Genome Wide Analysis Points towards Subtype-Specific Diseases in Different Genetic Forms of Amyotrophic Lateral Sclerosis




Abstract
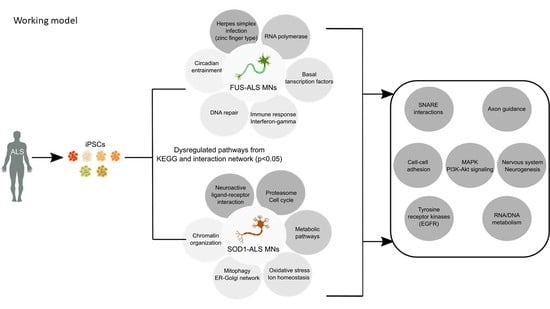
:
1. Introduction
2. Results
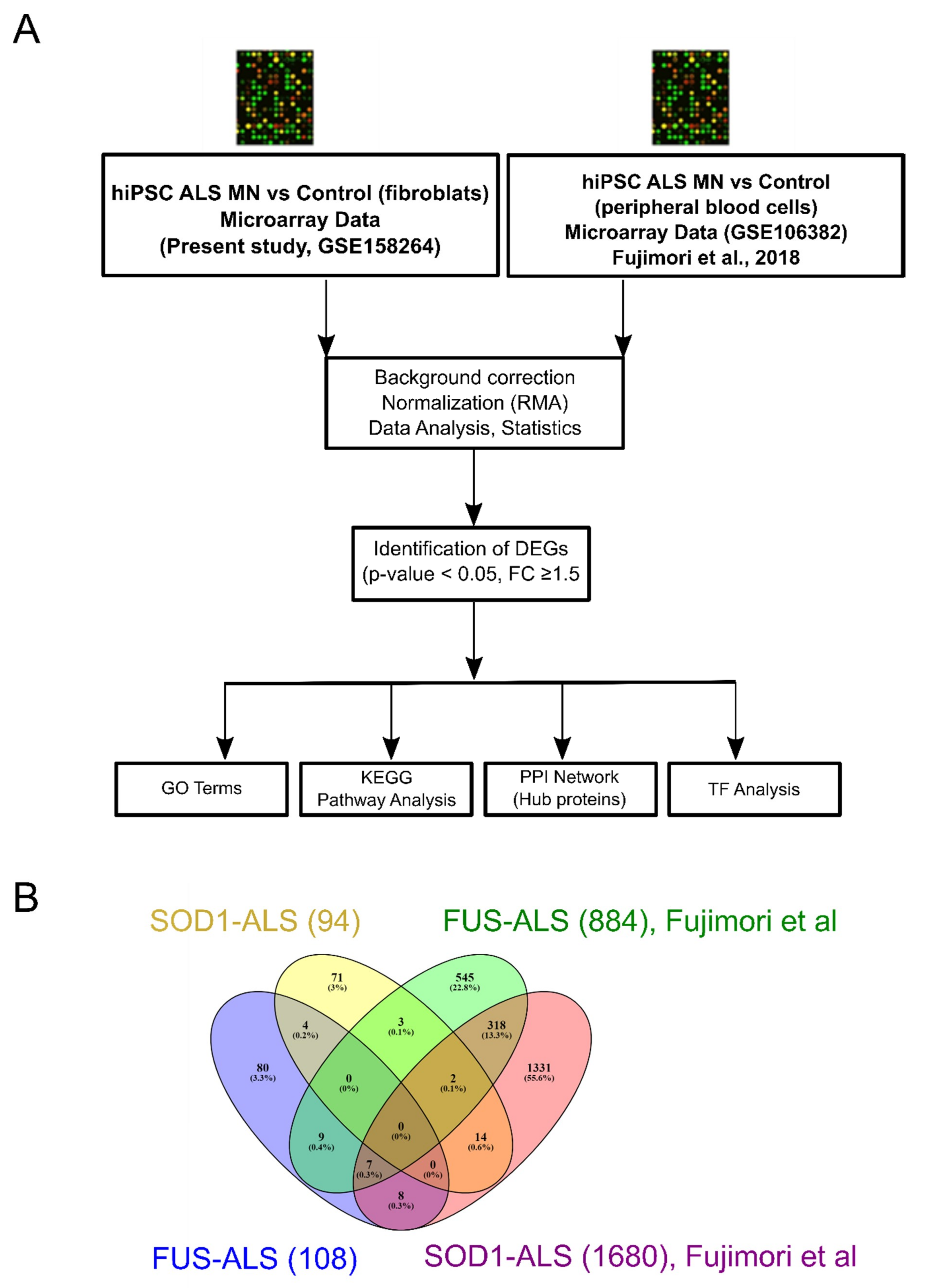
2.1. Analysis of Individual Datasets
2.2. Identification of DEGs between FUS- and SOD1-ALS iPSC MNs in Both Datasets
2.3. Gene Ontology (GO) and KEGG Pathway Enrichment Analysis of DEGs
2.4. DEGs-Transcription Factor Interaction Analysis
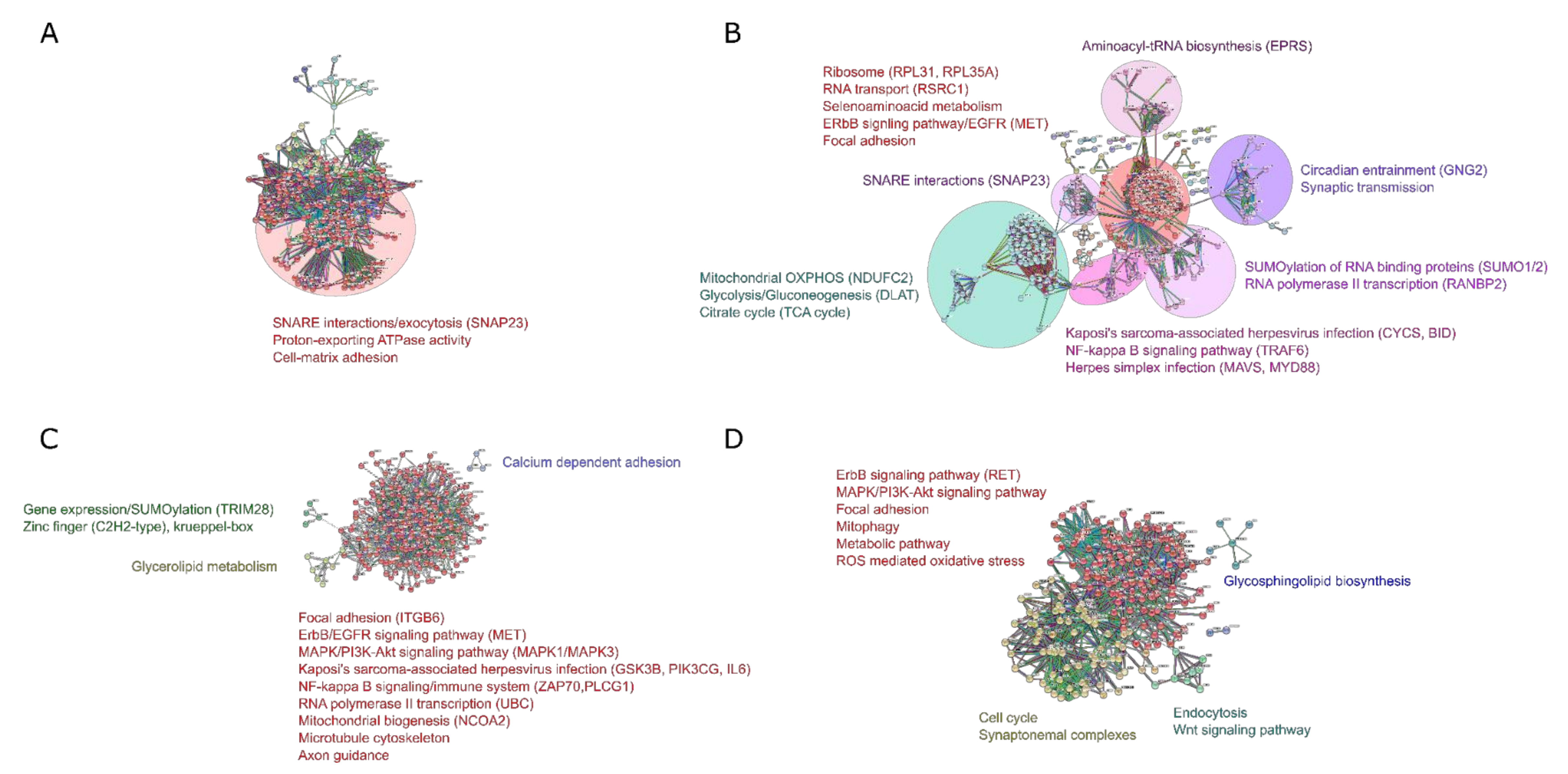
2.5. Protein–Protein Interactome (PPI) Analysis
3. Discussion
4. Materials and Methods
4.1. Patient Characteristics
4.2. Generation and Expansion of Cell Lines
4.3. RNA Isolation and Microarray Hybridization
4.4. Gene-Expression Validation Data Set
4.5. Data Analysis and Identification of DEGs
4.6. GO and Pathway Enrichment Analysis of DEGs
4.7. Protein–Protein Interaction Network and Hub Gene Analysis
4.8. Gene-Transcription Factor Interaction Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| fALS | Familial ALS |
| sALS | Sporadic ALS |
| MN | Motor neuron |
| FUS | Fused in sarcoma |
| SOD1 | Superoxide dismutase 1 |
| C9ORF72 | Chromosome 9 open reading frame 72 |
| TARDBP | TAR DNA-binding protein 43 |
| iPSC | human patient-derived induced pluripotent stem cell |
| NPC | Neural progenitor cells |
| MPC | Motor neuron precursor cell |
| DEG | Differentially expressed genes |
| GEO | Gene expression omnibus |
| DSBR | Double stranded break repair |
| ECM | Extracellular matrix |
| HSV | Herpes simplex virus |
| KEGG | Kyto encyclopedia of genes and genomes |
| STRING | The search tool for the retrieval of interacting genes/proteins |
| PGS | Partek genomic suite (Partek Inc) |
| GO | Gene ontology |
| ENCODE | Encyclopedia of DNA elements |
| ChEA | ChIP-X database |
| MAPK | Mitogen activated protein kinase |
| PPI | Protein-protein interactome analysis |
| PPARG | Peroxisome proliferator activated receptor gamma |
| PAR | poly-ADP-ribose |
| SNAP23 | Synaptosome associated protein 23 |
| SNARE | Soluble N-ethylmaleimide-sensitive factor attachment protein receptors |
| SMN | Survival motor neuron protein |
| SMA | Spinal muscular atrophy |
| CAM | Cell adhesion molecules |
| TOE1 | Target of EGR1 |
| EGFR | Early growth response protein |
| ZNF | Zinc finger proteins |
| KRAB | Kruppel-associated box |
| TF | Transcription factors |
| WT | Whole transcript |
| OXPHOS | Oxidative phosphorylation |
References
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Riva, N.; Agosta, F.; Lunetta, C.; Filippi, M.; Quattrini, A. Recent advances in amyotrophic lateral sclerosis. J. Neurol. 2016, 263, 1241–1254. [Google Scholar] [CrossRef] [Green Version]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 1–19. [Google Scholar] [CrossRef]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chió, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E. Incidence of amyotrophic lateral sclerosis in Europe. J. Neurol. Neurosurg. Psychiatry 2010, 81, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in als and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Beleza-Meireles, A.; Al-Chalabi, A. Genetic studies of amyotrophic lateral sclerosis: Controversies and perspectives. Amyotroph. Lateral Scler. 2009, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Brenner, D.; Weydt, P.; Meyer, T.; Grehl, T.; Petri, S.; Grosskreutz, J.; Schuster, J.; Volk, A.E.; Borck, G.; et al. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J. Neurol. Neurosurg. Psychiatry 2018, 89, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.C.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Sierra, H.; Cordova, M.; Chen, C.S.J.; Rajadhyaksha, M. Confocal imaging-guided laser ablation of basal cell carcinomas: An ex vivo study. J. Investig. Dermatol. 2015, 135, 612–615. [Google Scholar] [CrossRef] [Green Version]
- Bunney, P.E.; Zink, A.N.; Holm, A.A.; Billington, C.J.; Kotz, C.M. Orexin activation counteracts decreases in nonexercise activity thermogenesis (NEAT) caused by high-fat diet. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Paré, B.; Lehmann, M.; Beaudin, M.; Nordström, U.; Saikali, S.; Julien, J.P.; Gilthorpe, J.D.; Marklund, S.L.; Cashman, N.R.; Andersen, P.M.; et al. Misfolded SOD1 pathology in sporadic Amyotrophic Lateral Sclerosis. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Sama, R.R.K.; Ward, C.L.; Bosco, D.A. Functions of FUS/TLS from DNA repair to stress response: Implications for ALS. ASN Neuro 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- Rhoads, S.N.; Monahan, Z.T.; Yee, D.S.; Leung, A.Y.; Newcombe, C.G.; O’Meally, R.N.; Cole, R.N.; Shewmaker, F.P. The prionlike domain of FUS is multiphosphorylated following DNA damage without altering nuclear localization. Mol. Biol. Cell 2018, 29, 1786–1797. [Google Scholar] [CrossRef]
- Martinez-Macias, M.I.; Moore, D.A.Q.; Green, R.L.; Gomez-Herreros, F.; Naumann, M.; Hermann, A.; Van Damme, P.; Hafezparast, M.; Caldecott, K.W. FUS (fused in sarcoma) is a component of the cellular response to topoisomerase I–induced DNA breakage and transcriptional stress. Life Sci. Alliance 2019, 2, 1–14. [Google Scholar] [CrossRef]
- Vance, C.; Lehmann, R.; Broihier, H.T.; Moore, L.A.; Lehmann, R.; Lehmann, R.; Davey, J.; Nielsen, O.; Varshavsky, A.; Hamon, Y.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, T.J.; Bosco, D.A.; LeClerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.A.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Hulsen, T.; de Vlieg, J.; Alkema, W. BioVenn—A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 2008, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.C.; Feuer, R.; Cashman, N.; Luo, H. Enteroviral infection: The forgotten link to amyotrophic lateral sclerosis? Front. Mol. Neurosci. 2018, 11, 1–8. [Google Scholar] [CrossRef]
- Su, L.N.; Song, X.Q.; Xue, Z.X.; Zheng, C.Q.; Yin, H.F.; Wei, H.P. Network analysis of microRNAs, transcription factors, and target genes involved in axon regeneration. J. Zhejiang Univ. Sci. B 2018, 19, 293–304. [Google Scholar] [CrossRef]
- Shastri, A.; Bonifati, D.M.; Kishore, U. Innate immunity and neuroinflammation. Mediators Inflamm. 2013. [Google Scholar] [CrossRef] [PubMed]
- Farhadian, S.; Patel, P.; Spudich, S. Neurological Complications of HIV Infection. Curr. Infect. Dis. Rep. 2017, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, F. Role of neuroinflammation in amyotrophic lateral sclerosis: Cellular mechanisms and therapeutic implications. Front. Immunol. 2017. [Google Scholar] [CrossRef] [Green Version]
- Helmken, C.; Hofmann, Y.; Schoenen, F.; Oprea, G.; Raschke, H.; Rudnik-Schöneborn, S.; Zerres, K.; Wirth, B. Evidence for a modifying pathway in SMA discordant families: Reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum. Genet. 2003, 114, 11–21. [Google Scholar] [CrossRef]
- Zeitler, B.; Froelich, S.; Marlen, K.; Shivak, D.A.; Yu, Q.; Li, D.; Pearl, J.R.; Miller, J.C.; Zhang, L.; Paschon, D.E.; et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat. Med. 2019, 25, 1131–1142. [Google Scholar] [CrossRef]
- Xia, J.; Gill, E.E.; Hancock, R.E.W. NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc. 2015, 10, 823–844. [Google Scholar] [CrossRef]
- Myers, R.M.; Stamatoyannopoulos, J.; Snyder, M.; Dunham, I.; Hardison, R.C.; Bernstein, B.E.; Gingeras, T.R.; Kent, W.J.; Birney, E.; Wold, B.; et al. A user’s guide to the Encyclopedia of DNA elements (ENCODE). PLoS Biol. 2011, 9. [Google Scholar] [CrossRef]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.I.; Mazloom, A.R.; Ma’ayan, A. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; Van Der Lee, R.; Bessy, A.; Chèneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D260–D266. [Google Scholar] [CrossRef] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, S.; Fang, M.; Ni, B.; Lu, D.; Martin, B.; Maudsley, S. Central role of the EGF receptor in neurometabolic aging. Int. J. Endocrinol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrada, C.; Villalobo, A. Epidermal Growth Factor Receptor in the Adult Brain. Cell Cycle Cent. Nerv. Syst. 2008, 265–277. [Google Scholar] [CrossRef]
- Teixeira, A.L.; Gomes, M.; Medeiros, R. EGFR signaling pathway and related-miRNAs in age-related diseases: The example of miR-221 and miR-222. Front. Genet. 2012, 3, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Lai, Y.; Kim, S.; Varkey, J.; Lou, X.; Song, J.K.; Diao, J.; Langen, R.; Shin, Y.K. Nonaggregated α-synuclein influences snare-dependent vesicle docking via membrane binding. Biochemistry 2014, 53, 3889–3896. [Google Scholar] [CrossRef] [PubMed]
- Sibilia, M.; Steinbach, J.P.; Stingl, L.; Aguzzi, A.; Wagner, E.F. A strain-independent postnatal neurodegeneration in mice lacking the EGF receptor. EMBO J. 1998, 17, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.; Bullock, M.R.; Altememi, N.; Zhou, Z.; Hagood, S.; Rolfe, A.; McGinn, M.J.; Hamm, R.; Colello, R.J. The effect of epidermal growth factor in the injured brain after trauma in rats. J. Neurotrauma 2010, 27, 923–938. [Google Scholar] [CrossRef] [Green Version]
- Offen, D.; Barhum, Y.; Melamed, E.; Embacher, N.; Schindler, C.; Ransmayr, G. Spinal cord mRNA profile in patients with ALS: Comparison with transgenic mice expressing the human SOD-1 mutant. J. Mol. Neurosci. 2009, 38, 85–93. [Google Scholar] [CrossRef]
- Bellmann, J.; Monette, A.; Tripathy, V.; Sójka, A.; Abo-Rady, M.; Janosh, A.; Bhatnagar, R.; Bickle, M.; Mouland, A.J.; Sterneckert, J. Viral Infections Exacerbate FUS-ALS Phenotypes in iPSC-Derived Spinal Neurons in a Virus Species-Specific Manner. Front. Cell. Neurosci. 2019, 13, 1–18. [Google Scholar] [CrossRef]
- Park, J.H.; Park, H.S.; Hong, S.; Kang, S. Motor neurons derived from ALS-related mouse iPS cells recapitulate pathological features of ALS. Exp. Mol. Med. 2016, 48, e276. [Google Scholar] [CrossRef]
- Li, W.; Lee, M.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; von Geldern, G.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153. [Google Scholar] [CrossRef]
- Waldron, D. An endogenous retrovirus contributes to ALS. Nat. Rev. Microbiol. 2015, 13, 661. [Google Scholar] [CrossRef]
- Cermelli, C.; Vinceti, M.; Beretti, F.; Pietrini, V.; Nacci, G.; Pietrosemoli, P.; Bartoletti, A.; Guidetti, D.; Sola, P.; Bergomi, M.; et al. Risk of sporadic amyotrophic lateral sclerosis associated with seropositivity for herpesviruses and echovirus-7. Eur. J. Epidemiol. 2003, 18, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Sola, P.; Bedin, R.; Casoni, F.; Barozzi, P.; Mandrioli, J.; Merelli, E. New insights into the viral theory of amyotrophic lateral sclerosis: Study on the possible role of Kaposi’s sarcoma-associated virus/human herpesvirus 8. Eur. Neurol. 2002, 47, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Duarte, L.F.; Farías, M.A.; Álvarez, D.M.; Bueno, S.M.; Riedel, C.A.; González, P.A. Herpes simplex virus type 1 infection of the central nervous system: Insights into proposed interrelationships with neurodegenerative disorders. Front. Cell. Neurosci. 2019, 13, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunker, W.; Song, Y.; Zhao, Y.; Karijolich, J. FUS negatively regulates Kaposi’s sarcoma-associated herpesvirus gene expression. Viruses 2018, 10, 359. [Google Scholar] [CrossRef] [Green Version]
- Bowen, L.N.; Tyagi, R.; Li, W.; Alfahad, T.; Smith, B.; Wright, M.; Singer, E.J.; Nath, A. HIV-associated motor neuron disease: HERV-K activation and response to antiretroviral therapy. Neurology 2016, 87, 1756–1762. [Google Scholar] [CrossRef] [Green Version]
- Lehnardt, S. Innate immunity and neuroinflammation in the CNS: The role of microglia in toll-like receptor-mediated neuronal injury. Glia 2010, 58, 253–263. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, T.; Wang, H.; Wang, T.; Qin, M.; Bao, P.; Wang, R.; Liu, Y.; Chang, H.C.; Yan, J.; et al. Neurodegeneration-associated FUS is a novel regulator of circadian gene expression. Transl. Neurodegener. 2018, 7, 1–11. [Google Scholar] [CrossRef]
- Wang, R.; Jiang, X.; Bao, P.; Qin, M.; Xu, J. Circadian control of stress granules by oscillating EIF2α. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Celona, B.; Von Dollen, J.; Vatsavayai, S.C.; Kashima, R.; Johnson, J.R.; Tang, A.A.; Hata, A.; Miller, B.L.; Huang, E.J.; Krogan, N.J.; et al. Suppression of c9orf72 RNA repeat-induced neurotoxicity by the ALS-associated RNA-binding protein Zfp106. Elife 2017, 6, 1–17. [Google Scholar] [CrossRef]
- Naumann, M.; Peikert, K.; Günther, R.; van der Kooi, A.J.; Aronica, E.; Hübers, A.; Danel, V.; Corcia, P.; Pan-Montojo, F.; Cirak, S.; et al. Phenotypes and malignancy risk of different FUS mutations in genetic amyotrophic lateral sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 2384–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genestine, M.; Caricati, E.; Fico, A.; Richelme, S.; Hassani, H.; Sunyach, C.; Lamballe, F.; Panzica, G.C.; Pettmann, B.; Helmbacher, F.; et al. Enhanced neuronal Met signalling levels in ALS mice delay disease onset. Cell Death Dis. 2011, 2, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sama, R.R.K.; Fallini, C.; Gatto, R.; McKeon, J.E.; Song, Y.; Rotunno, M.S.; Penaranda, S.; Abdurakhmanov, I.; Landers, J.E.; Morfini, G.; et al. ALS-linked FUS exerts a gain of toxic function involving aberrant p38 MAPK activation. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Lunn, J.S.; Sakowski, S.A.; Kim, B.; Rosenberg, A.A.; Feldman, E.L. Induced Motor Neuron Degeneration. Dev. Neurobiol. 2009, 69, 871–884. [Google Scholar] [CrossRef] [Green Version]
- Tank, E.M.; Figueroa-Romero, C.; Hinder, L.M.; Bedi, K.; Archbold, H.C.; Li, X.; Weskamp, K.; Safren, N.; Paez-Colasante, X.; Pacut, C.; et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Togashi, H.; Sakisaka, T.; Takai, Y. Cell adhesion molecules in the central nervous system. Cell Adhes. Migr. 2009, 3, 29–35. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, M.; Sanes, J.R.; Weiner, J.A. Synaptic adhesion molecules. Curr. Opin. Cell Biol. 2003, 15, 621–632. [Google Scholar] [CrossRef]
- Washbourne, P.; Dityatev, A.; Scheiffele, P.; Biederer, T.; Weiner, J.A.; Christopherson, K.S.; El-Husseini, A. Cell adhesion molecules in synapse formation. J. Neurosci. 2004, 24, 9244–9249. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, K.L.; Kalmar, B.; Rhymes, E.R.; Fellows, A.D.; Ahmed, M.; Whiting, P.; Davies, C.H.; Greensmith, L.; Schiavo, G. Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS article. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Mhatrea, M.; Floyd, R.A.; Hensley, K. Oxidative stress and neuroinflammation in Alzheimer’s disease and amyotrophic lateral sclerosis: Common links and potential therapeutic targets. J. Alzheimer Dis. 2004, 6, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Imbeault, M.; Helleboid, P.Y.; Trono, D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017. [Google Scholar] [CrossRef]
- Herquel, B.; Ouararhni, K.; Khetchoumian, K.; Ignat, M.; Teletin, M.; Mark, M.; Béchade, G.; Van Dorsselaer, A.; Sanglier-Cianférani, S.; Hamiche, A.; et al. Transcription cofactors TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes that suppress murine hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valle-García, D.; Qadeer, Z.A.; McHugh, D.S.; Ghiraldini, F.G.; Chowdhury, A.H.; Hasson, D.; Dyer, M.A.; Recillas-Targa, F.; Bernstein, E. ATRX binds to atypical chromatin domains at the 3′ exons of zinc finger genes to preserve H3K9me3 enrichment. Epigenetics 2016, 11, 398–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wezyk, M.; Spólnicka, M.; Pośpiech, E.; Pepłońska, B.; Zbieć-Piekarska, R.; Ilkowski, J.; Styczyńska, M.; Barczak, A.; Zboch, M.; Filipek-Gliszczynska, A.; et al. Hypermethylation of TRIM59 and KLF14 influences cell death signaling in familial Alzheimer’s disease. Oxid. Med. Cell. Longev. 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Rousseaux, M.W.C.; Revelli, J.P.; Vázquez-Vełez, G.E.; Kim, J.Y.; Craigen, E.; Gonzales, K.; Beckinghausen, J.; Zoghbi, H.Y. Depleting trim28 in adult mice is well tolerated and reduces levels of α-synuclein and tau. Elife 2018, 7, 1–16. [Google Scholar] [CrossRef]
- Bunch, H. RNA polymerase II pausing and transcriptional regulation of the HSP70 expression. Eur. J. Cell Biol. 2017, 96, 739–745. [Google Scholar] [CrossRef]
- Grassi, D.A.; Jönsson, M.E.; Brattås, P.L.; Jakobsson, J. TRIM28 and the control of transposable elements in the brain. Brain Res. 2019, 1705, 43–47. [Google Scholar] [CrossRef]
- Sun, R.; Liang, D.; Gao, Y.; Lan, K. Kaposi’s Sarcoma-Associated Herpesvirus-Encoded LANA Interacts with Host KAP1 To Facilitate Establishment of Viral Latency. J. Virol. 2014, 88, 7331–7344. [Google Scholar] [CrossRef] [Green Version]
- Chien, H.C.; Wang, H.Y.; Su, Y.N.; Lai, K.Y.; Lu, L.C.; Chen, P.C.; Tsai, S.F.; Wu, C.I.; Hsieh, W.S.; Shen, C.K.J. Targeted Disruption in Mice of a Neural Stem Cell-Maintaining, KRAB-Zn Finger-Encoding Gene That Has Rapidly Evolved in the Human Lineage. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Tonchev, A.B.; Yamashima, T. “Transcribing” postischemic neurogenesis: A tale revealing hopes of adult brain repair. J. Mol. Med. 2007, 85, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, I.; Blackmore, M.G. Selecting optimal combinations of transcription factors to promote axon regeneration: Why mechanisms matter. Neurosci. Lett. 2017, 652, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.R.; Islam, T.; Zaman, T.; Shahjaman, M.; Karim, M.R.; Huq, F.; Quinn, J.M.W.; Holsinger, R.M.D.; Gov, E.; Moni, M.A. Identification of molecular signatures and pathways to identify novel therapeutic targets in Alzheimer’s disease: Insights from a systems biomedicine perspective. Genomics 2020, 112, 1290–1299. [Google Scholar] [CrossRef] [PubMed]
- Kiaei, M. Peroxisome proliferator-activated receptor-γ in amyotrophic lateral sclerosis and Huntington’s disease. PPAR Res. 2008, 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, R.; Bing, G. Agonism of Peroxisome Proliferator Receptor-Gamma may have Therapeutic Potential for Neuroinflammation and Parkinsons Disease. Curr. Neuropharmacol. 2007, 5, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Ceyzériat, K.; de Sauvage, M.A.C.; Aubry, F.; Auregan, G.; Guillermier, M.; Ruiz, M.; Petit, F.; Houitte, D.; Faivre, E.; et al. The JAK/STAT3 pathway is a common inducer of astrocyte reactivity in Alzheimer’s and Huntington’s diseases. J. Neurosci. 2015, 35, 2817–2829. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.-C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Deng, T.; Huang, Y.; Weng, K.; Lin, S.; Li, Y.; Shi, G.; Chen, Y.; Huang, J.; Liu, D.; Ma, W.; et al. TOE1 acts as a 3′ exonuclease for telomerase RNA and regulates telomere maintenance. Nucleic Acids Res. 2019, 47, 391–405. [Google Scholar] [CrossRef]
- Schütz, B.; Reimann, J.; Dumitrescu-Ozimek, L.; Kappes-Horn, K.; Landreth, G.E.; Schürmann, B.; Zimmer, A.; Heneka, M.T. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J. Neurosci. 2005, 25, 7805–7812. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Albertz, J.; Guo, Z.; Peng, Q.; Rudow, G.; Troncoso, J.C.; Ross, C.A.; Duan, W. Neuroprotective effects of PPAR-γ agonist rosiglitazone in N171-82Q mouse model of Huntington’s disease. J. Neurochem. 2013. [Google Scholar] [CrossRef]
- Hennig, S.; Kong, G.; Mannen, T.; Sadowska, A.; Kobelke, S.; Blythe, A.; Knott, G.J.; Iyer, S.S.; Ho, D.; Newcombe, E.A.; et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J. Cell Biol. 2015, 210, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Maldonado, M.A.; Bokov, A.F.; Majumder, S.; Oddo, S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 22687–22692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, R.H.; Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000, 14, 1553–1577. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.; Ko, S.Y.; Lee, I.Y.; Wang, S.E.; Lee, S.H.; Oh, D.H.; Kim, Y.S.; Son, H. Carbamylated erythropoietin promotes neurite outgrowth and neuronal spine formation in association with CBP/p300. Biochem. Biophys. Res. Commun. 2014, 446, 79–84. [Google Scholar] [CrossRef]
- Reinhardt, P.; Glatza, M.; Hemmer, K.; Tsytsyura, Y.; Thiel, C.S.; Höing, S.; Moritz, S.; Parga, J.A.; Wagner, L.; Bruder, J.M.; et al. Derivation and Expansion Using Only Small Molecules of Human Neural Progenitors for Neurodegenerative Disease Modeling. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Reinhardt, P.; Schmid, B.; Burbulla, L.F.; Schöndorf, D.C.; Wagner, L.; Glatza, M.; Höing, S.; Hargus, G.; Heck, S.A.; Dhingra, A.; et al. Genetic correction of a lrrk2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 2013, 12, 354–367. [Google Scholar] [CrossRef] [Green Version]
- Bursch, F.; Kalmbach, N.; Naujock, M.; Staege, S.; Eggenschwiler, R.; Abo-Rady, M.; Japtok, J.; Guo, W.; Hensel, N.; Reinhardt, P.; et al. Altered calcium dynamics and glutamate receptor properties in iPSC-derived motor neurons from ALS patients with C9orf72, FUS, SOD1 or TDP43 mutations. Hum. Mol. Genet. 2019, 28, 2835–2850. [Google Scholar] [CrossRef]
- Naujock, M.; Stanslowsky, N.; Bufler, S.; Naumann, M.; Reinhardt, P.; Sterneckert, J.; Kefalakes, E.; Kassebaum, C.; Bursch, F.; Lojewski, X.; et al. 4-Aminopyridine Induced Activity Rescues Hypoexcitable Motor Neurons from Amyotrophic Lateral Sclerosis Patient-Derived Induced. Stem Cells 2016, 34, 1563–1575. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Sel. Work. Terry Speed 2012, 601–616. [Google Scholar] [CrossRef] [Green Version]
- Oliveros, J.C. Venny. An Interactive Tool for Comparing Lists with Venn’s Diagrams. 2007. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 15 July 2020).
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brohée, S.; van Helden, J. Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinform. 2006, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Phenotype Type | Brain Region | Platform | Reference |
|---|---|---|---|---|
| GSE106382 | SOD1-ALS (versus Control) | MN | HG-U133_Plus_2 | [23] |
| GSM2836942 | SOD1-ALS-patient 1 (H46R-1SOD1-4) | MN | Affymetrix GeneChip Human HG_U133 Plus 2.0 | [23] |
| GSM2836943 | SOD1-ALS-patient 2 (H46R-2SOD1-4) | MN | Affymetrix GeneChip Human HG_U133 Plus 2.0 | [23] |
| GSM2836934 | Control | MN | Affymetrix GeneChip Human HG_U133 Plus 2.0 | [23] |
| GSM2836935 | Control | MN | Affymetrix GeneChip Human HG_U133 Plus 2.0 | [23] |
| GSM2836936 | Control | MN | Affymetrix GeneChip Human HG_U133 Plus 2.0 | [23] |
| GSM2836937 | Control | MN | Affymetrix GeneChip Human HG_U133 Plus 2.0 | [23] |
| GSM2836938 | FUS-ALS-patient 1 (H517D-FALS2e3)) | MN | Affymetrix GeneChip Human HG_U133 Plus 2.0 | [23] |
| GSM2836939 | FUS-ALS-patient 1 (H517D-FALS2e23) | MN | Affymetrix GeneChip Human HG_U133 Plus 2.0 | [23] |
| In this study; GSE158264 | FUS-, SOD1-ALS (versus Control) | Spinal MN | HuGene-2_0-st | |
| In this study; GSE158264 | FUS-ALS-patient 1 (R521L-clone 1) | Spinal MN | Affymetrix GeneChip Human Gene 2.0 ST Array | |
| In this study; GSE158264 | FUS-ALS-patient 1 (R521L-clone 2) | Spinal MN | Affymetrix GeneChip Human Gene 2.0 ST Array | |
| In this study; GSE158264 | FUS-ALS-patient 2 (R521C-clone 1) | Spinal MN | Affymetrix GeneChip Human Gene 2.0 ST Array | |
| In this study; GSE158264 | FUS-ALS-patient 2 (R521C-clone 2) | Spinal MN | Affymetrix GeneChip Human Gene 2.0 ST Array | |
| In this study; GSE158264 | SOD1-ALS-patient 1 (D90A-clone 1) | Spinal MN | Affymetrix GeneChip Human Gene 2.0 ST Array | |
| In this study; GSE158264 | SOD1-ALS-patient 1 (D90A-clone 2) | Spinal MN | Affymetrix GeneChip Human Gene 2.0 ST Array | |
| In this study; GSE158264 | SOD1-ALS-patient 2 (R115G-clone 1) | Spinal MN | Affymetrix GeneChip Human Gene 2.0 ST Array | |
| In this study; GSE158264 | Control 1 (proband 1-clone 1) | Spinal MN | Affymetrix GeneChip Human Gene 2.0 ST Array | |
| In this study; GSE158264 | Control 2 (proband 2-clone 1) | Spinal MN | Affymetrix GeneChip Human Gene 2.0 ST Array | |
| In this study; GSE158264 | Control 3 (proband 3-clone 1) | Spinal Mn | Affymetrix GeneChip Human Gene 2.0 ST Array | |
| In this study; GSE158264 | Control 4 (proband 2-clone 2) | Spinal MN | Affymetrix GeneChip Human Gene 2.0 ST Array |
| Genotype | Cell Culture Model | Sex | Age at Biopsy (Years) | Mutation | Family History | Age of Disease Onset | Clinical Phenotype | Disease Duration (Months) |
|---|---|---|---|---|---|---|---|---|
| Controls | hiPSC | |||||||
| female | 48 | - | - | - | - | - | ||
| male | 60 | - | - | - | - | - | ||
| female | 45 | - | - | - | - | - | ||
| female | 50 | - | - | - | - | - | ||
| FUS-ALS | hiPSC | |||||||
| female | 58 | p.R521C * | Pos. for ALS | 57 | spinal | 7 | ||
| female | 65 | p.R521L * | Pos. for ALS | 61 | spinal | 60 | ||
| SOD1-ALS | hiPSC | |||||||
| Male | 59 | p.R115G | Pos. for ALS | n.a | spinal | n.a | ||
| female | 46 | p.D90A * | Pos. for ALS | n.a | Spinal | n.a |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dash, B.P.; Naumann, M.; Sterneckert, J.; Hermann, A. Genome Wide Analysis Points towards Subtype-Specific Diseases in Different Genetic Forms of Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2020, 21, 6938. https://doi.org/10.3390/ijms21186938
Dash BP, Naumann M, Sterneckert J, Hermann A. Genome Wide Analysis Points towards Subtype-Specific Diseases in Different Genetic Forms of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2020; 21(18):6938. https://doi.org/10.3390/ijms21186938
Chicago/Turabian StyleDash, Banaja P., Marcel Naumann, Jared Sterneckert, and Andreas Hermann. 2020. "Genome Wide Analysis Points towards Subtype-Specific Diseases in Different Genetic Forms of Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 21, no. 18: 6938. https://doi.org/10.3390/ijms21186938
APA StyleDash, B. P., Naumann, M., Sterneckert, J., & Hermann, A. (2020). Genome Wide Analysis Points towards Subtype-Specific Diseases in Different Genetic Forms of Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 21(18), 6938. https://doi.org/10.3390/ijms21186938