The ALS-Related σ1R E102Q Mutant Eludes Ligand Control and Exhibits Anomalous Response to Calcium


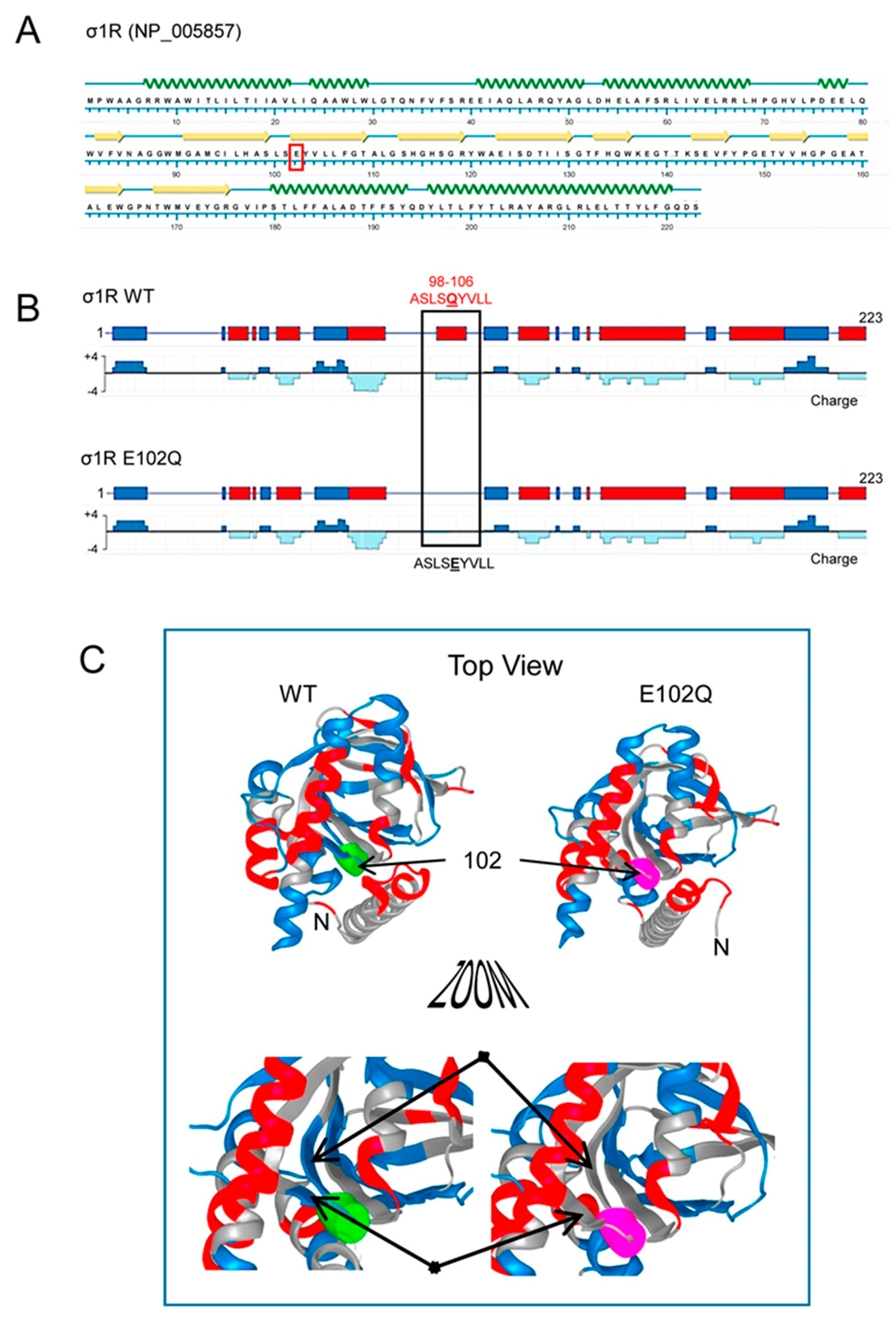{kind=link}
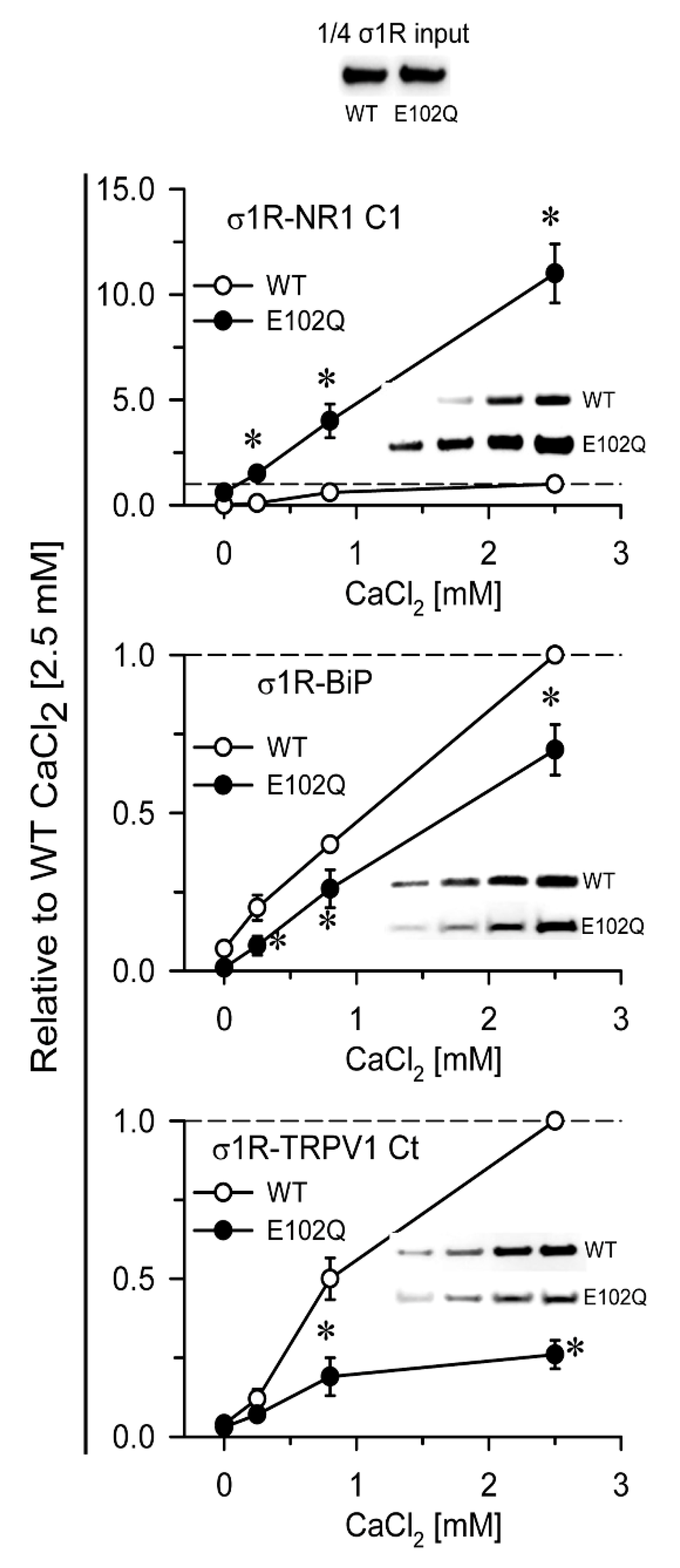{kind=link}
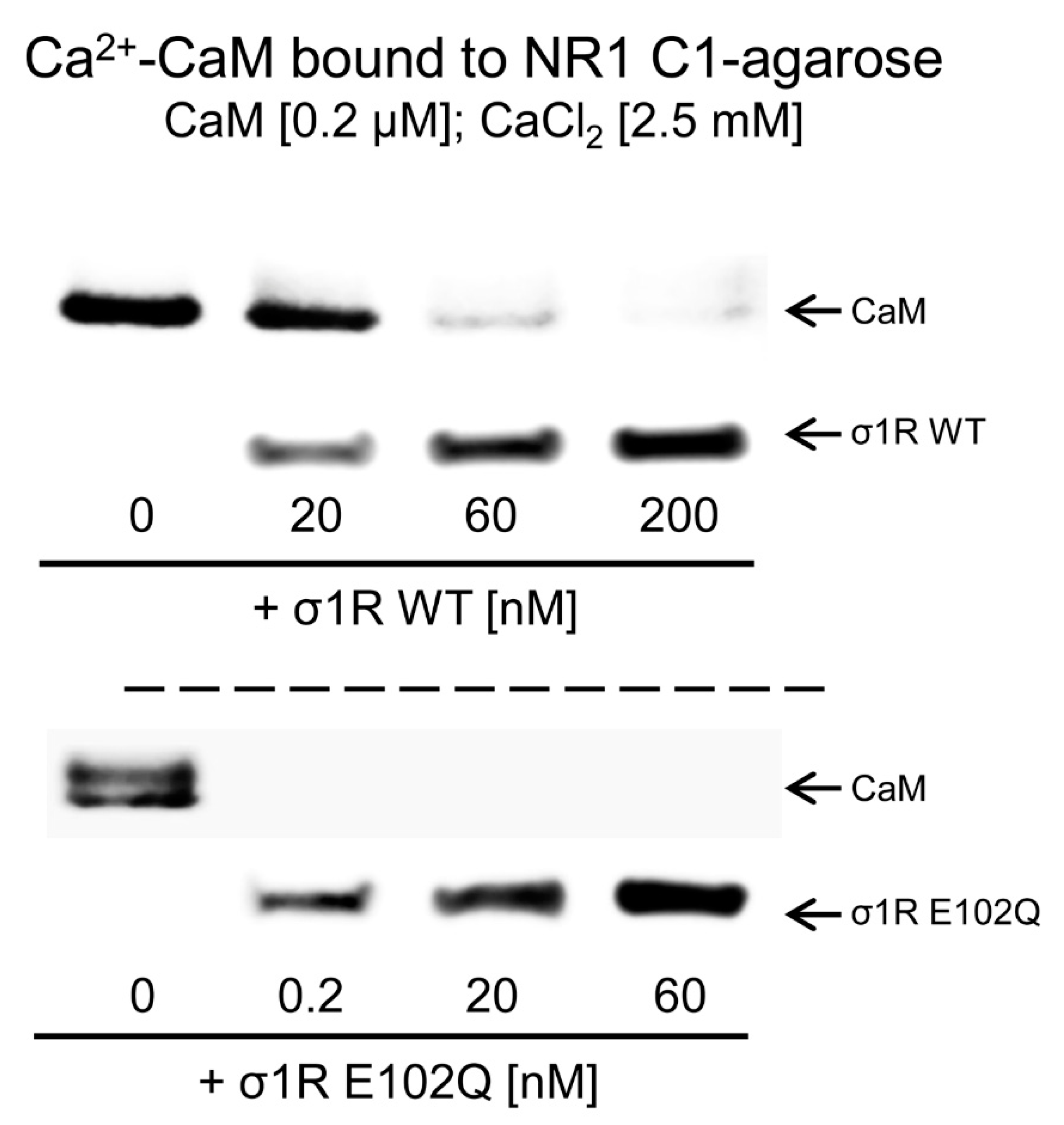{kind=link}
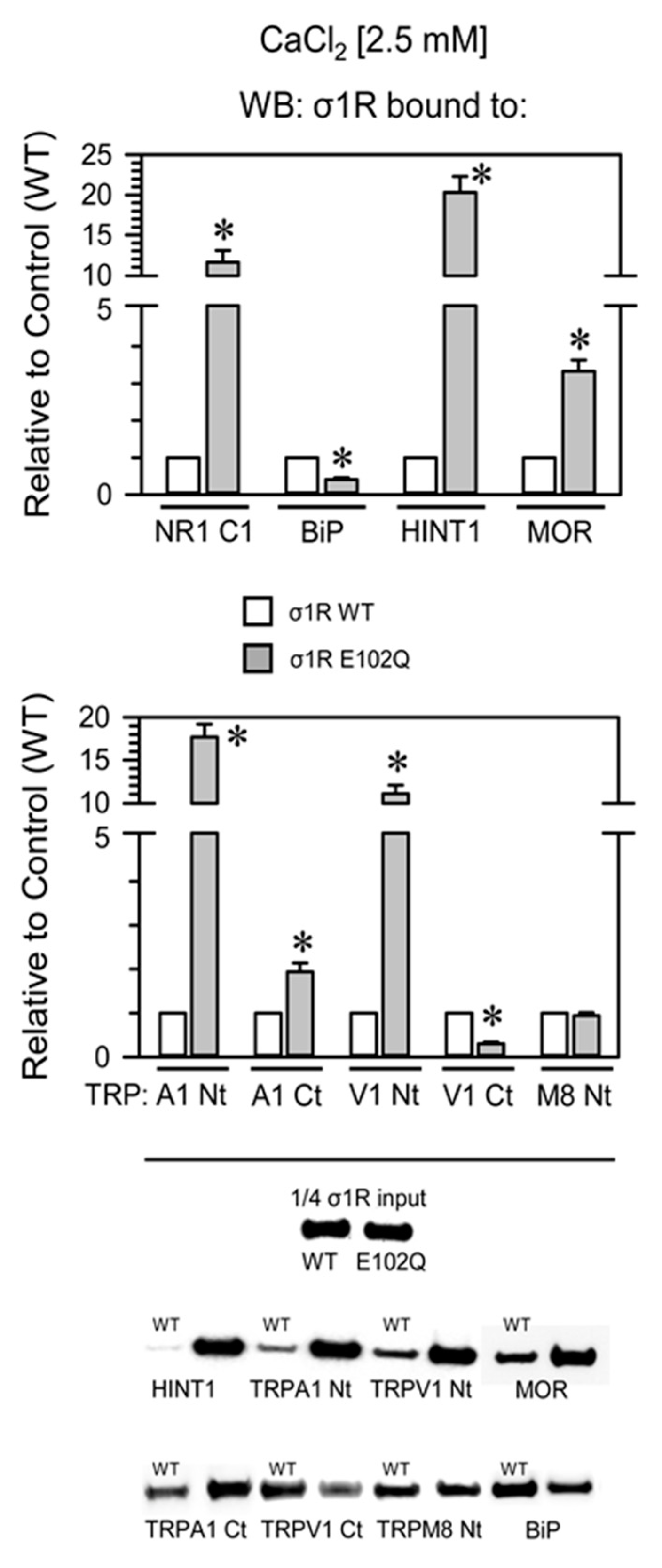{kind=link}
{kind=link}
Abstract
:1. Introduction
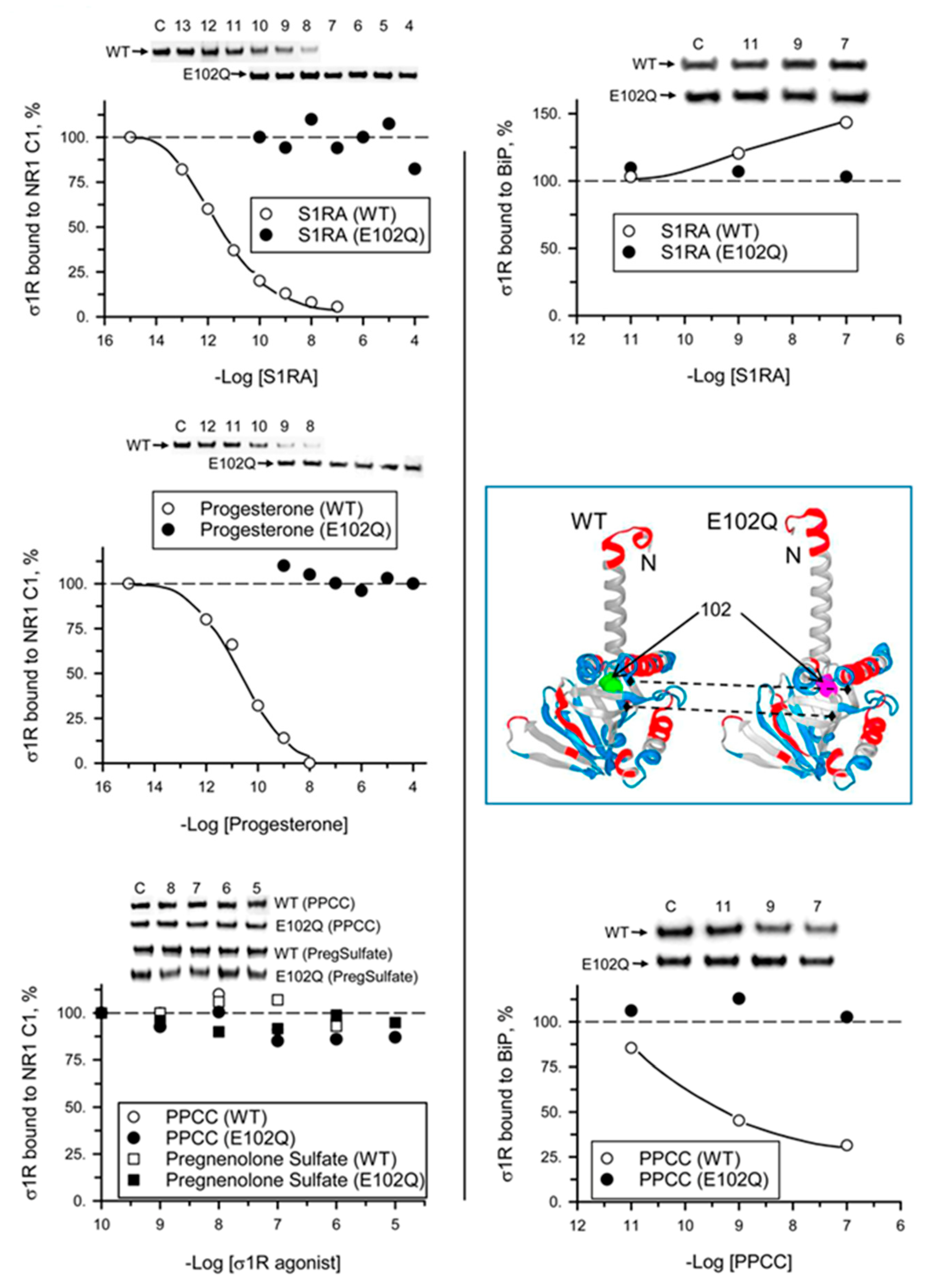
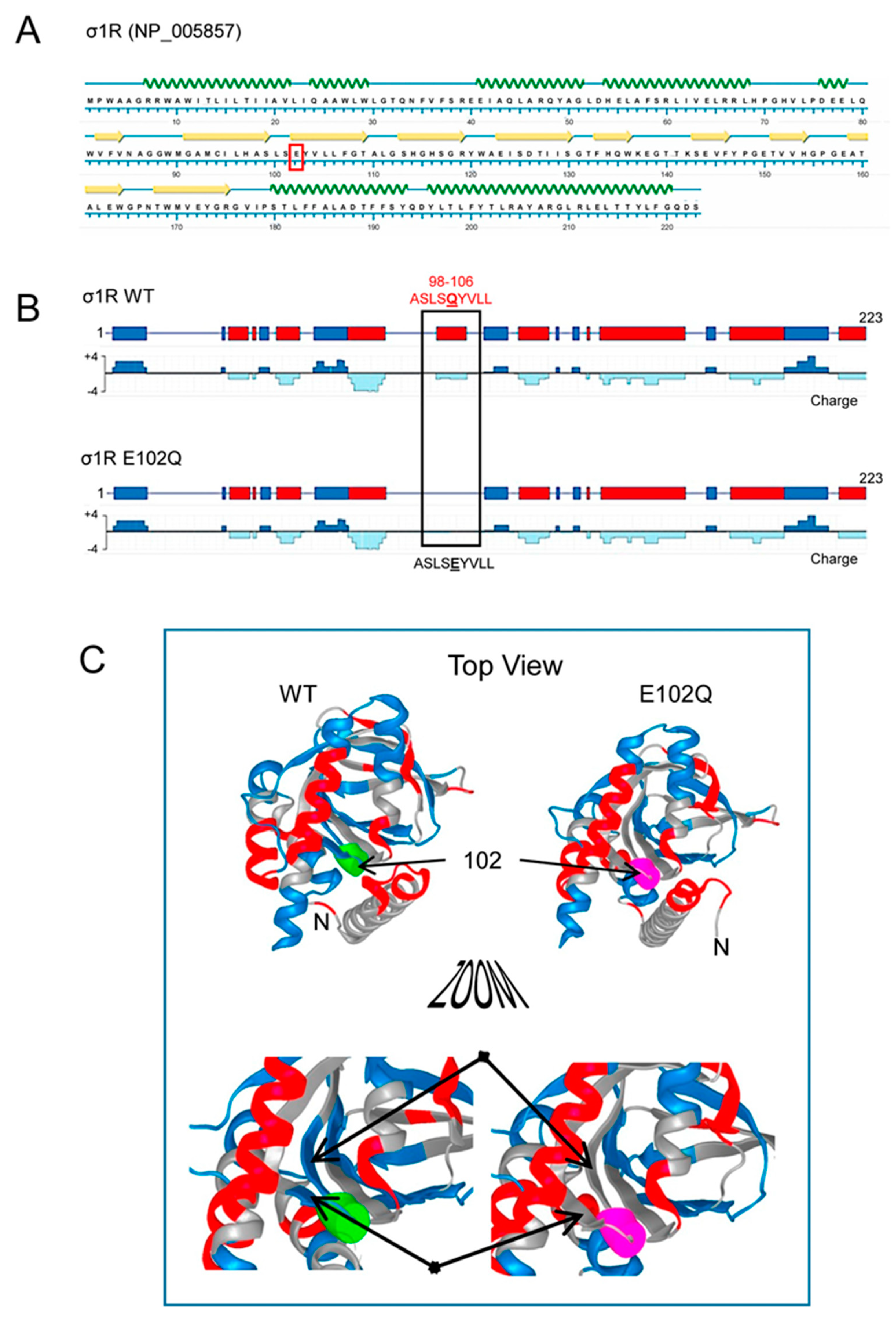
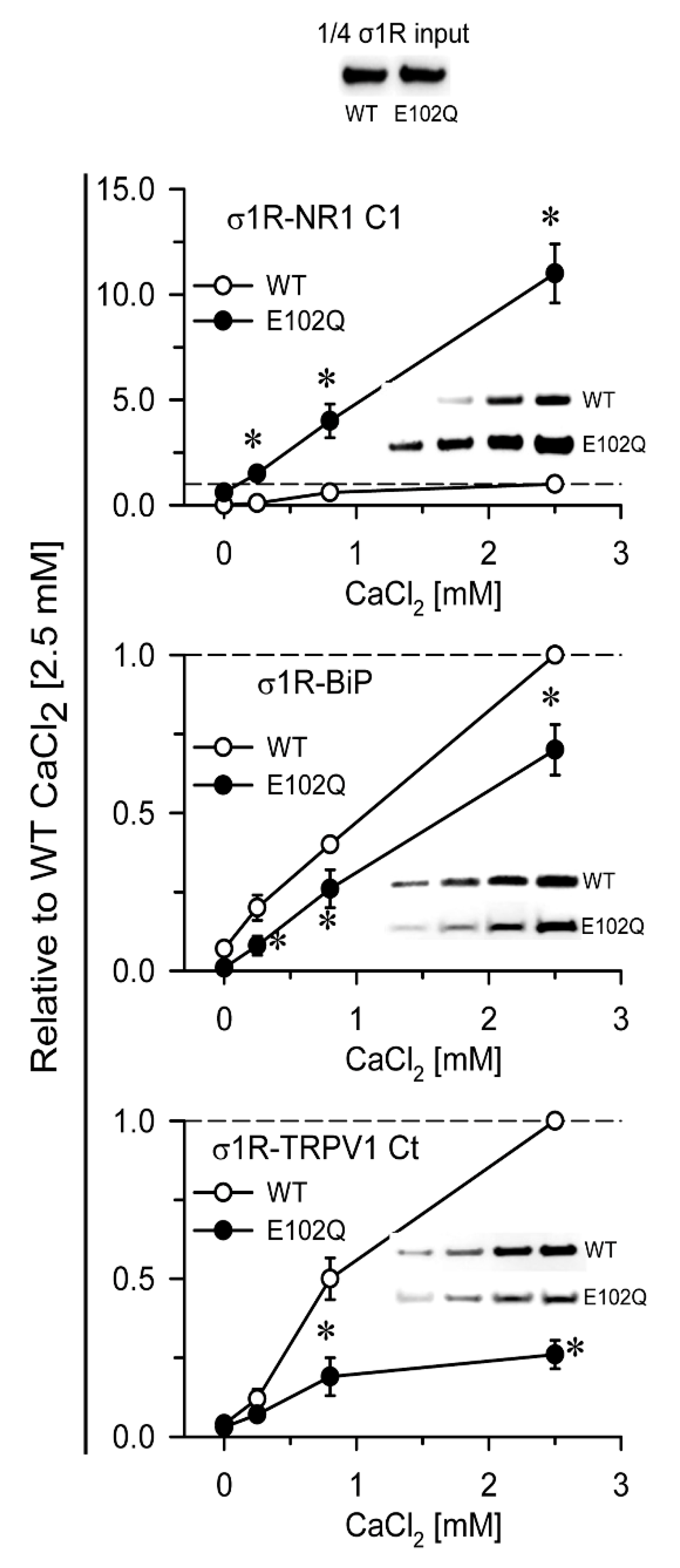
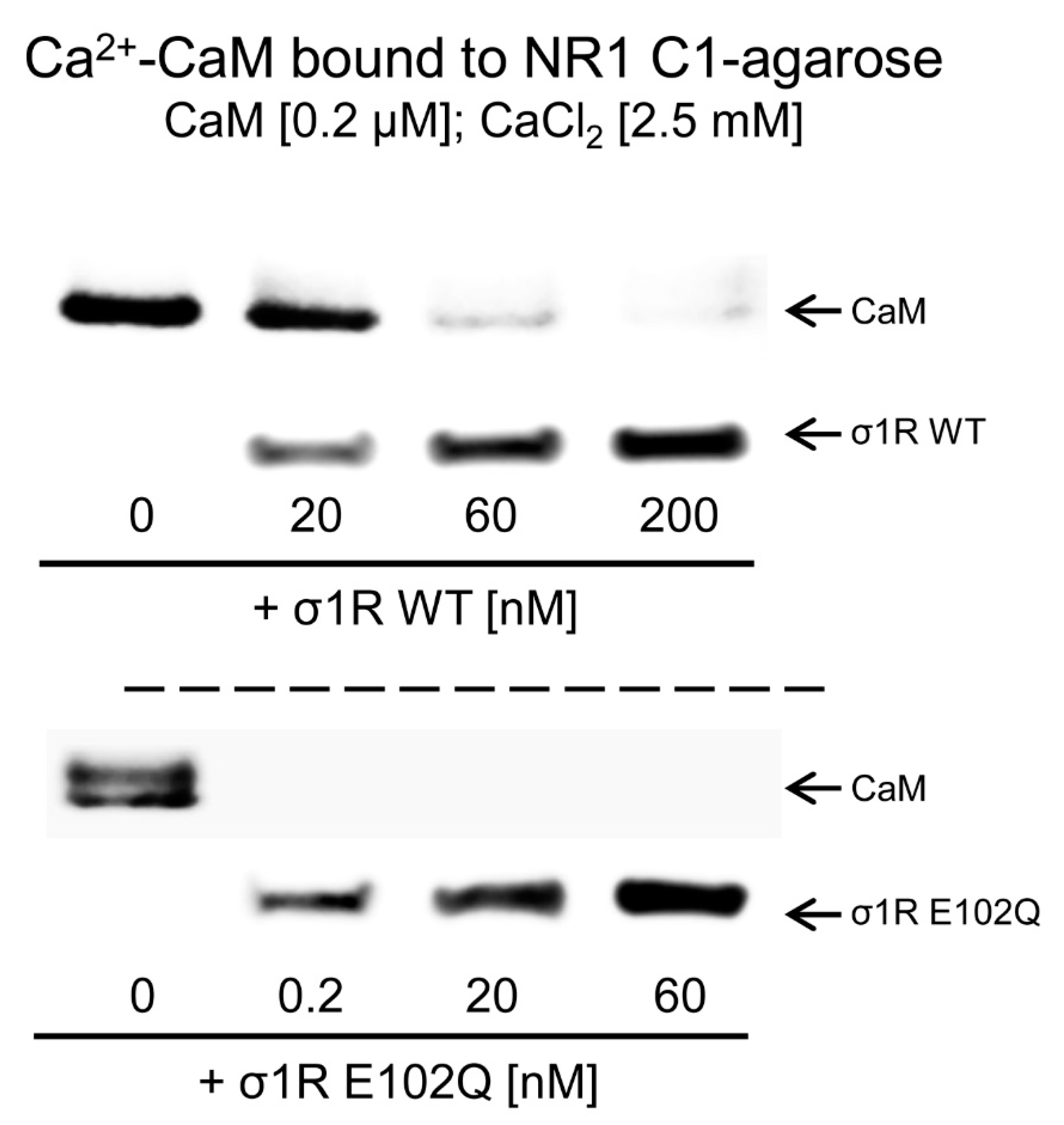
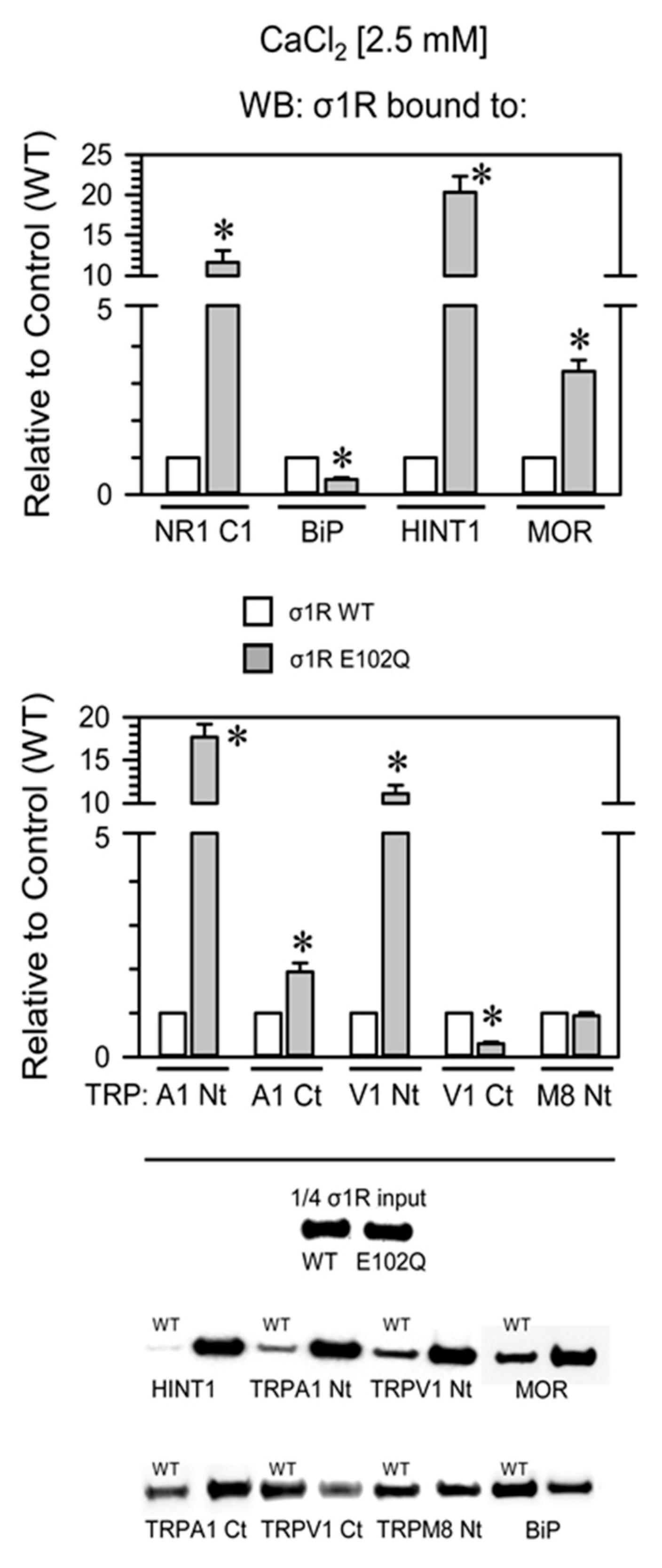
2. Results
3. Discussion
4. Materials and Methods
4.1. Recombinant Protein Expression
4.2. In Vitro Interactions between Recombinant Proteins: Pull-Down of Recombinant Proteins
4.3. Western Blotting
4.4. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| BiP | Binding immunoglobulin protein |
| CaM | Calmodulin |
| CHAPS | 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate |
| ER | Endoplasmic reticulum |
| FTLD | Frontotemporal lobar degeneration |
| GPCR | G-protein coupled receptor |
| HINT1 | Histidine triad nucleotide-binding protein 1 |
| IP3R3 | Inositol 1,4,5-triphosphate receptor type 3 |
| MAM | Mitochondria-associated ER membranes |
| MOR | Mu-opioid receptor |
| NMDAR | N-methyl-D-aspartate receptor |
| PPCC | 2-[(4-Hydroxy-4-phenyl-1-piperidinyl)methyl]-1-(4-methylphenyl)-cyclopropanecarboxylic acid |
| S1RA | 4-[2-[[5-methyl-1-(2-naphthalenyl)-1H-pyrazol-3-yl]oxy]ethyl] morpholine |
| σ1R | Sigma 1 receptor |
| TRP | Transient receptor potential calcium channel |
| WT | Wild-type |
References
- Gundlach, A.L.; Largent, B.L.; Snyder, S.H. Autoradiographic localization of sigma receptor binding sites in guinea pig and rat central nervous system with (+)3H-3-(3-hydroxyphenyl)-N-(1-propyl)piperidine. J. Neurosci 1986, 6, 1757–1770. [Google Scholar] [CrossRef] [PubMed]
- Largent, B.L.; Gundlach, A.L.; Snyder, S.H. Pharmacological and autoradiographic discrimination of sigma and phencyclidine receptor binding sites in brain with (+)-[3H]SKF 10,047, (+)-[3H]-3-[3-hydroxyphenyl]-N-(1-propyl)piperidine and [3H]-1-[1-(2-thienyl)cyclohexyl]piperidine. J. Pharm. Exp. 1986, 238, 739–748. [Google Scholar]
- Zukin, S.R.; Tempel, A.; Gardner, E.L.; Zukin, R.S. Interaction of [3H](-)-SKF-10,047 with brain sigma receptors: Characterization and autoradiographic visualization. J. Neurochem 1986, 46, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Bowen, W.D.; Goldstein, S.R.; Roberts, A.H.; Patrick, S.L.; Hohmann, A.G.; DeCosta, B. Autoradiographic distribution of [3H](+)-pentazocine and [3H]1,3-di-o-tolylguanidine (DTG) binding sites in guinea pig brain: A comparative study. Brain Res. 1992, 581, 33–38. [Google Scholar] [CrossRef]
- Kim, F.J.; Kovalyshyn, I.; Burgman, M.; Neilan, C.; Chien, C.C.; Pasternak, G.W. Sigma 1 receptor modulation of G-protein-coupled receptor signaling: Potentiation of opioid transduction independent from receptor binding. Mol. Pharm. 2010, 77, 695–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, G.; Moreno, E.; Aymerich, M.; Marcellino, D.; McCormick, P.J.; Mallol, J.; Cortes, A.; Casado, V.; Canela, E.I.; Ortiz, J.; et al. Direct involvement of sigma-1 receptors in the dopamine D1 receptor-mediated effects of cocaine. Proc. Natl Acad Sci USA 2010, 107, 18676–18681. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Herrero-Labrador, R.; Burgueño, J.; Zamanillo, D.; Garzón, J. The calcium-sensitive Sigma-1 receptor prevents cannabinoids from provoking glutamate NMDA receptor hypofunction: Implications in antinociception and psychotic diseases. Int J. Neuropsychopharmacol 2014, 17, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 receptors (sigma(1) binding sites) form raft-like microdomains and target lipid droplets on the endoplasmic reticulum: Roles in endoplasmic reticulum lipid compartmentalization and export. J. Pharm. Exp. 2003, 306, 718–725. [Google Scholar] [CrossRef] [Green Version]
- Pal, A.; Fontanilla, D.; Gopalakrishnan, A.; Chae, Y.K.; Markley, J.L.; Ruoho, A.E. The sigma-1 receptor protects against cellular oxidative stress and activates antioxidant response elements. Eur. J. Pharm. 2012, 682, 12–20. [Google Scholar] [CrossRef] [Green Version]
- Tsai, S.Y.; Chuang, J.Y.; Tsai, M.S.; Wang, X.F.; Xi, Z.X.; Hung, J.J.; Chang, W.C.; Bonci, A.; Su, T.P. Sigma-1 receptor mediates cocaine-induced transcriptional regulation by recruiting chromatin-remodeling factors at the nuclear envelope. Proc Natl Acad Sci USA 2015, 112, E6562–E6570. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Su, T.P. The potential role of sigma-1 receptors in lipid transport and lipid raft reconstitution in the brain: Implication for drug abuse. Life Sci 2005, 77, 1612–1624. [Google Scholar] [CrossRef] [PubMed]
- Maurice, T.; Su, T.P. The pharmacology of sigma-1 receptors. Pharmacology 2009, 124, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, J.; Pasternak, G.W. Molecular cloning and pharmacological characterization of the rat sigma1 receptor. Biochem. Pharm. 2001, 62, 349–355. [Google Scholar] [CrossRef]
- Sabino, V.; Cottone, P.; Parylak, S.L.; Steardo, L.; Zorrilla, E.P. Sigma-1 receptor knockout mice display a depressive-like phenotype. Behav Brain Res. 2009, 198, 472–476. [Google Scholar] [CrossRef] [Green Version]
- Ishiguro, H.; Ohtsuki, T.; Toru, M.; Itokawa, M.; Aoki, J.; Shibuya, H.; Kurumaji, A.; Okubo, Y.; Iwawaki, A.; Ota, K.; et al. Association between polymorphisms in the type 1 sigma receptor gene and schizophrenia. Neurosci Lett 1998, 257, 45–48. [Google Scholar] [CrossRef]
- Sánchez-Blázquez, P.; Pozo-Rodrigálvarez, A.; Merlos, M.; Garzón, J. The Sigma-1 Receptor Antagonist, S1RA, Reduces Stroke Damage, Ameliorates Post-Stroke Neurological Deficits and Suppresses the Overexpression of MMP-9. Mol. Neurobiol 2018, 55, 4940–4951. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Cui, X.; Roon, P.; Saul, A.; Smith, S.B. The Role of Sigma1R in Mammalian Retina. Adv. Exp. Med. Biol. 2017, 964, 267–284. [Google Scholar] [CrossRef]
- Mavlyutov, T.A.; Nickells, R.W.; Guo, L.W. Accelerated retinal ganglion cell death in mice deficient in the Sigma-1 receptor. Mol. Vis. 2011, 17, 1034–1043. [Google Scholar]
- Villard, V.; Espallergues, J.; Keller, E.; Vamvakides, A.; Maurice, T. Anti-amnesic and neuroprotective potentials of the mixed muscarinic receptor/sigma 1 (sigma1) ligand ANAVEX2-73, a novel aminotetrahydrofuran derivative. J. Psychopharmacol 2011, 25, 1101–1117. [Google Scholar] [CrossRef]
- Francardo, V.; Bez, F.; Wieloch, T.; Nissbrandt, H.; Ruscher, K.; Cenci, M.A. Pharmacological stimulation of sigma-1 receptors has neurorestorative effects in experimental parkinsonism. Brain 2014, 137, 1998–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyrskyluoto, A.; Pulli, I.; Tornqvist, K.; Ho, T.H.; Korhonen, L.; Lindholm, D. Sigma-1 receptor agonist PRE084 is protective against mutant huntingtin-induced cell degeneration: Involvement of calpastatin and the NF-kappaB pathway. Cell. Death Dis. 2013, 4, e646. [Google Scholar] [CrossRef] [PubMed]
- Luty, A.A.; Kwok, J.B.; Dobson-Stone, C.; Loy, C.T.; Coupland, K.G.; Karlstrom, H.; Sobow, T.; Tchorzewska, J.; Maruszak, A.; Barcikowska, M.; et al. Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann. Neurol 2010, 68, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hu, Z.; Liu, L.; Xie, Y.; Zhan, Y.; Zi, X.; Wang, J.; Wu, L.; Xia, K.; Tang, B.; et al. A SIGMAR1 splice-site mutation causes distal hereditary motor neuropathy. Neurology 2015, 84, 2430–2437. [Google Scholar] [CrossRef] [PubMed]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol 2011, 70, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. Embo Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef] [PubMed]
- Mavlyutov, T.A.; Epstein, M.L.; Andersen, K.A.; Ziskind-Conhaim, L.; Ruoho, A.E. The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons. An anatomical and behavioral study. Neuroscience 2010, 167, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Bernard-Marissal, N.; Medard, J.J.; Azzedine, H.; Chrast, R. Dysfunction in endoplasmic reticulum-mitochondria crosstalk underlies SIGMAR1 loss of function mediated motor neuron degeneration. Brain 2015, 138, 875–890. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human sigma1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef]
- Dreser, A.; Vollrath, J.T.; Sechi, A.; Johann, S.; Roos, A.; Yamoah, A.; Katona, I.; Bohlega, S.; Wiemuth, D.; Tian, Y.; et al. The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ. 2017, 24, 1655–1671. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Herrero-Labrador, R.; Martínez-Murillo, R.; Merlos, M.; Vela, J.M.; Garzón, J. The sigma1 receptor engages the redox-regulated HINT1 protein to bring opioid analgesia under NMDA receptor negative control. Antioxid. Redox Signal. 2015, 22, 799–818. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Muñoz, M.; Cortés-Montero, E.; Pozo-Rodrigálvarez, A.; Sánchez-Blázquez, P.; Garzón-Niño, J. The ON:OFF switch, sigma1R-HINT1 protein, controls GPCR-NMDA receptor cross-regulation: Implications in neurological disorders. Oncotarget 2015, 6, 35458–35477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortés-Montero, E.; Sánchez-Blázquez, P.; Onetti, Y.; Merlos, M.; Garzón, J. Ligands Exert Biased Activity to Regulate Sigma 1 Receptor Interactions With Cationic TRPA1, TRPV1, and TRPM8 Channels. Front. Pharm. 2019, 10, 634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Le, W.D.; Xie, Y.Y.; Wang, X.P. Current Therapy of Drugs in Amyotrophic Lateral Sclerosis. Curr. Neuropharmacol. 2016, 14, 314–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Garzón, J. Fenfluramine diminishes NMDA receptor-mediated seizures via its mixed activity at serotonin 5HT2A and type 1 sigma receptors. Oncotarget 2018, 9, 23373–23389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Su, T.P. Regulating ankyrin dynamics: Roles of sigma-1 receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, M.D.; Zhang, S.; Bernhadt, J.P.; Huganir, R.L. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell 1996, 84, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.Y.; Hristova, E.; Ahlskog, N.; Tasse, L.A.; Ngsee, J.K.; Chudalayandi, P.; Bergeron, R. Aberrant Subcellular Dynamics of Sigma-1 Receptor Mutants Underlying Neuromuscular Diseases. Mol. Pharm. 2016, 90, 238–253. [Google Scholar] [CrossRef] [Green Version]
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170. [Google Scholar] [CrossRef]
- Cortés-Montero, E.; Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Garzón, J. The Axonal Motor Neuropathy-Related HINT1 Protein Is a Zinc- and Calmodulin-Regulated Cysteine SUMO Protease. Antioxid. Redox Signal. 2019, 31, 503–520. [Google Scholar] [CrossRef]
- Zimon, M.; Baets, J.; Almeida-Souza, L.; De, V.E.; Nikodinovic, J.; Parman, Y.; Battaloglu, E.; Matur, Z.; Guergueltcheva, V.; Tournev, I.; et al. Loss-of-function mutations in HINT1 cause axonal neuropathy with neuromyotonia. Nat. Genet. 2012, 44, 1080–1083. [Google Scholar] [CrossRef] [PubMed]
- Weiske, J.; Huber, O. The histidine triad protein Hint1 interacts with Pontin and Reptin and inhibits TCF-beta-catenin-mediated transcription. J. Cell Sci. 2005, 118, 3117–3129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leamey, C.A.; Sawatari, A. Teneurins: Mediators of Complex Neural Circuit Assembly in Mammals. Front. Neurosci. 2019, 13, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosca, T.J.; Hong, W.; Dani, V.S.; Favaloro, V.; Luo, L. Trans-synaptic Teneurin signalling in neuromuscular synapse organization and target choice. Nature 2012, 484, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, N.; Fukushi, M.; Kosaki, K.; Doyle, A.D.; de Vega, S.; Yoshizaki, K.; Akazawa, C.; Arikawa-Hirasawa, E.; Yamada, Y. Teneurin-4 is a novel regulator of oligodendrocyte differentiation and myelination of small-diameter axons in the CNS. J. Neurosci. 2012, 32, 11586–11599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almendra, L.; Laranjeira, F.; Fernandez-Marmiesse, A.; Negrao, L. SIGMAR1 gene mutation causing Distal Hereditary Motor Neuropathy in a Portuguese family. Acta Myol. 2018, 37, 2–4. [Google Scholar] [PubMed]
- Gregianin, E.; Pallafacchina, G.; Zanin, S.; Crippa, V.; Rusmini, P.; Poletti, A.; Fang, M.; Li, Z.; Diano, L.; Petrucci, A.; et al. Loss-of-function mutations in the SIGMAR1 gene cause distal hereditary motor neuropathy by impairing ER-mitochondria tethering and Ca2+ signalling. Hum. Mol. Genet. 2016, 25, 3741–3753. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.I.; Ahmad, A.; Raza, S.I.; Amar, A.; Ali, A.; Bhatti, A.; John, P.; Mohyuddin, A.; Ahmad, W.; Hassan, M.J. In silico analysis of SIGMAR1 variant (rs4879809) segregating in a consanguineous Pakistani family showing amyotrophic lateral sclerosis without frontotemporal lobar dementia. Neurogenetics 2015, 16, 299–306. [Google Scholar] [CrossRef]
- Fukunaga, K.; Shinoda, Y.; Tagashira, H. The role of SIGMAR1 gene mutation and mitochondrial dysfunction in amyotrophic lateral sclerosis. J. Pharm. Sci. 2015, 127, 36–41. [Google Scholar] [CrossRef] [Green Version]
- Ryskamp, D.A.; Zhemkov, V.; Bezprozvanny, I. Mutational Analysis of Sigma-1 Receptor’s Role in Synaptic Stability. Front. Neurosci. 2019, 13, 1012. [Google Scholar] [CrossRef] [Green Version]
- Su, T.P.; Hayashi, T.; Maurice, T.; Buch, S.; Ruoho, A.E. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharm. Sci. 2010, 31, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramyan, A.M.; Yano, H.; Xu, M.; Liu, L.; Naing, S.; Fant, A.D.; Shi, L. The Glu102 mutation disrupts higher-order oligomerization of the sigma 1 receptor. Comput. Struct. Biotechnol. J. 2020, 18, 199–206. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Muñoz, M.; Cortés-Montero, E.; Garzón-Niño, J.; Sánchez-Blázquez, P. The ALS-Related σ1R E102Q Mutant Eludes Ligand Control and Exhibits Anomalous Response to Calcium. Int. J. Mol. Sci. 2020, 21, 7339. https://doi.org/10.3390/ijms21197339
Rodríguez-Muñoz M, Cortés-Montero E, Garzón-Niño J, Sánchez-Blázquez P. The ALS-Related σ1R E102Q Mutant Eludes Ligand Control and Exhibits Anomalous Response to Calcium. International Journal of Molecular Sciences. 2020; 21(19):7339. https://doi.org/10.3390/ijms21197339
Chicago/Turabian StyleRodríguez-Muñoz, María, Elsa Cortés-Montero, Javier Garzón-Niño, and Pilar Sánchez-Blázquez. 2020. "The ALS-Related σ1R E102Q Mutant Eludes Ligand Control and Exhibits Anomalous Response to Calcium" International Journal of Molecular Sciences 21, no. 19: 7339. https://doi.org/10.3390/ijms21197339
APA StyleRodríguez-Muñoz, M., Cortés-Montero, E., Garzón-Niño, J., & Sánchez-Blázquez, P. (2020). The ALS-Related σ1R E102Q Mutant Eludes Ligand Control and Exhibits Anomalous Response to Calcium. International Journal of Molecular Sciences, 21(19), 7339. https://doi.org/10.3390/ijms21197339