J.; et al. Mutation of neuron-specific chromatin remodeling subunit BAF53b: Rescue of plasticity and memory by manipulating actin remodeling. Learn. Mem. 2017, 24, 199–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppikofer, M.; Bai, T.; Gan, Y.; Haley, B.; Liu, P.; Sandoval, W.; Ciferri, C.; Cochran, A.G. Expansion of the ISWI chromatin remodeler family with new active complexes. EMBO Rep. 2017, 18, 1697–1706. [Google Scholar] [CrossRef] [PubMed]
- Bartholomew, B. Regulating the Chromatin Landscape: Structural and Mechanistic Perspectives. Annu. Rev. Biochem. 2014, 83, 671–696. [Google Scholar] [CrossRef] [Green Version]
- Flaus, A.; Owen-Hughes, T. Mechanisms for ATP-dependent chromatin remodelling: The means to the end. FEBS J. 2011, 278, 3579–3595. [Google Scholar] [CrossRef] [Green Version]
- Lazzaro, M.A.; Todd, M.A.; Lavigne, P.; Vallee, D.; De Maria, A.; Picketts, D.J. Characterization of novel isoforms and evaluation of SNF2L/SMARCA1 as a candidate gene for X-linked mental retardation in 12 families linked to Xq25-26. BMC Med. Genet. 2018, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Deuring, R.; Fanti, L.; Armstrong, J.A.; Sarte, M.; Papoulas, O.; Prestel, M.; Daubresse, G.; Verardo, M.; Moseley, S.L.; Berloco, M.; et al. The ISWI Chromatin-Remodeling Protein Is Required for Gene Expression and the Maintenance of Higher Order Chromatin Structure In Vivo. Mol. Cell 2000, 5, 355–365. [Google Scholar] [CrossRef]
- Boyer, L.A.; Latek, R.R.; Peterson, C.L. Opinion: The SANT domain: A unique histone-tail-binding module? Nat. Rev. Mol. Cell Biol. 2004, 5, 158–163. [Google Scholar] [CrossRef]
- Banting, G.S.; Barak, O.; Ames, T.M.; Burnham, A.C.; Kardel, M.D.; Cooch, N.S.; Davidson, C.E.; Godbout, R.; McDermid, H.E.; Shiekhattar, R. CECR2, a protein involved in neurulation, forms a novel chromatin remodeling complex with SNF2L. Hum. Mol. Genet. 2005, 14, 513–524. [Google Scholar] [CrossRef] [Green Version]
- De La Cruz, X.; Lois, S.; Sánchez-Molina, S.; Martínez-Balbás, M.A. Do protein motifs read the histone code? BioEssays 2005, 27, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Hendrich, B.; Guy, J.; Ramsahoye, B.; Wilson, V.A.; Bird, A. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 2001, 15, 710–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.B.; Zhang, Y. Mi2, an auto-antigen for dermatomyositis, is an ATP-dependent nucleosome remodeling factor. Nucleic Acids Res. 2001, 29, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
- Ahringer, J. NuRD and SIN3: Histone deacetylase complexes in development. Trends Genet. 2000, 16, 351–356. [Google Scholar] [CrossRef]
- Denslow, S.A.; Wade, P.A. The human Mi-2/NuRD complex and gene regulation. Oncogene 2007, 26, 5433–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, Y. The Expanding Mi-2/NuRD Complexes: A Schematic Glance. Proteom. Insights 2010, 3, 79–109. [Google Scholar] [CrossRef]
- Corona, D.F.V.; Tamkun, J.W. Multiple roles for ISWI in transcription, chromosome organization and DNA replication. Biochim. Biophys. Acta Gene Struct. Expr. 2004, 1677, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Narlikar, G.J.; Sundaramoorthy, R.; Owen-Hughes, T. Mechanisms and functions of ATP-dependent chromatin-remodeling enzymes. Cell 2013, 154, 490–503. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.H.; Hamana, N.; Nezu, J.I.; Shimane, M. A novel family of bromodomain genes. Genomics 2000, 63, 40–45. [Google Scholar] [CrossRef]
- White, A.O.; Wood, M.A. Does stress remove the HDAC brakes for the formation and persistence of long-term memory? Neurobiol. Learn. Mem. 2014, 112, 61–67. [Google Scholar] [CrossRef]
- Li, J.; Zhao, G.; Gao, X. Development of neurodevelopmental disorders: A regulatory mechanism involving bromodomain-containing proteins. J. Neurodev. Disord. 2013, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sartor, G.C.; Powell, S.K.; Brothers, S.P.; Wahlestedt, C. Epigenetic readers of lysine acetylation regulate cocaine-induced plasticity. J. Neurosci. 2015, 35, 15062–15072. [Google Scholar] [CrossRef] [Green Version]
- Phan, M.L.; Bieszczad, K.M. Sensory Cortical Plasticity Participates in the Epigenetic Regulation of Robust Memory Formation. Neural Plast. 2016. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Wang, L.; Krokhotin, A.; Walsh, J.J.; Li, H.; Shoji, H.; Ghosh, S.; George, R.D.; Miller, E.L.; Elias, L.; et al. ACF chromatin-remodeling complex mediates stress-induced depressive-like behavior. Nat. Med. 2015, 21, 1146–1153. [Google Scholar] [CrossRef] [Green Version]
- Yoo, M.; Barak, O.; Ames, T.M.; Burnham, A.C.; Kardel, M.D.; Cooch, N.S.; Davidson, C.E.; Godbout, R.; McDermid, H.E.; Shiekhattar, R.; et al. BAF53b, a neuron-specific nucleosome remodeling factor, is induced after learning and facilitates long-term memory consolidation. J. Neurosci. 2017, 37, 3686–3697. [Google Scholar] [CrossRef] [PubMed]
- Olave, I.A.; Reck-peterson, S.L.; Crabtree, G.R. Nuclear Actin and Actin-related proteins in chromatin remodeling. Annu. Rev. Biochem. 2002, 71, 755–781. [Google Scholar] [CrossRef]
- Park, J.; Wood, M.A.; Cole, M.D. BAF53 forms distinct nuclear complexes and functions as a critical c-Myc-interacting nuclear cofactor for oncogenic transformation. Mol. Cell. Biol. 2002, 22, 1307–1316. [Google Scholar] [CrossRef] [Green Version]
- Harata, M.; Oma, Y.; Mizuno, S.; Jiang, Y.W.; Stillman, D.J.; Wintersberger, U. The nuclear actin-related protein of Saccharomyces cerevisiae, Act3p/Arp4, interacts with core histones. Mol. Biol. Cell 1999, 10, 2595–2605. [Google Scholar] [CrossRef] [Green Version]
- Aizawa, H.; Kobayashi, M.; Tanaka, S.; Fukai, T.; Okamoto, H. Molecular characterization of the subnuclei in rat habenula. J. Comp. Neurol. 2012, 520, 4051–4066. [Google Scholar] [CrossRef]
- Roozendaal, B.; Hernandez, A.; Cabrera, S.M.; Hagewoud, R.; Malvaez, M.; Stefanko, D.P.; Haettig, J.; Wood, M.A. Membrane-associated glucocorticoid activity is necessary for modulation of long-term memory via chromatin modification. J. Neurosci. 2010, 30, 5037–5046. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.A.; Marshall, J.F. Molecular substrates for retrieval and reconsolidation of cocaine-associated contextual memory. Neuron 2005, 47, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Torregrossa, M.M.; Corlett, P.R.; Taylor, J.R. Aberrant learning and memory in addiction. Neurobiol. Learn. Mem. 2011, 96, 609–623. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.R.; Horger, B.A.; Taylor, J.R.; Horger, B.A. Enhanced responding for conditioned reward produced by intra-accumbens amphetamine is potentiated after cocaine sensitization. Psychopharmacology 1999, 142, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Di Chiara, G. Drug addiction as dopamine-dependent associative learning disorder. Eur. J. Pharmacol. 1999, 375, 13–30. [Google Scholar] [CrossRef]
- Childress, A.R.; Mozley, P.D.; McElgin, W.; Fitzgerald, J.; Reivich, M.; O’Brien, C.P. Limbic activation during cue-induced cocaine craving. Am. J. Psychiatry 1999, 156, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Childress, A.; Ehrman, R.; McLellan, A.T.; O’Brien, C. Conditioned craving and arousal in cocaine addiction: A preliminary report. NIDA Res. Monogr. Ser. 1988, 88, 74–80. [Google Scholar]
- Miller, C.A.; Marshall, J.F. Altered Fos expression in neural pathways underlying cue-elicited drug seeking in the rat. Eur. J. Neurosci. 2005, 21, 1385–1393. [Google Scholar]
- Wolf, M.E. Synaptic mechanisms underlying persistent cocaine craving. Nat. Rev. Neurosci. 2016, 17, 351–365. [Google Scholar]
- Kauer, J.A.; Malenka, R.C. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007, 8, 844–858. [Google Scholar]
- Shaham, Y.; Shalev, U.; Lu, L.; De Wit, H.; Stewart, J. The reinstatement model of drug relapse: History, methodology and major findings. Psychopharmacology 2003, 168, 3–20. [Google Scholar] [CrossRef]
- Murray, J.E.; Belin-Rauscent, A.; Simon, M.; Giuliano, C.; Benoit-Marand, M.; Everitt, B.J.; Belin, D. Basolateral and central amygdala differentially recruit and maintain dorsolateral striatumdependent cocaine-seeking habits. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Jordi, E.; Heiman, M.; Marion-Poll, L.; Guermonprez, P.; Cheng, S.K.; Nairn, A.C.; Greengard, P.; Girault, J.-A. Differential effects of cocaine on histone posttranslational modifications in identified populations of striatal neurons. Proc. Natl. Acad. Sci. USA 2013, 110, 9511–9516. [Google Scholar]
- Kumar, A.; Choi, K.H.; Renthal, W.; Tsankova, N.M.; Theobald, D.E.H.; Truong, H.T.; Russo, S.J.; LaPlant, Q.; Sasaki, T.S.; Whistler, K.N. Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 2005, 48, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Levine, A.A.; Guan, Z.; Barco, A.; Xu, S.; Kandel, E.R.; Schwartz, J.H. CREB-binding protein controls response to cocaine by acetylating histones at the fosB promoter in the mouse striatum. Proc. Natl. Acad. Sci. USA 2005, 102, 19186–19191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertran-Gonzalez, J.; Håkansson, K.; Borgkvist, A.; Irinopoulou, T.; Brami-Cherrier, K.; Usiello, A.; Greengard, P.; Hervé, D.; Girault, J.A.; Valjent, E.; et al. Histone H3 phosphorylation is under the opposite tonic control of dopamine D2 and adenosine A2A receptors in striatopallidal neurons. Neuropsychopharmacology 2009, 34, 1710–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchcock, L.N.; Raybuck, J.D.; Wood, M.A.; Lattal, K.M. Effects of a histone deacetylase 3 inhibitor on extinction and reinstatement of cocaine self-administration in rats. Psychopharmacology 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Romieu, P.; Host, L.; Gobaille, S.; Sandner, G.; Aunis, D.; Zwiller, J. Histone Deacetylase Inhibitors Decrease Cocaine But Not Sucrose Self-Administration in Rats. J. Neurosci. 2008, 28, 9342–9348. [Google Scholar] [CrossRef]
- Sun, J.; Wang, L.; Jiang, B.; Hui, B.; Lv, Z.; Ma, L. The effects of sodium butyrate, an inhibitor of histone deacetylase, on the cocaine- and sucrose-maintained self-administration in rats. Neurosci. Lett. 2008, 441, 72–76. [Google Scholar] [CrossRef]
- Malvaez, M.; Greenfield, V.Y.; Matheos, D.P.; Angelillis, N.A.; Murphy, M.D.; Kennedy, P.J.; Wood, M.A.; Wassum, K.M. Habits Are Negatively Regulated by Histone Deacetylase 3 in the Dorsal Striatum. Biol. Psychiatry 2018, 84, 383–392. [Google Scholar] [CrossRef]
- Maze, I.; Covingtoni, H.E.; Dietz, D.M.; Laplant, Q.; Renthal, W.; Russo, S.J.; Mechanic, M.; Mouzon, E.; Neve, R.L.; Haggarty, S.J. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 2010, 327, 213–216. [Google Scholar] [CrossRef] [Green Version]
- Anderson, E.M.; Larson, E.B.; Guzman, D.; Marie Wissman, A.; Neve, R.L.; Nestler, E.J.; Self, D.W. Overexpression of the histone dimethyltransferase G9a in nucleus accumbens shell increases cocaine self-administration, stress-induced reinstatement, and anxiety. J. Neurosci. 2018, 38, 803–813. [Google Scholar] [CrossRef]
- Anderson, E.M.; Sun, H.; Guzman, D.; Taniguchi, M.; Cowan, C.W.; Maze, I.; Nestler, E.J.; Self, D.W. Knockdown of the histone di-methyltransferase G9a in nucleus accumbens shell decreases cocaine self-administration, stress-induced reinstatement, and anxiety. Neuropsychopharmacology 2019, 44, 1370–1376. [Google Scholar] [CrossRef]
- Scherma, M.; Qvist, J.S.; Asok, A.; Huang, S.S.C.; Masia, P.; Deidda, M.; Wei, Y.B.; Soni, R.K.; Fratta, W.; Fadda, P.; et al. Cannabinoid exposure in rat adolescence reprograms the initial behavioral, molecular, and epigenetic response to cocaine. Proc. Natl. Acad. Sci. USA 2020, 117, 9991–10002. [Google Scholar] [CrossRef] [Green Version]
- Walker, D.M.; Cates, H.M.; Loh, Y.-H.E.; Purushothaman, I.; Ramakrishnan, A.; Cahill, K.M.; Lardner, C.K.; Godino, A.; Kronman, H.G.; Rabkin, J.; et al. Cocaine self-administration alters transcriptome-wide responses in the brain’s reward circuitry. Biol. Psychiatry 2018, 84, 867–880. [Google Scholar] [CrossRef]
- Sun, H.S.; Damez-Werno, D.M.; Scobie, K.N.; Shao, N.-Y.Y.; Dias, C.; Rabkin, J.; Wright, K.N.; Mouzon, E.; Kabbaj, M.; Neve, R.; et al. Regulation of BAZ1A and nucleosome positioning in the nucleus accumbens in response to cocaine. Neuroscience 2017, 353, 1–6. [Google Scholar] [CrossRef]
- Egervari, G.; Landry, J.; Callens, J.; Fullard, J.F.; Roussos, P.; Keller, E.; Hurd, Y.L. Striatal H3K27 Acetylation Linked to Glutamatergic Gene Dysregulation in Human Heroin Abusers Holds Promise as Therapeutic Target. Biol. Psychiatry 2017, 81, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Gancarz, A.M.; Wang, Z.J.; Schroeder, G.L.; Damez-Werno, D.; Braunscheidel, K.M.; Mueller, L.E.; Humby, M.S.; Caccamise, A.; Martin, J.A.; Dietz, K.C.; et al. Activin receptor signaling regulates cocaine-primed behavioral and morphological plasticity. Nat. Neurosci. 2015, 18, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Renthal, W.; Kumar, A.; Xiao, G.; Wilkinson, M.; Covington, H.E.; Maze, I.; Sikder, D.; Robison, A.J.; LaPlant, Q.; Dietz, D.M.; et al. Genome-wide Analysis of Chromatin Regulation by Cocaine Reveals a Role for Sirtuins. Neuron 2009, 62, 335–348. [Google Scholar] [CrossRef] [Green Version]
- McClung, C.A.; Nestler, E.J. Neuroplasticity mediated by altered gene expression. Neuropsychopharmacology 2008, 33, 3–17. [Google Scholar] [CrossRef]
- McClung, C.A.; Nestler, E.J. Regulation of gene expression and cocaine reward by CREB and ΔFosB. Nat. Neurosci. 2003, 6, 1208–1215. [Google Scholar] [CrossRef]
- Mathies, L.D.; Aliev, F.; Davies, A.G.; Dick, D.M.; Bettinger, J.C. Variation in SWI/SNF Chromatin Remodeling Complex Proteins is Associated with Alcohol Dependence and Antisocial Behavior in Human Populations. Alcohol. Clin. Exp. Res. 2017, 41, 2033–2040. [Google Scholar] [CrossRef] [PubMed]
- Burrowes, S.G.; Salem, N.A.; Tseng, A.M.; Balaraman, S.; Pinson, M.R.; Garcia, C.; Miranda, R.C. The BAF (BRG1/BRM-Associated Factor) chromatin-remodeling complex exhibits ethanol sensitivity in fetal neural progenitor cells and regulates transcription at the miR-9-2 encoding gene locus. Alcohol 2017, 60, 149–158. [Google Scholar] [CrossRef] [Green Version]
- Alaghband, Y.; Kwapis, X.J.L.; Kim, X.E.S.; Hemstedt, T.J.; Lo, A.J.; White, O.; Al-kachak, X.A.; Aimiuwu, X.O.V.; Bodinayake, K.K.; Oparaugo, N.C.; et al. CREST in the Nucleus Accumbens Core Regulates Cocaine Conditioned Place Preference, Cocaine-Seeking Behavior, and Synaptic Plasticity. J. Neurosci. 2018, 38, 9514–9526. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.J.; Martin, J.A.; Mueller, L.E.; Caccamise, A.; Werner, C.T.; Neve, R.L.; Gancarz, A.M.; Li, J.X.; Dietz, D.M. BRG1 in the Nucleus Accumbens Regulates Cocaine-Seeking Behavior. Biol. Psychiatry 2016, 80, 652–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, G.; Kramár, E.A.; López, A.J.; Huynh, G.; Wood, M.A.; Kwapis, J.L. Deleting HDAC3 rescues long-term memory impairments induced by disruption of the neuron-specific chromatin remodeling subunit BAF53b_AN022018. Learn. Mem. 2018, 25, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Green, J.; Das Neves, R.P.; Wallace, H.A.C.; Smith, A.J.H.; Hughes, J.; Gray, N.; Taylor, S.; Wood, W.G.; Higgs, D.R.; et al. Association between active genes occurs at nuclear speckles and is modulated by chromatin environment. J. Cell Biol. 2008, 182, 1083–1097. [Google Scholar] [CrossRef] [Green Version]
- Galganski, L.; Urbanek, M.O.; Krzyzosiak, W.J. Nuclear speckles: Molecular organization, biological function and role in disease. Nucleic Acids Res. 2017, 45, 10350–10368. [Google Scholar] [CrossRef] [Green Version]
- Ito, K.; Takizawa, T. Nuclear architecture in the nervous system: Development, function, and neurodevelopmental diseases. Front. Genet. 2018, 9, 1–8. [Google Scholar] [CrossRef]
| Histone Modifying Enzyme Class | Member | Target Residues | Effect on Long-Term Memory | References |
|---|---|---|---|---|
| Acetyltransferase | KAT2A (GCN5)/KAT2B (PCAF) CBP (KAT3A)/p300 (KAT3B) | H3: K9, K14, K18, K23 H4: K8, K12 H2A:K5; H2B: K12, K15; H3: K14; H4: K5, K8 | Permissive Permissive | [2,29,31,48,49,50,51,52,53,54,55] |
| Histone Deacetylase | HDAC1 | H2A: All; H2B: All; H3: All; H4: All | Repressive | [2,50,51,56,57,58] |
| HDAC2 | H2A: All; H2B: All; H3: All; H4: All | |||
| HDAC3 | H2A: All; H2B: All; H3: All; H4: All | |||
| HDAC4 | Enzymatically Non-Deacetylating | |||
| HDAC5 | Enzymatically Non-Deacetylating | |||
| HDAC6 | H4: K5, K8 | |||
| Lysine Methyltransferase | KMT1C (G9a) | H3: K9 | Both | [2,33,34,50,51,59,60,61,62] |
| KMT1D (GLP) | H3: K9 | Both | ||
| KMT2A (MII1) | H3: K4 | Permissive | ||
| KMT2B (MII2) | H3: K4 | Repressive | ||
| KMT6A (EZH2) | H3: K27 | Repressive | ||
| Lysine Demethylase | KDM1 | H3: K4, K9; H4: K20 | Permissive | [2,37,50,51,59] |
| KDM4B | H3: K4 K9 | Both | ||
| KDM5C | H3: K9 | Both | ||
| KDM6A | H3: K27 | Repressive |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
López, A.J.; Hecking, J.K.; White, A.O. The Emerging Role of ATP-Dependent Chromatin Remodeling in Memory and Substance Use Disorders. Int. J. Mol. Sci. 2020, 21, 6816. https://doi.org/10.3390/ijms21186816
López AJ, Hecking JK, White AO. The Emerging Role of ATP-Dependent Chromatin Remodeling in Memory and Substance Use Disorders. International Journal of Molecular Sciences. 2020; 21(18):6816. https://doi.org/10.3390/ijms21186816
Chicago/Turabian StyleLópez, Alberto J., Julia K. Hecking, and André O. White. 2020. "The Emerging Role of ATP-Dependent Chromatin Remodeling in Memory and Substance Use Disorders" International Journal of Molecular Sciences 21, no. 18: 6816. https://doi.org/10.3390/ijms21186816
APA StyleLópez, A. J., Hecking, J. K., & White, A. O. (2020). The Emerging Role of ATP-Dependent Chromatin Remodeling in Memory and Substance Use Disorders. International Journal of Molecular Sciences, 21(18), 6816. https://doi.org/10.3390/ijms21186816