Astaxanthin Suppresses PM2.5-Induced Neuroinflammation by Regulating Akt Phosphorylation in BV-2 Microglial Cells

Abstract
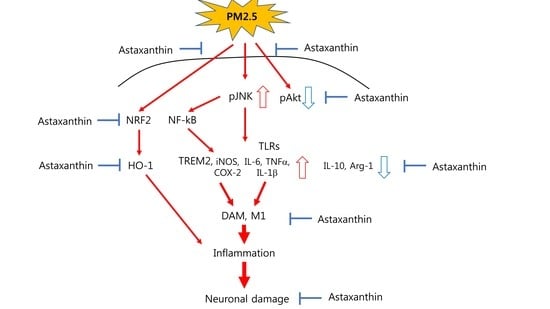
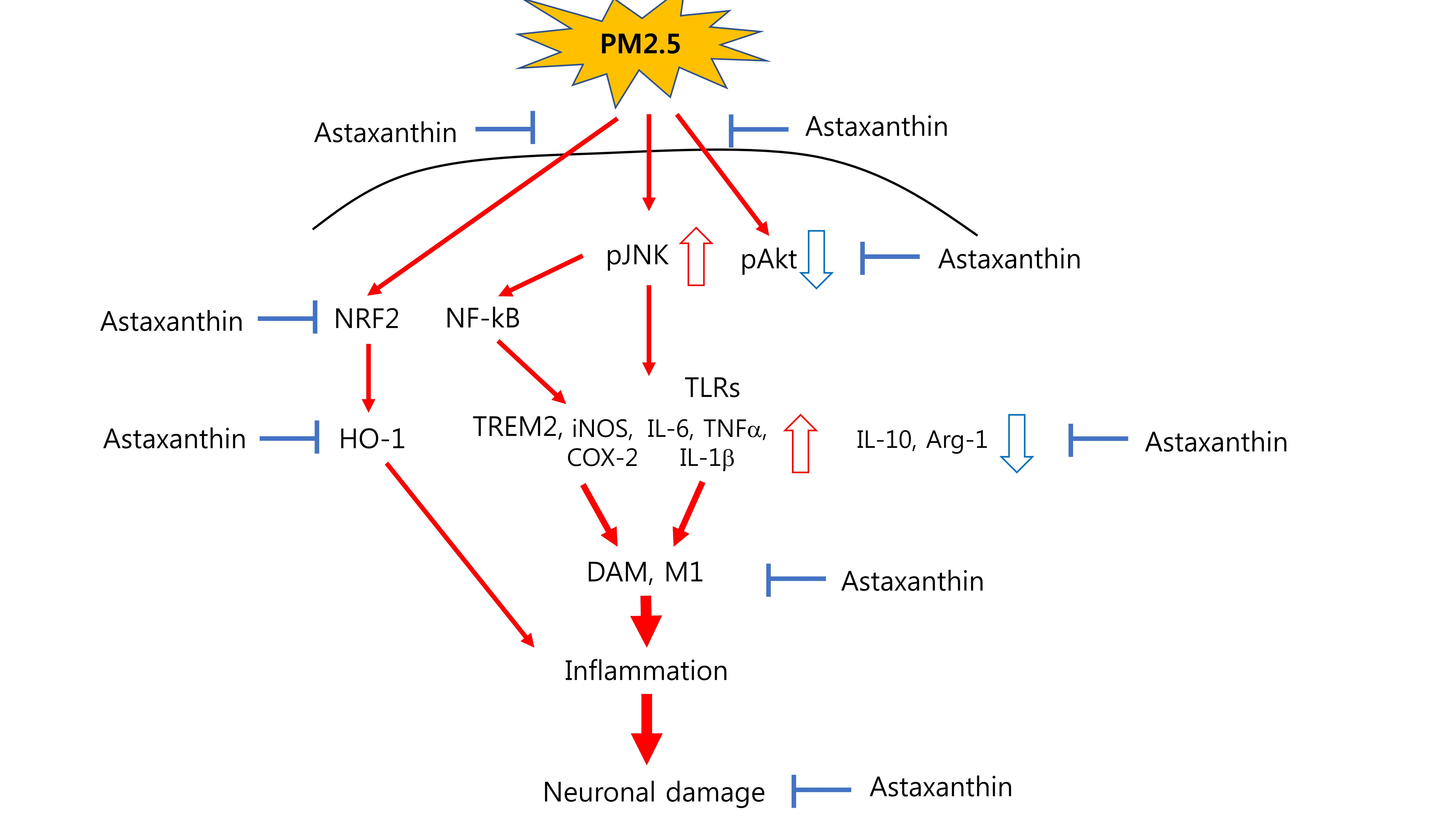
1. Introduction
2. Results
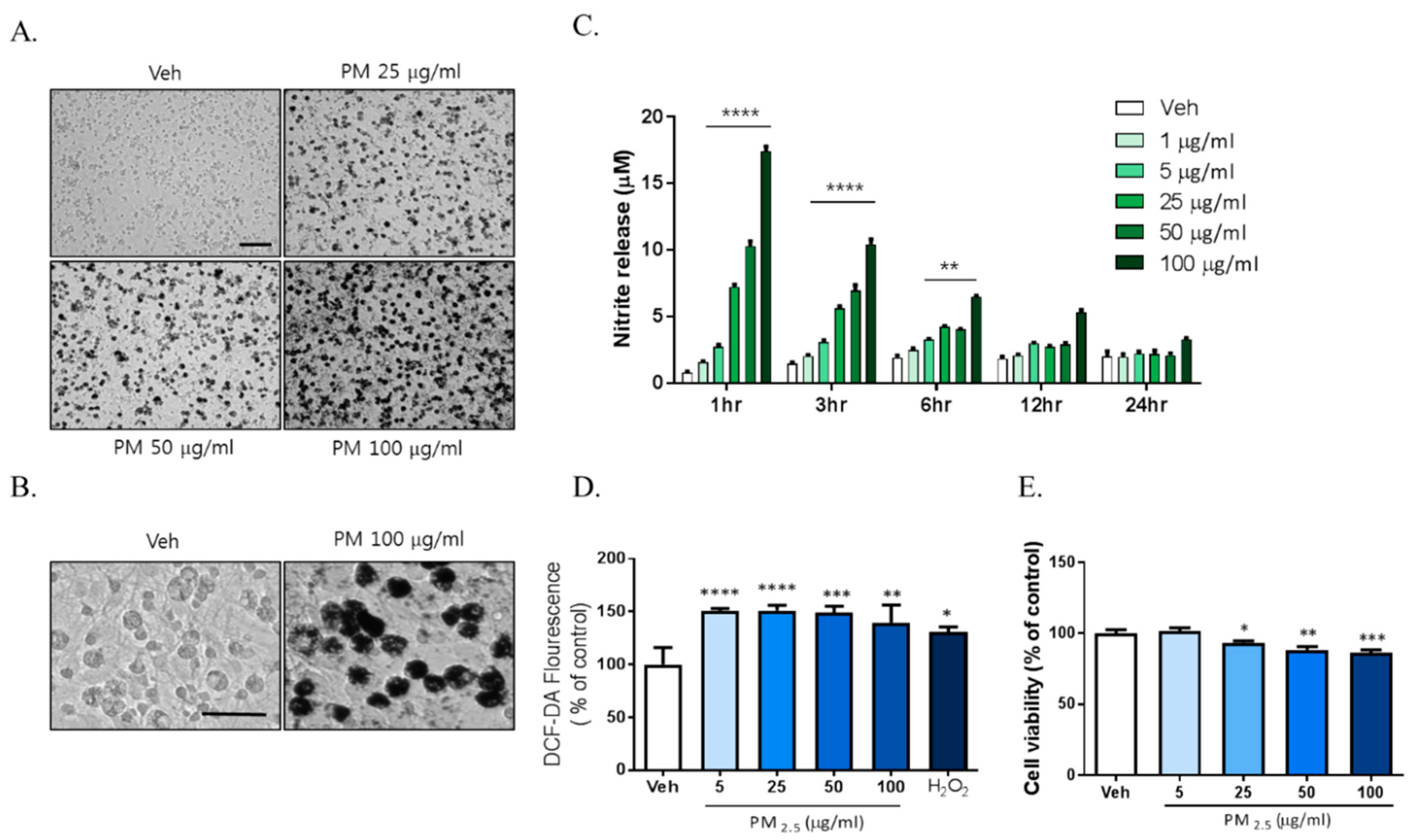
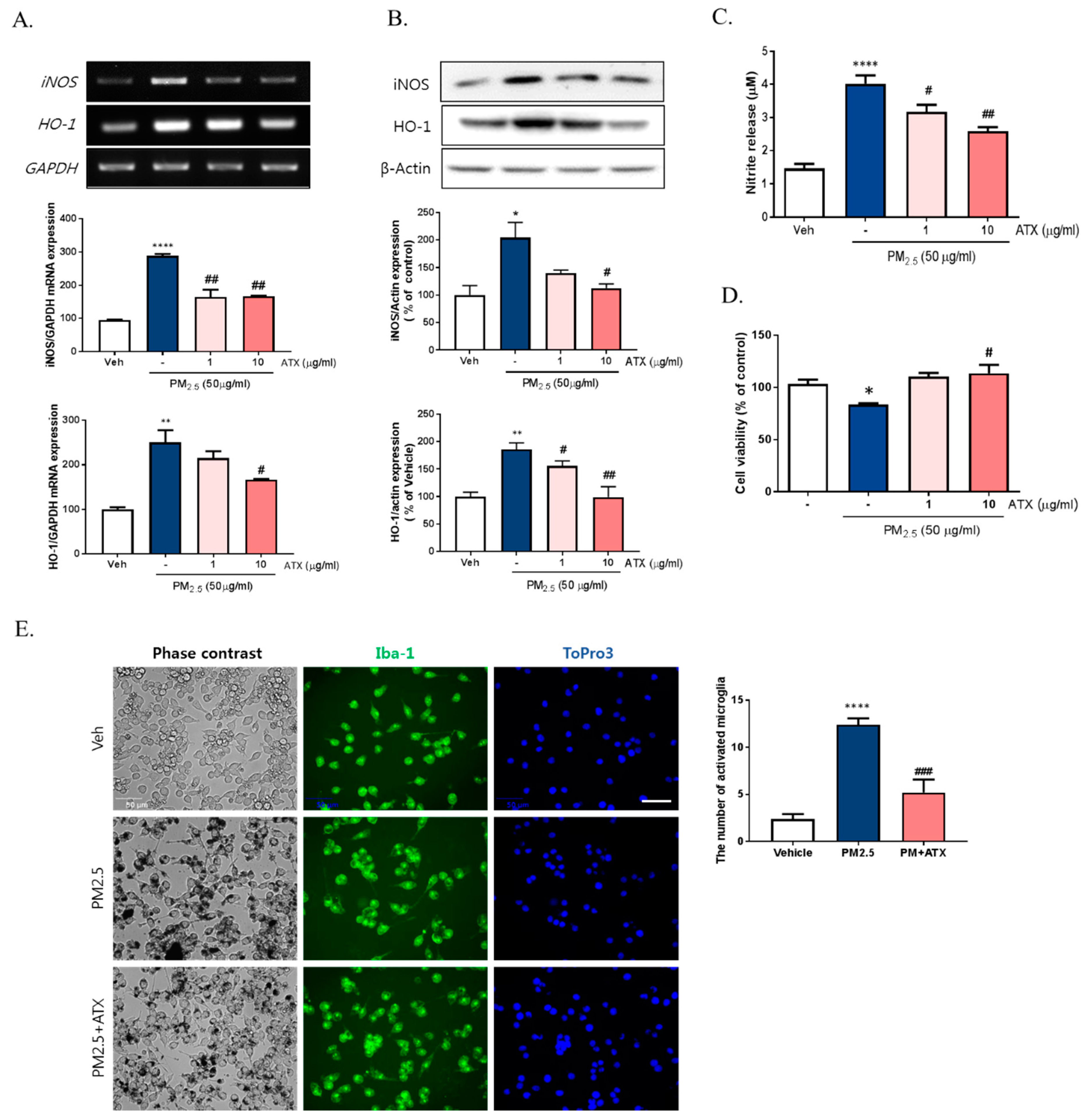
2.1. PM2.5 Increased Neuroinflammation via Microglial Activation
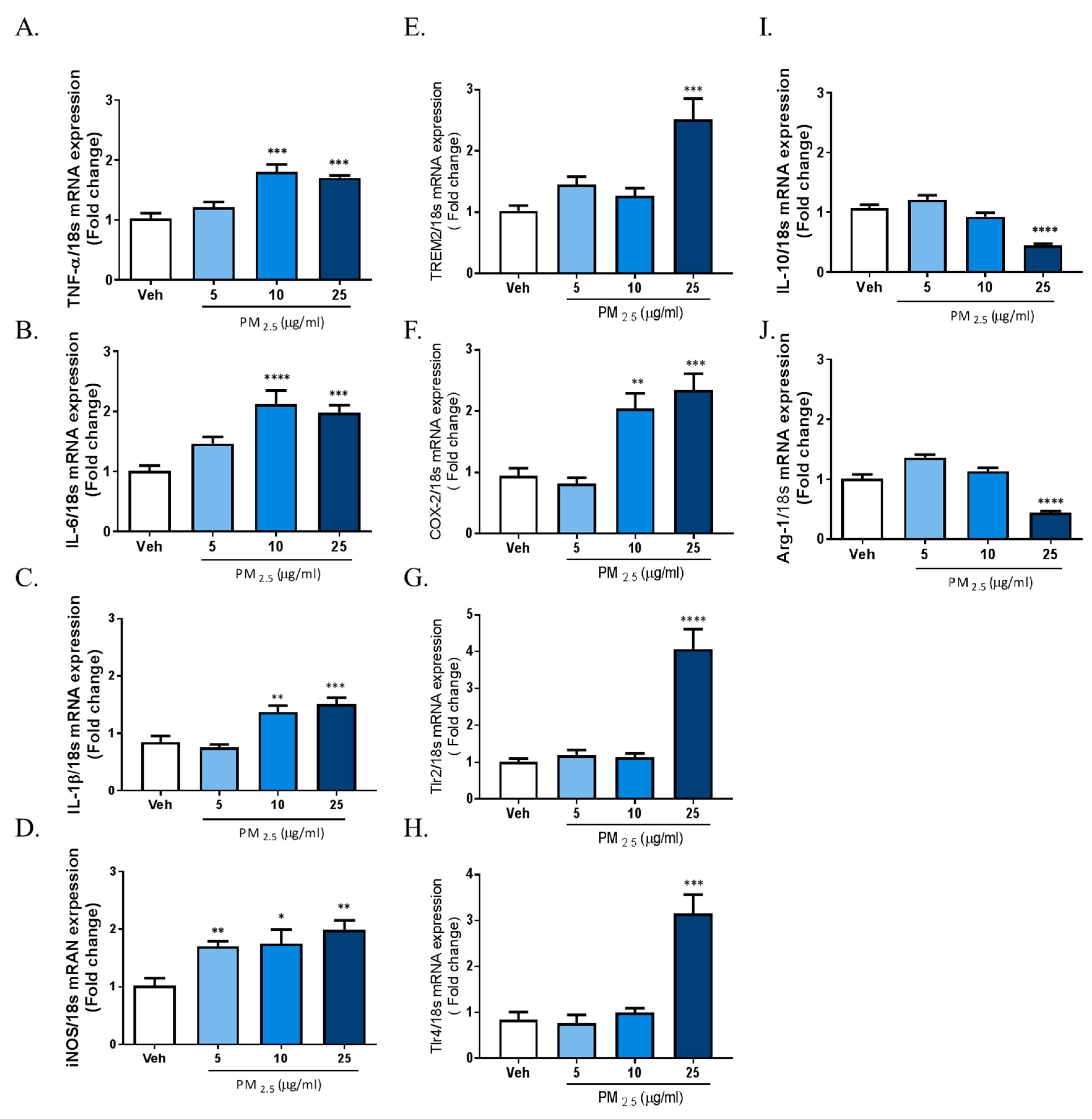
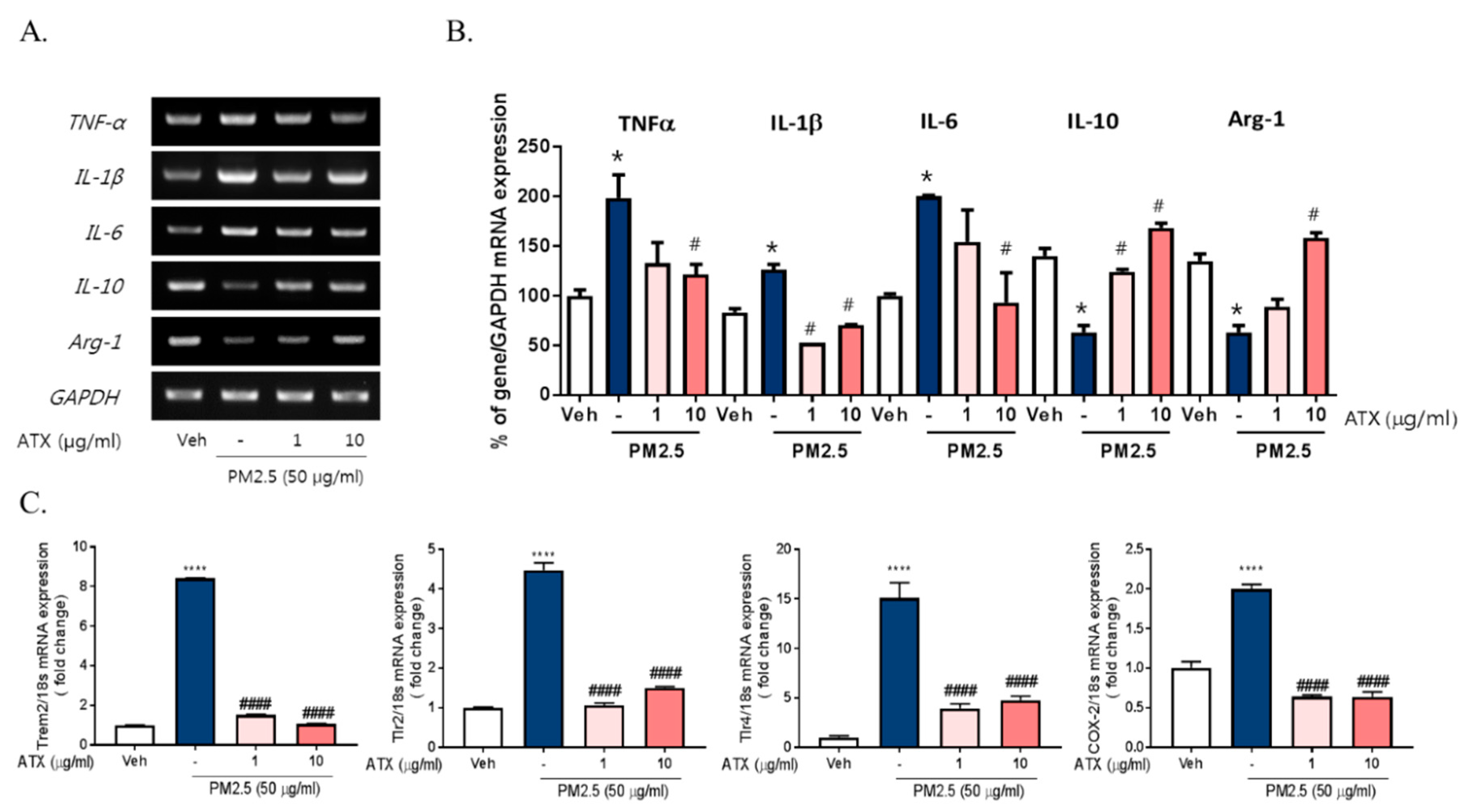
2.2. PM2.5 Increased the Transcription of Proinflammatory M1 and DAM Phenotype Molecules, and Decreased the Expression of Anti-Inflammatory Genes
2.3. ATX Treatment Inhibited PM2.5-Induced Neuroinflammation in BV-2 Microglial Cells
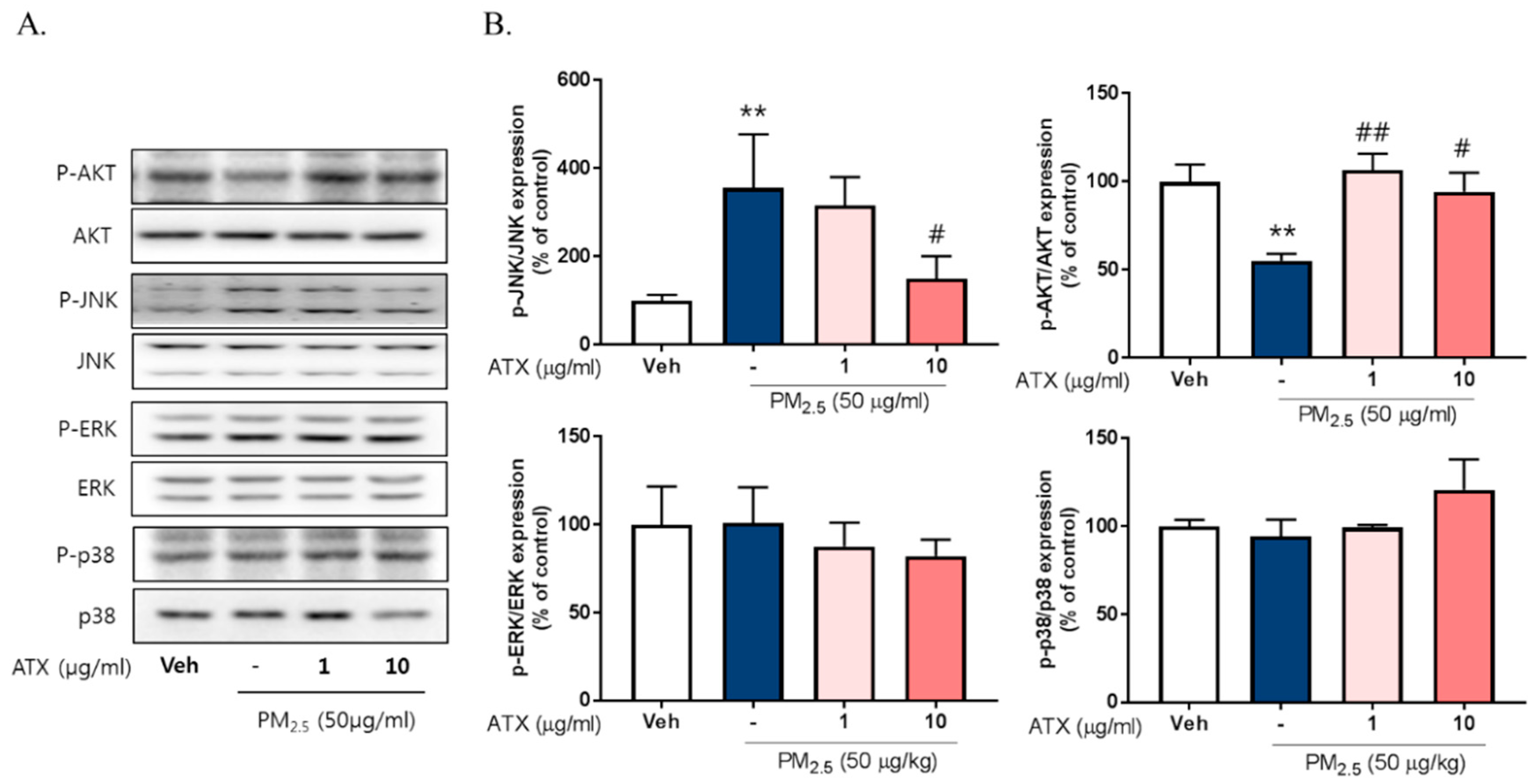
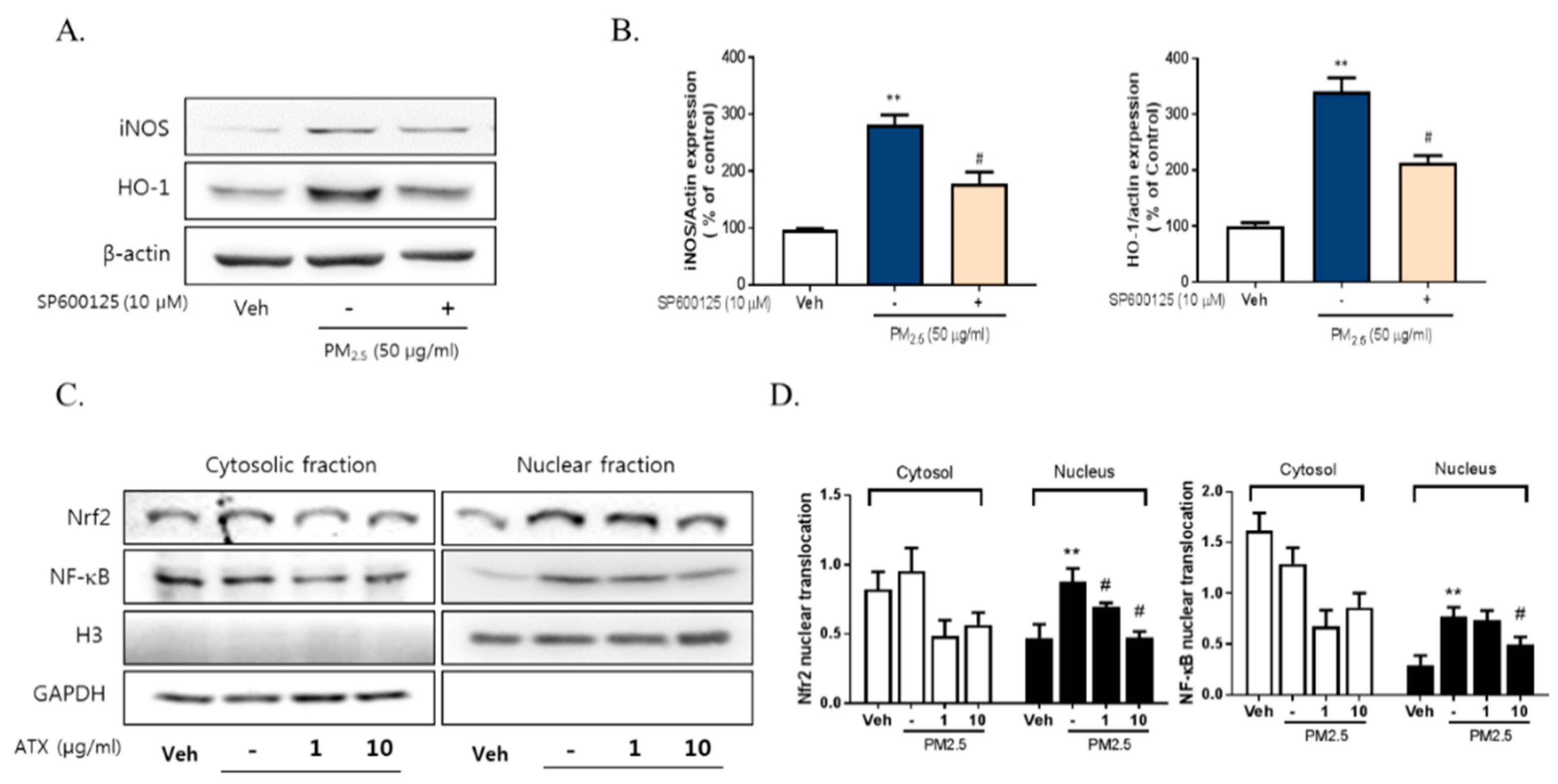
2.4. ATX Decreased PM2.5-Induced pJNK Activation and Increased Akt Phosphorylation
2.5. Astaxanthin Inhibited Neuronal Cell Death through PM2.5-Induced Microglial Activation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Drug Treatment
4.3. Determination of Nitrite Concentration
4.4. Measurement of Intracellular ROS
4.5. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.6. Western Blot Analysis
4.7. Measurement of Cell Viability
4.8. Immunocytochemistry
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shou, Y.; Huang, Y.; Zhu, X.; Liu, C.; Hu, Y.; Wang, H. A review of the possible associations between ambient PM2.5 exposures and the development of Alzheimer’s diseaseEcotoxicol. Environ. Saf. 2019, 174, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Calderon-Garciduenas, L. Air pollution: Mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 2009, 32, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Garciduenas, L.; Franco-Lira, M.; Henriquez-Roldan, C.; Osnaya, N.; Gonzalez-Maciel, A.; Reynoso-Robles, R.; Villarreal-Calderon, R.; Herritt, L.; Brooks, D.; Keefe, S.; et al. Urban air pollution: Influences on olfactory function and pathology in exposed children and young adults. Exp. Toxicol. Pathol. 2010, 62, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Graeber, M.B. Changing face of microglia. Science 2010, 330, 783–788. [Google Scholar] [CrossRef]
- Rangaraju, S.; Dammer, E.B.; Raza, S.A.; Rathakrishnan, P.; Xiao, H.; Gao, T.; Duong, D.M.; Pennington, M.W.; Lah, J.J.; Seyfried, N.T.; et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegener. 2018, 13, 24. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Remolina Serrano, J.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef]
- Bemiller, S.M.; McCray, T.J.; Allan, K.; Formica, S.V.; Xu, G.; Wilson, G.; Kokiko-Cochran, O.N.; Crish, S.D.; Lasagna-Reeves, C.A.; Ransohoff, R.M.; et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 2017, 12, 74. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef]
- Higuera-Ciapara, I.; Felix-Valenzuela, L.; Goycoolea, F.M. Astaxanthin: A review of its chemistry and applications. Crit. Rev. Food. Sci. Nutr. 2006, 46, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Hussein, G.; Sankawa, U.; Goto, H.; Matsumoto, K.; Watanabe, H. Astaxanthin, a carotenoid with potential in human health and nutrition. J. Nat. Prod. 2006, 69, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Farruggia, C.; Kim, M.B.; Bae, M.; Lee, Y.; Pham, T.X.; Yang, Y.; Han, M.J.; Park, Y.K.; Lee, J.Y. Astaxanthin exerts anti-inflammatory and antioxidant effects in macrophages in NRF2-dependent and independent manners. J. Nutr. Biochem. 2018, 62, 202–209. [Google Scholar] [CrossRef]
- Wu, H.; Niu, H.; Shao, A.; Wu, C.; Dixon, B.J.; Zhang, J.; Yang, S.; Wang, Y. Astaxanthin as a Potential Neuroprotective Agent for Neurological Diseases. Mar. Drugs 2015, 13, 5750–5766. [Google Scholar] [CrossRef]
- Sztretye, M.; Dienes, B.; Gonczi, M.; Czirjak, T.; Csernoch, L.; Dux, L.; Szentesi, P.; Keller-Pinter, A. Astaxanthin: A Potential Mitochondrial-Targeted Antioxidant Treatment in Diseases and with Aging. Oxid. Med. Cell. Longev. 2019, 2019, 3849692. [Google Scholar] [CrossRef]
- Kim, S.H.; Kim, H. Inhibitory Effect of Astaxanthin on Oxidative Stress-Induced Mitochondrial Dysfunction-A Mini-Review. Nutrients 2018, 10, 1137. [Google Scholar] [CrossRef]
- Ambati, R.R.; Phang, S.M.; Ravi, S.; Aswathanarayana, R.G. Astaxanthin: Sources, extraction, stability, biological activities and its commercial applications—A review. Mar. Drugs 2014, 12, 128–152. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Bai, S.K.; Lee, K.S.; Namkoong, S.; Na, H.J.; Ha, K.S.; Han, J.A.; Yim, S.V.; Chang, K.; Kwon, Y.G.; et al. Astaxanthin inhibits nitric oxide production and inflammatory gene expression by suppressing I(kappa)B kinase-dependent NF-kappaB activation. Moll. Cells 2003, 16, 97–105. [Google Scholar]
- Campoio, T.R.; Oliveira, F.A.; Otton, R. Oxidative stress in human lymphocytes treated with fatty acid mixture: Role of carotenoid astaxanthin. Toxicol. In Vitro 2011, 25, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.; Xuan, R.; Jiang, L.; Wu, W.; Zhen, Z.; Song, Y.; Hong, L.; Zheng, K.; Zhang, J.; Xu, Q.; et al. Astaxanthin Induces the Nrf2/HO-1 Antioxidant Pathway in Human Umbilical Vein Endothelial Cells by Generating Trace Amounts of ROS. J. Agric. Food Chem. 2018, 66, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Haghani, A.; Johnson, R.; Safi, N.; Zhang, H.; Thorwald, M.; Mousavi, A.; Woodward, N.C.; Shirmohammadi, F.; Coussa, V.; Wise, J.P.; et al. Toxicity of urban air pollution particulate matter in developing and adult mouse brain: Comparison of total and filter-eluted nanoparticles. Environ. Int. 2020, 136, 105510. [Google Scholar] [CrossRef]
- Peterson, B.S.; Rauh, V.A.; Bansal, R.; Hao, X.; Toth, Z.; Nati, G.; Walsh, K.; Miller, R.L.; Arias, F.; Semanek, D.; et al. Effects of prenatal exposure to air pollutants (polycyclic aromatic hydrocarbons) on the development of brain white matter, cognition, and behavior in later childhood. JAMA Psychiatry 2015, 72, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Peters, R.; Ee, N.; Peters, J.; Booth, A.; Mudway, I.; Anstey, K.J. Air Pollution and Dementia: A Systematic Review. J. Alzheimers Dis. 2019, 70, S145–S163. [Google Scholar] [CrossRef]
- Power, M.C.; Adar, S.D.; Yanosky, J.D.; Weuve, J. Exposure to air pollution as a potential contributor to cognitive function, cognitive decline, brain imaging, and dementia: A systematic review of epidemiologic research. Neurotoxicology 2016, 56, 235–253. [Google Scholar] [CrossRef]
- Calderon-Garciduenas, L.; Reynoso-Robles, R.; Gonzalez-Maciel, A. Combustion and friction-derived nanoparticles and industrial-sourced nanoparticles: The culprit of Alzheimer and Parkinson’s diseases. Environ. Res. 2019, 176, 108574. [Google Scholar] [CrossRef]
- Maher, B.A.; Ahmed, I.A.; Karloukovski, V.; MacLaren, D.A.; Foulds, P.G.; Allsop, D.; Mann, D.M.; Torres-Jardon, R.; Calderon-Garciduenas, L. Magnetite pollution nanoparticles in the human brain. Proc. Natl. Acad. Sci. USA 2016, 113, 10797–10801. [Google Scholar] [CrossRef]
- Cole, T.B.; Coburn, J.; Dao, K.; Roque, P.; Chang, Y.C.; Kalia, V.; Guilarte, T.R.; Dziedzic, J.; Costa, L.G. Sex and genetic differences in the effects of acute diesel exhaust exposure on inflammation and oxidative stress in mouse brain. Toxicology 2016, 374, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Woodward, N.C.; Levine, M.C.; Haghani, A.; Shirmohammadi, F.; Saffari, A.; Sioutas, C.; Morgan, T.E.; Finch, C.E. Toll-like receptor 4 in glial inflammatory responses to air pollution in vitro and in vivo. J. Neuroinflamm. 2017, 14, 84. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.L.; Liu, X.; Pelkowski, S.; Palmer, B.; Conrad, K.; Oberdorster, G.; Weston, D.; Mayer-Proschel, M.; Cory-Slechta, D.A. Early postnatal exposure to ultrafine particulate matter air pollution: Persistent ventriculomegaly, neurochemical disruption, and glial activation preferentially in male mice. Environ. Health Perspect. 2014, 122, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Levesque, S.; Surace, M.J.; McDonald, J.; Block, M.L. Air pollution & the brain: Subchronic diesel exhaust exposure causes neuroinflammation and elevates early markers of neurodegenerative disease. J. Neuroinflamm. 2011, 8, 105. [Google Scholar]
- Cacciottolo, M.; Wang, X.; Driscoll, I.; Woodward, N.; Saffari, A.; Reyes, J.; Serre, M.L.; Vizuete, W.; Sioutas, C.; Morgan, T.E.; et al. Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models. Transl. Psychiatry 2017, 7, e1022. [Google Scholar] [CrossRef]
- Morgan, T.E.; Davis, D.A.; Iwata, N.; Tanner, J.A.; Snyder, D.; Ning, Z.; Kam, W.; Hsu, Y.T.; Winkler, J.W.; Chen, J.C.; et al. Glutamatergic neurons in rodent models respond to nanoscale particulate urban air pollutants in vivo and in vitro. Environ. Health Perspect. 2011, 119, 1003–1009. [Google Scholar] [CrossRef]
- Bolton, J.L.; Smith, S.H.; Huff, N.C.; Gilmour, M.I.; Foster, W.M.; Auten, R.L.; Bilbo, S.D. Prenatal air pollution exposure induces neuroinflammation and predisposes offspring to weight gain in adulthood in a sex-specific manner. FASEB J. 2012, 26, 4743–4754. [Google Scholar] [CrossRef]
- Klocke, C.; Allen, J.L.; Sobolewski, M.; Blum, J.L.; Zelikoff, J.T.; Cory-Slechta, D.A. Exposure to fine and ultrafine particulate matter during gestation alters postnatal oligodendrocyte maturation, proliferation capacity, and myelination. Neurotoxicology 2018, 65, 196–206. [Google Scholar] [CrossRef]
- Woodward, N.C.; Haghani, A.; Johnson, R.G.; Hsu, T.M.; Saffari, A.; Sioutas, C.; Kanoski, S.E.; Finch, C.E.; Morgan, T.E. Prenatal and early life exposure to air pollution induced hippocampal vascular leakage and impaired neurogenesis in association with behavioral deficits. Transl. Psychiatry 2018, 8, 261. [Google Scholar] [CrossRef]
- Davis, D.A.; Bortolato, M.; Godar, S.C.; Sander, T.K.; Iwata, N.; Pakbin, P.; Shih, J.C.; Berhane, K.; McConnell, R.; Sioutas, C.; et al. Prenatal exposure to urban air nanoparticles in mice causes altered neuronal differentiation and depression-like responses. PLoS ONE 2013, 8, e64128. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Block, M.L.; Wu, X.; Pei, Z.; Li, G.; Wang, T.; Qin, L.; Wilson, B.; Yang, J.; Hong, J.S.; Veronesi, B. Nanometer size diesel exhaust particles are selectively toxic to dopaminergic neurons: The role of microglia, phagocytosis, and NADPH oxidase. FASEB J. 2004, 18, 1618–1620. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.Y.; Ma, J.K. The dual effect of the particulate and organic components of diesel exhaust particles on the alteration of pulmonary immune/inflammatory responses and metabolic enzymes. J. Environ. Sci. Health Part C. Environ. Carcinog. Ecotoxicol. Rev. 2002, 20, 117–147. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Liao, J.; Hu, Y.; Wang, Y.; Sun, C.; Zhang, C.; Wang, G. PM2.5 Upregulates MicroRNA-146a-3p and Induces M1 Polarization in RAW264.7 Cells by Targeting Sirtuin1. Int. J. Mol. Sci. 2019, 16, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Jaguin, M.; Fardel, O.; Lecureur, V. Exposure to diesel exhaust particle extracts (DEPe) impairs some polarization markers and functions of human macrophages through activation of AhR and Nrf2. PLoS ONE 2015, 10, e0116560. [Google Scholar] [CrossRef]
- Garcia-Revilla, J.; Alonso-Bellido, I.M.; Burguillos, M.A.; Herrera, A.J.; Espinosa-Oliva, A.M.; Ruiz, R.; Cruz-Hernandez, L.; Garcia-Dominguez, I.; Roca-Ceballos, M.A.; Santiago, M.; et al. Reformulating Pro-Oxidant Microglia in Neurodegeneration. J. Clin. Med. 2019, 8, 1719. [Google Scholar] [CrossRef]
- Jurga, A.M.; Paleczna, M.; Kuter, K.Z. Overview of General and Discriminating Markers of Differential Microglia Phenotypes. Front. Cell. Neurosci. 2020, 14, 198. [Google Scholar] [CrossRef]
- Gao, L.; Jiang, T.; Yao, X.; Yu, L.; Yang, X.; Li, Y. TREM2 and the Progression of Alzheimer’s Disease. Curr. Neurovasc. Res. 2017, 14, 177–183. [Google Scholar] [CrossRef]
- He, M.; Ichinose, T.; Yoshida, S.; Ito, T.; He, C.; Yoshida, Y.; Arashidani, K.; Takano, H.; Sun, G.; Shibamoto, T. PM2.5-induced lung inflammation in mice: Differences of inflammatory response in macrophages and type II alveolar cells. J. Appl. Toxicol. 2017, 37, 1203–1218. [Google Scholar] [CrossRef]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef]
- Dodd, C.A.; Filipov, N.M. Manganese potentiates LPS-induced heme-oxygenase 1 in microglia but not dopaminergic cells: Role in controlling microglial hydrogen peroxide and inflammatory cytokine output. Neurotoxicology 2011, 32, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Green, C.J.; Foresti, R. Regulation of heme oxygenase-1 by redox signals involving nitric oxide. Antioxid. Redox Signal. 2002, 4, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Otterbein, L.E.; Morse, D.; Choi, A.M. Heme oxygenase/carbon monoxide signaling pathways: Regulation and functional significance. Mol. Cell. Biochem. 2002, 234, 249–263. [Google Scholar] [CrossRef]
- Hassannia, B.; Wiernicki, B.; Ingold, I.; Qu, F.; Van Herck, S.; Tyurina, Y.Y.; Bayir, H.; Abhari, B.A.; Angeli, J.P.F.; Choi, S.M.; et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Investig. 2018, 128, 3341–3355. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Gall, T.; Balla, G.; Balla, J. Heme, Heme Oxygenase, and Endoplasmic Reticulum Stress-A New Insight into the Pathophysiology of Vascular Diseases. Int. J. Mol. Sci. 2019, 20, 3675. [Google Scholar] [CrossRef]
- Li, L.; Dong, H.; Song, E.; Xu, X.; Liu, L.; Song, Y. Nrf2/ARE pathway activation, HO-1 and NQO1 induction by polychlorinated biphenyl quinone is associated with reactive oxygen species and PI3K/AKT signaling. Chem. Biol. Interact. 2014, 209, 56–67. [Google Scholar] [CrossRef]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Li, N.; Alam, J.; Venkatesan, M.I.; Eiguren-Fernandez, A.; Schmitz, D.; Di Stefano, E.; Slaughter, N.; Killeen, E.; Wang, X.; Huang, A.; et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: Protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J. Immunol. 2004, 173, 3467–3481. [Google Scholar] [CrossRef]
- Sun, C.C.; Li, S.J.; Yang, C.L.; Xue, R.L.; Xi, Y.Y.; Wang, L.; Zhao, Q.L.; Li, D.J. Sulforaphane Attenuates Muscle Inflammation in Dystrophin-deficient mdx Mice via NF-E2-related Factor 2 (Nrf2)-mediated Inhibition of NF-kappaB Signaling Pathway. J. Biol. Chem. 2015, 290, 17784–17795. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Purdom-Dickinson, S.E.; Sheveleva, E.V.; Sun, H.; Chen, Q.M. Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol. Pharmacol. 2007, 72, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Giambanco, I.; Minelli, A.; Donato, R. Nrf2-Keap1 signaling in oxidative and reductive stress. Biochim. Biophys. Acta 2018, 1865, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Kwon, K.J.; Kim, J.N.; Kim, M.K.; Lee, J.; Ignarro, L.J.; Kim, H.J.; Shin, C.Y.; Han, S.H. Melatonin synergistically increases resveratrol-induced heme oxygenase-1 expression through the inhibition of ubiquitin-dependent proteasome pathway: A possible role in neuroprotection. J. Pineal Res. 2011, 50, 110–123. [Google Scholar] [CrossRef]
- Green, S.J.; Mellouk, S.; Hoffman, S.L.; Meltzer, M.S.; Nacy, C.A. Cellular mechanisms of nonspecific immunity to intracellular infection: Cytokine-induced synthesis of toxic nitrogen oxides from L-arginine by macrophages and hepatocytes. Immunol. Lett. 1990, 25, 15–19. [Google Scholar] [CrossRef]
- Shin, C.Y.; Choi, J.W.; Ryu, J.R.; Ko, K.H.; Choi, J.J.; Kim, H.S.; Lee, J.C.; Lee, S.J.; Kim, H.C.; Kim, W.K. Glucose deprivation decreases nitric oxide production via NADPH depletion in immunostimulated rat primary astrocytes. Glia 2002, 37, 268–274. [Google Scholar] [CrossRef]
- Kwon, K.J.; Cho, K.S.; Lee, S.H.; Kim, J.N.; Joo, S.H.; Ryu, J.H.; Ignarro, L.J.; Han, S.H.; Shin, C.Y. Regulation of tissue plasminogen activator/plasminogen activator inhibitor-1 by hydrocortisone in rat primary astrocytes. J. Neurosci. Res. 2011, 89, 1059–1069. [Google Scholar] [CrossRef]
- LeBel, C.P.; Ali, S.F.; McKee, M.; Bondy, S.C. Organometal-induced increases in oxygen reactive species: The potential of 2′,7′-dichlorofluorescin diacetate as an index of neurotoxic damage. Toxicol. Appl. Pharmacol. 1990, 104, 17–24. [Google Scholar] [CrossRef]
- Dutta, T.; Kaur, H.; Singh, S.; Mishra, A.; Tripathi, J.K.; Singh, N.; Pareek, A.; Singh, P. Developmental changes in storage proteins and peptidyl prolyl cis-trans isomerase activity in grains of different wheat cultivars. Food Chem. 2011, 128, 450–457. [Google Scholar] [CrossRef]
- Hansen, M.B.; Nielsen, S.E.; Berg, K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods 1989, 119, 203–210. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sense | Antisense | |
|---|---|---|
| Il-1β | AAA ATG CCT CGT GCT GTC TG | CTA TGT CCC GAC CAT TGC TG |
| Il-6 | TTG TGC AATGGC AAT TCT GA | TGG AAG TTG GGG TAG GAA GG |
| TNFα | TAG CCC ACG TCG TAG CAA AC | GGA GGC TGA CTT TCT CCT GG |
| iNOS | CTG GCT GCC TTG TTC AGC TA | AGT GTA GCG TTT CGG GAT CT |
| COX-2 | TGCTGTACAAGCAGTGGCAA | AGGTGCTCGGCTTCCAGTAT |
| HO-1 | TGTCACCCTGTGCTTGACCT | ATACCCGCTACCTGGGTGAC |
| IL-10 | AGGCGCTGTCATCGATTTCT | ATGGCCTTGTAGACACCTTGG |
| Arg-1 | ACAAGACAGGGCTCCTTTCAG | CGTTGAGTTCCGAAGCAAGC |
| Trem2 | GACCTCTCCACCAGTTTCTCC | TACATGACACCCTCAAGGACTG |
| Tlr2 | TGCTTTCCTGCTGGAGATTT | TGTAACGCAACAGCTTCAGG |
| Tlr4 | ACCTGGCTGGTTTACACGTC | CTGCCAGAGACATTGCAGAA |
| 18S rRNA | CATTAAATCAGTTATGTTGGTTCCTTTGD | TCGGCATGTATTAGCTCTAGAATTACC |
| Gapdh | GTG AAG GTC GGT GTG AAC GGA TTT | CAC AGT CTT CTG AGT GGC AGT GAT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, R.-E.; Shin, C.Y.; Han, S.-H.; Kwon, K.J. Astaxanthin Suppresses PM2.5-Induced Neuroinflammation by Regulating Akt Phosphorylation in BV-2 Microglial Cells. Int. J. Mol. Sci. 2020, 21, 7227. https://doi.org/10.3390/ijms21197227
Kim R-E, Shin CY, Han S-H, Kwon KJ. Astaxanthin Suppresses PM2.5-Induced Neuroinflammation by Regulating Akt Phosphorylation in BV-2 Microglial Cells. International Journal of Molecular Sciences. 2020; 21(19):7227. https://doi.org/10.3390/ijms21197227
Chicago/Turabian StyleKim, Ryeong-Eun, Chan Young Shin, Seol-Heui Han, and Kyoung Ja Kwon. 2020. "Astaxanthin Suppresses PM2.5-Induced Neuroinflammation by Regulating Akt Phosphorylation in BV-2 Microglial Cells" International Journal of Molecular Sciences 21, no. 19: 7227. https://doi.org/10.3390/ijms21197227
APA StyleKim, R.-E., Shin, C. Y., Han, S.-H., & Kwon, K. J. (2020). Astaxanthin Suppresses PM2.5-Induced Neuroinflammation by Regulating Akt Phosphorylation in BV-2 Microglial Cells. International Journal of Molecular Sciences, 21(19), 7227. https://doi.org/10.3390/ijms21197227