Investigating the Transition of Pre-Symptomatic to Symptomatic Huntington’s Disease Status Based on Omics Data

 , , ,
, , ,
Abstract
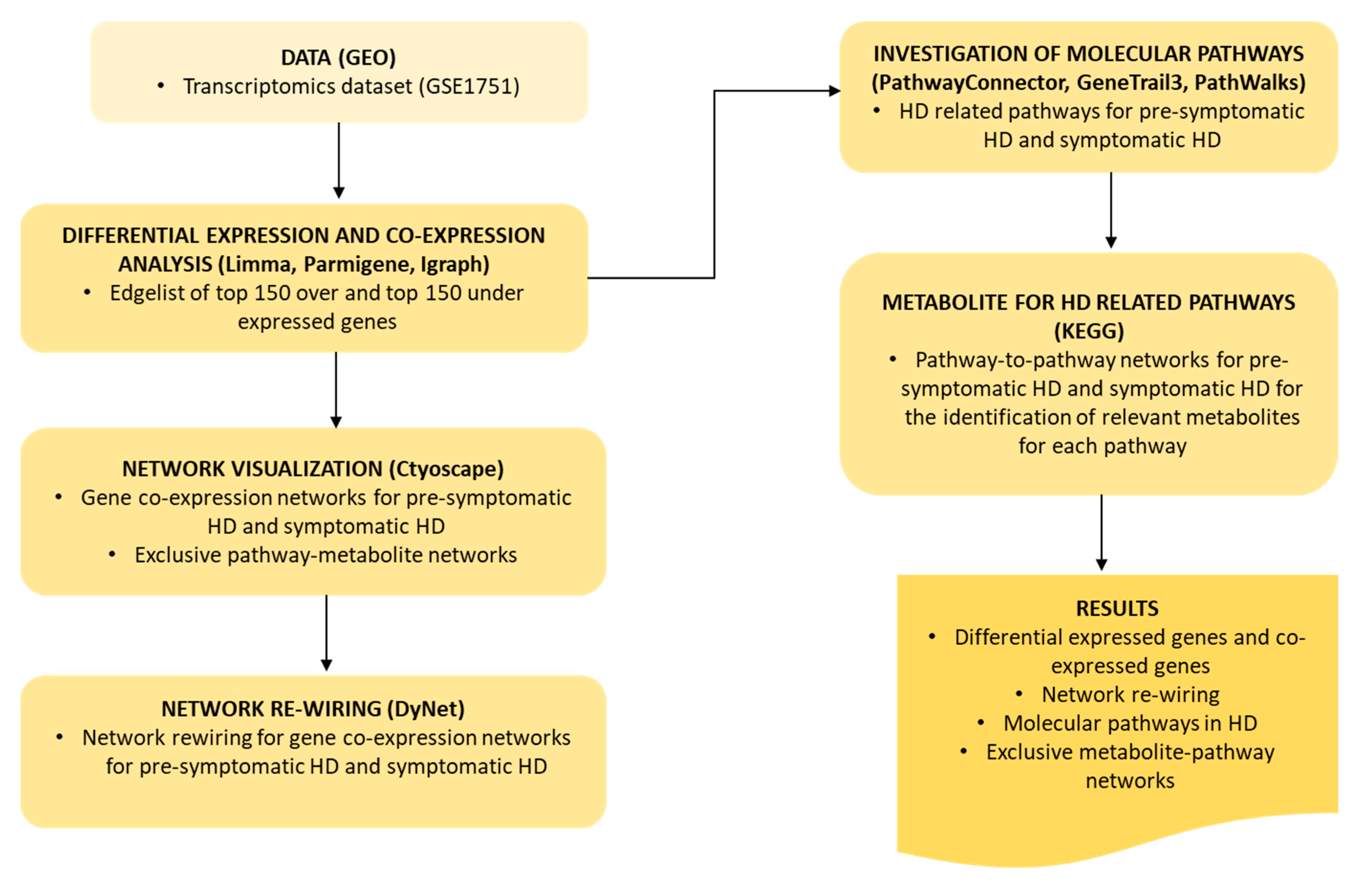
:1. Introduction
2. Results
2.1. Differentially Expressed Genes in Pre-Symptomatic and Symptomatic HD Patients
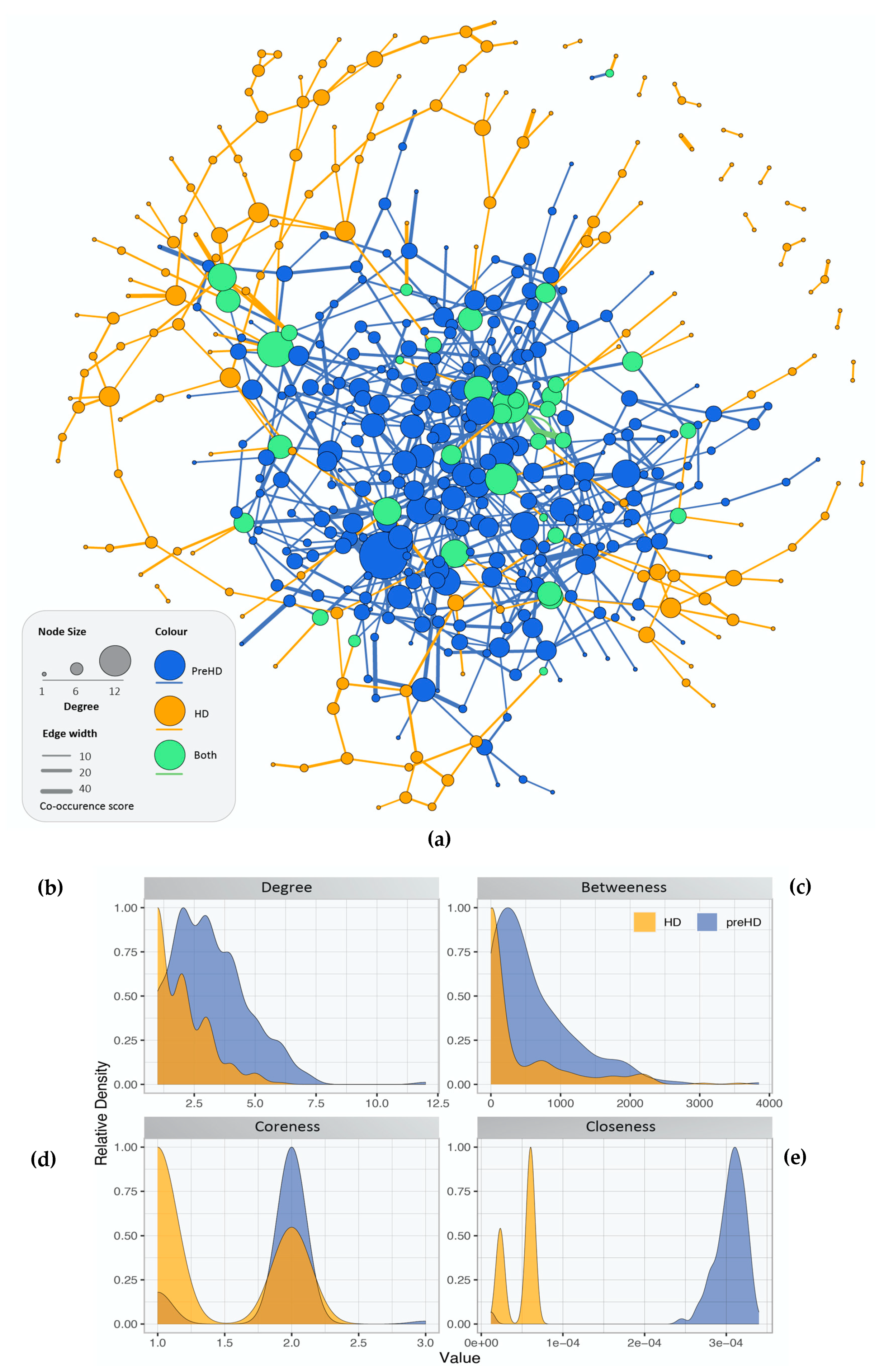
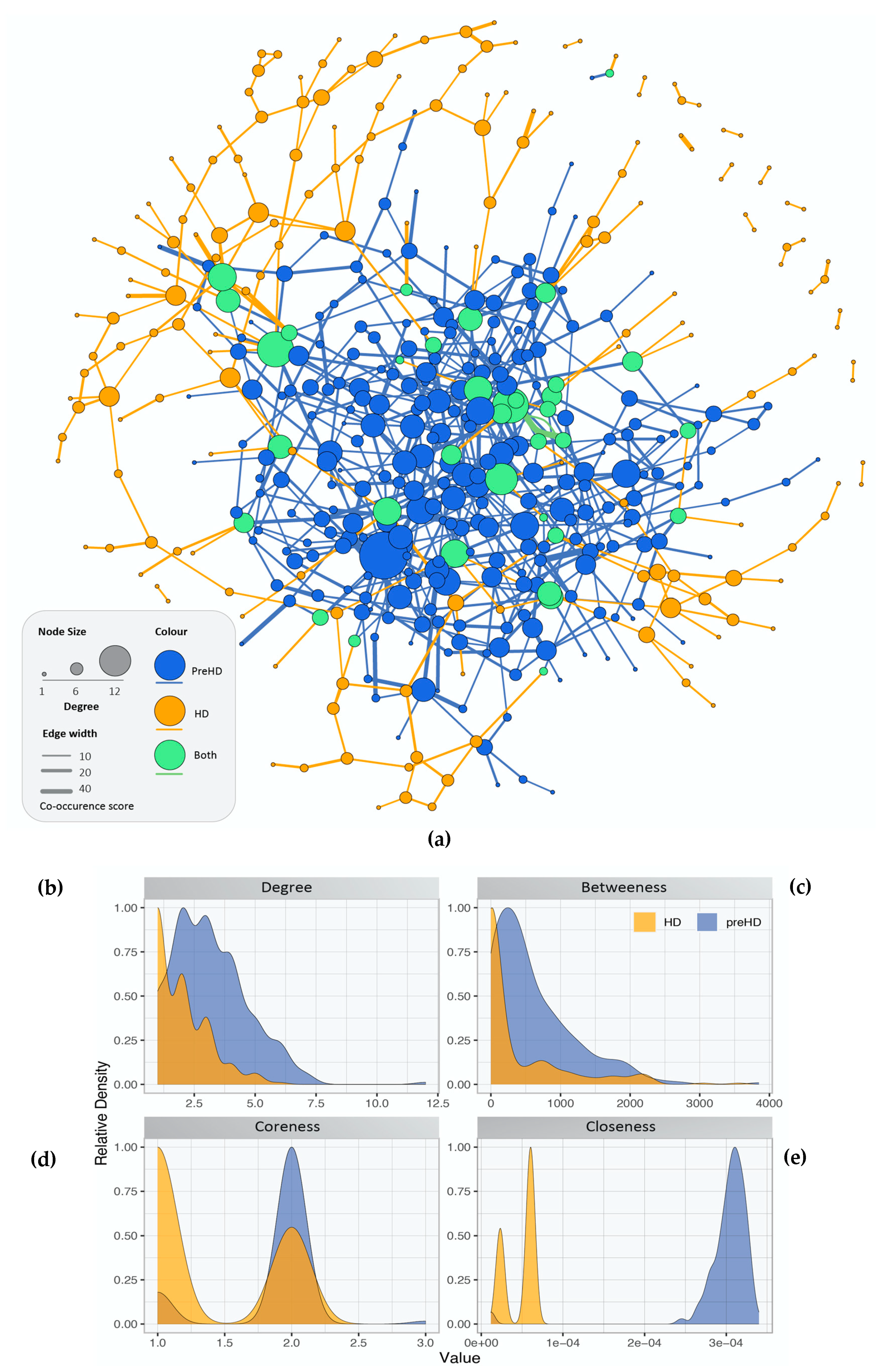
2.2. Gene Co-Expression Networks of Pre-Symptomatic and Symptomatic HD Patients
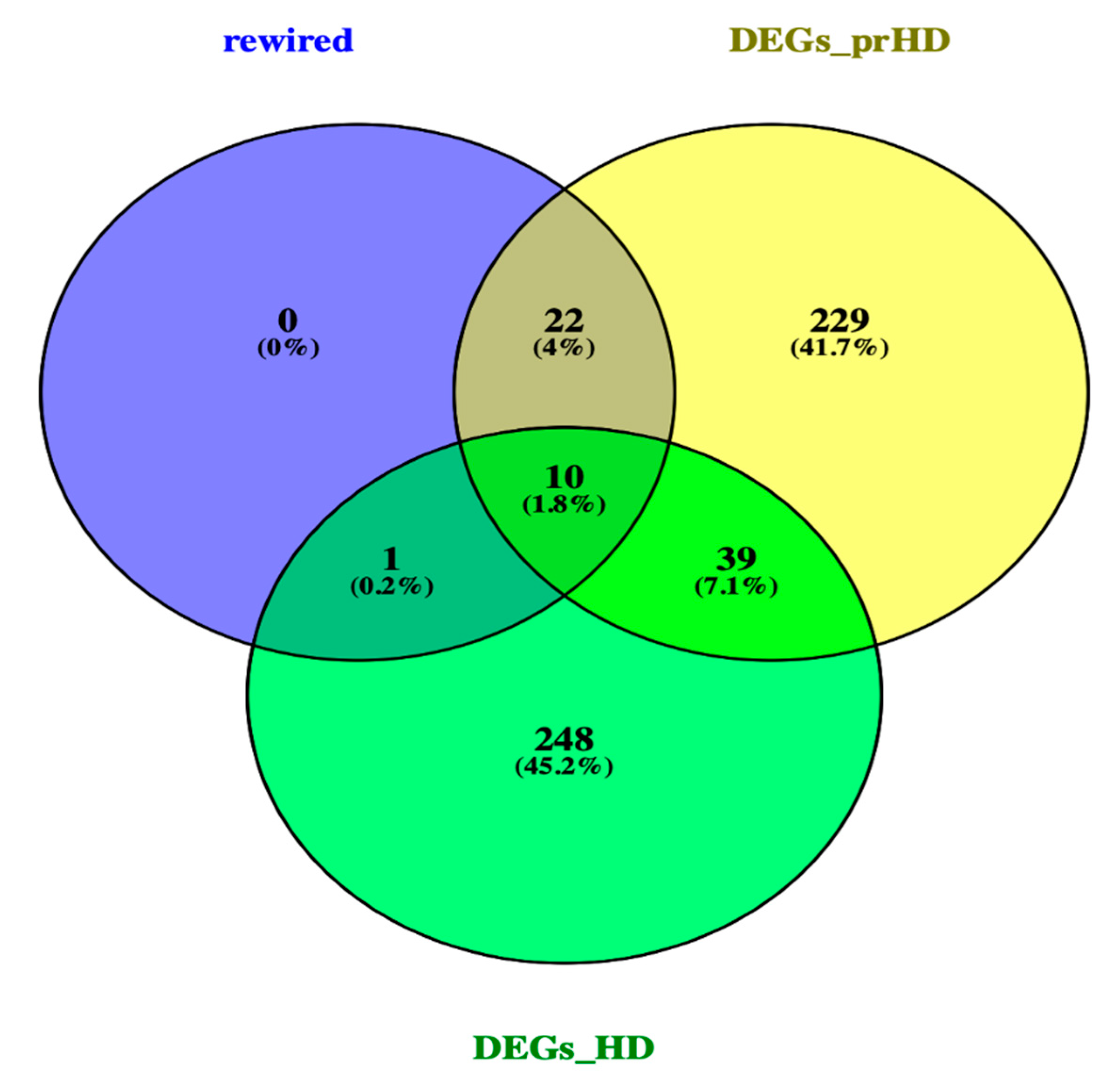
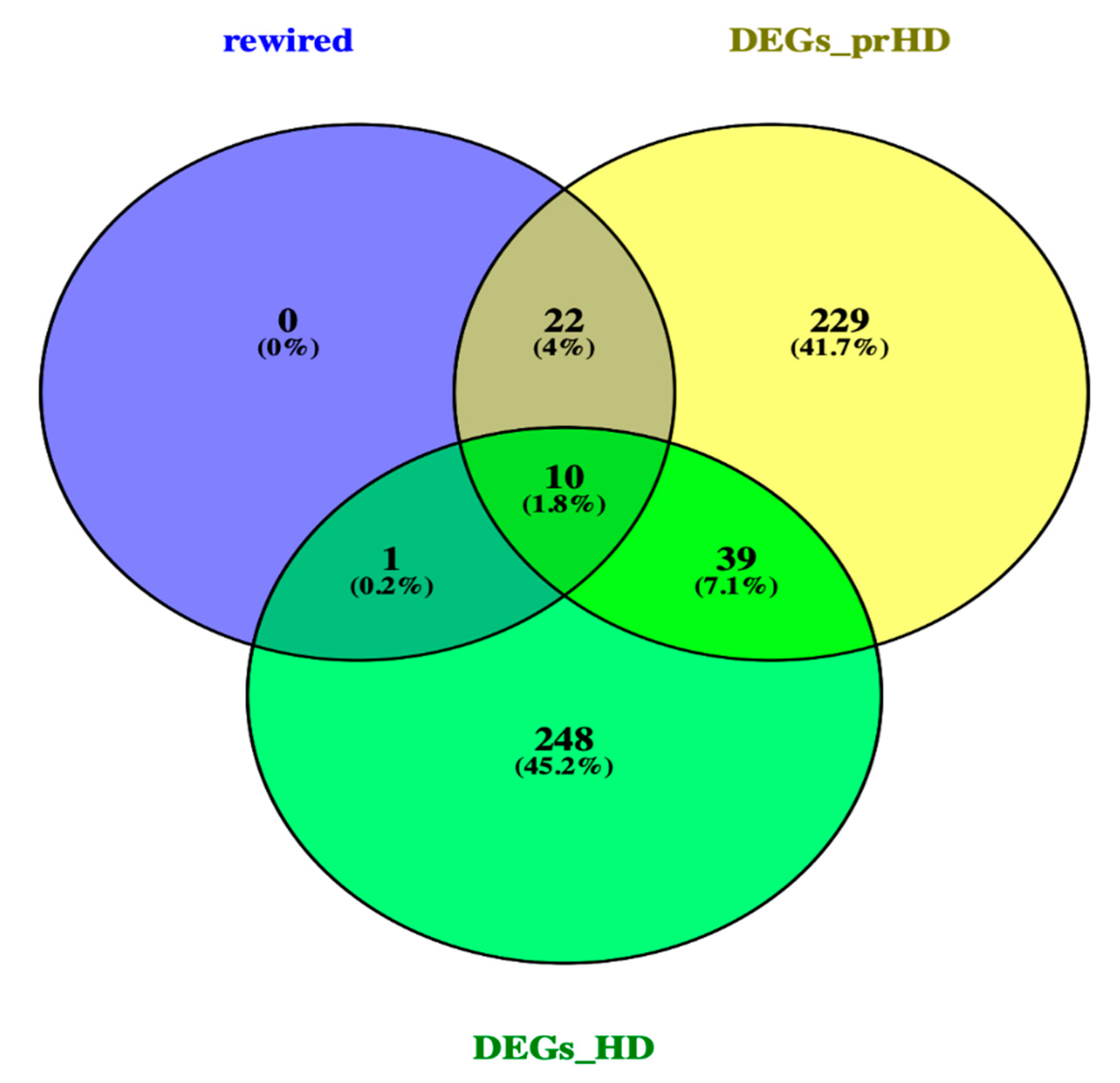
2.3. Network Rewiring between Gene Co-Expression Networks of Pre-Symptomatic and Symptomatic HD Patients Using DyNet
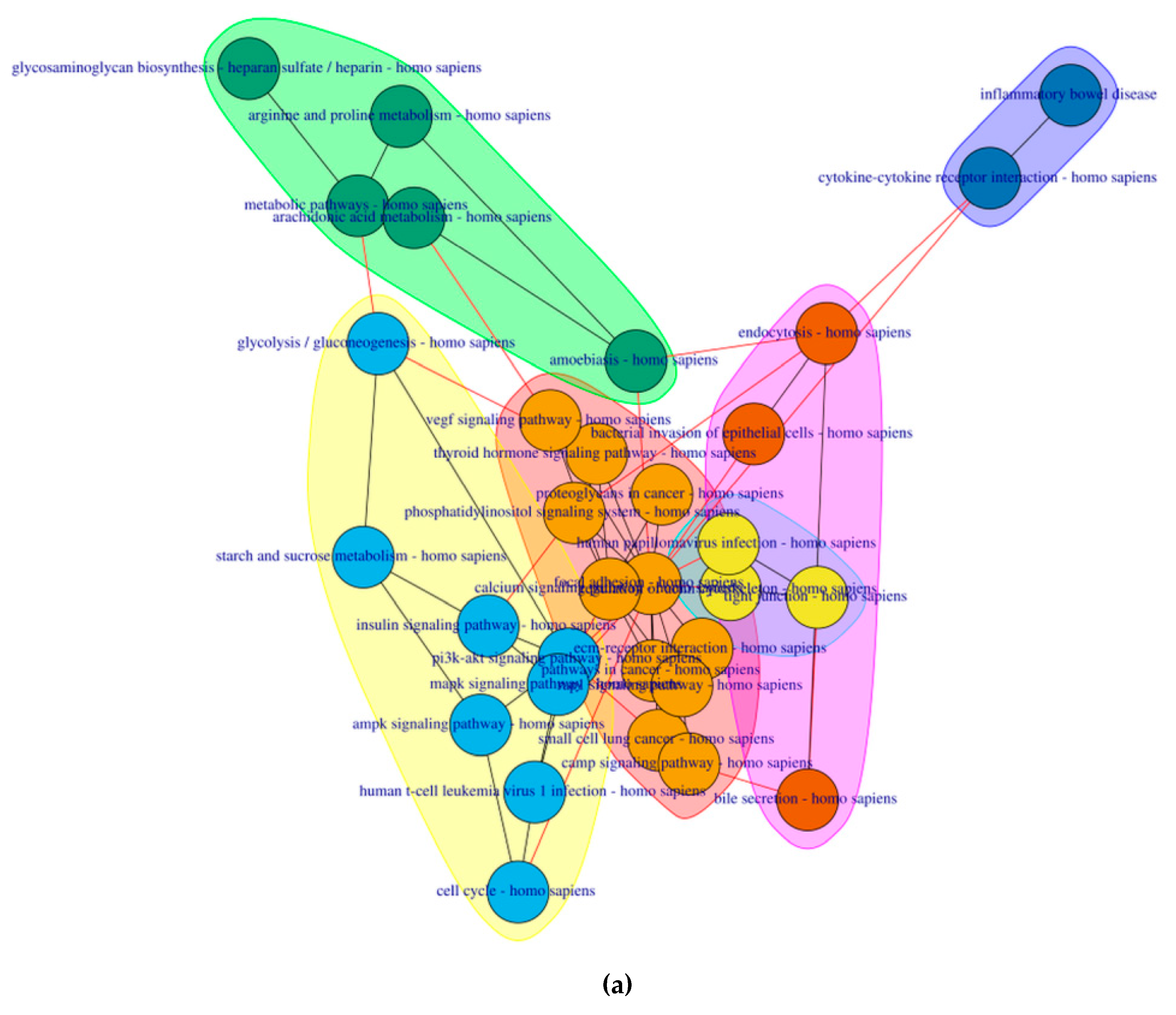
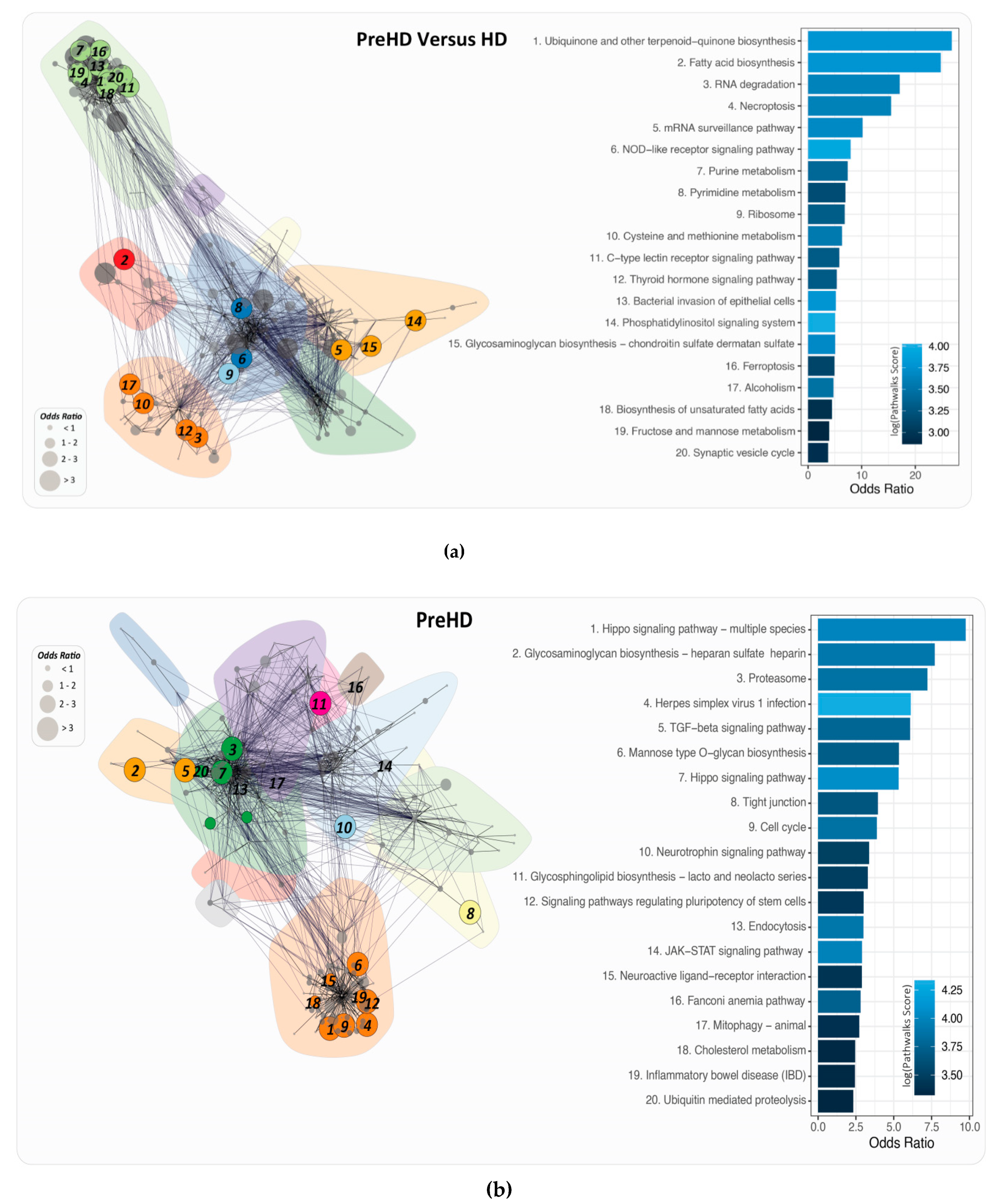
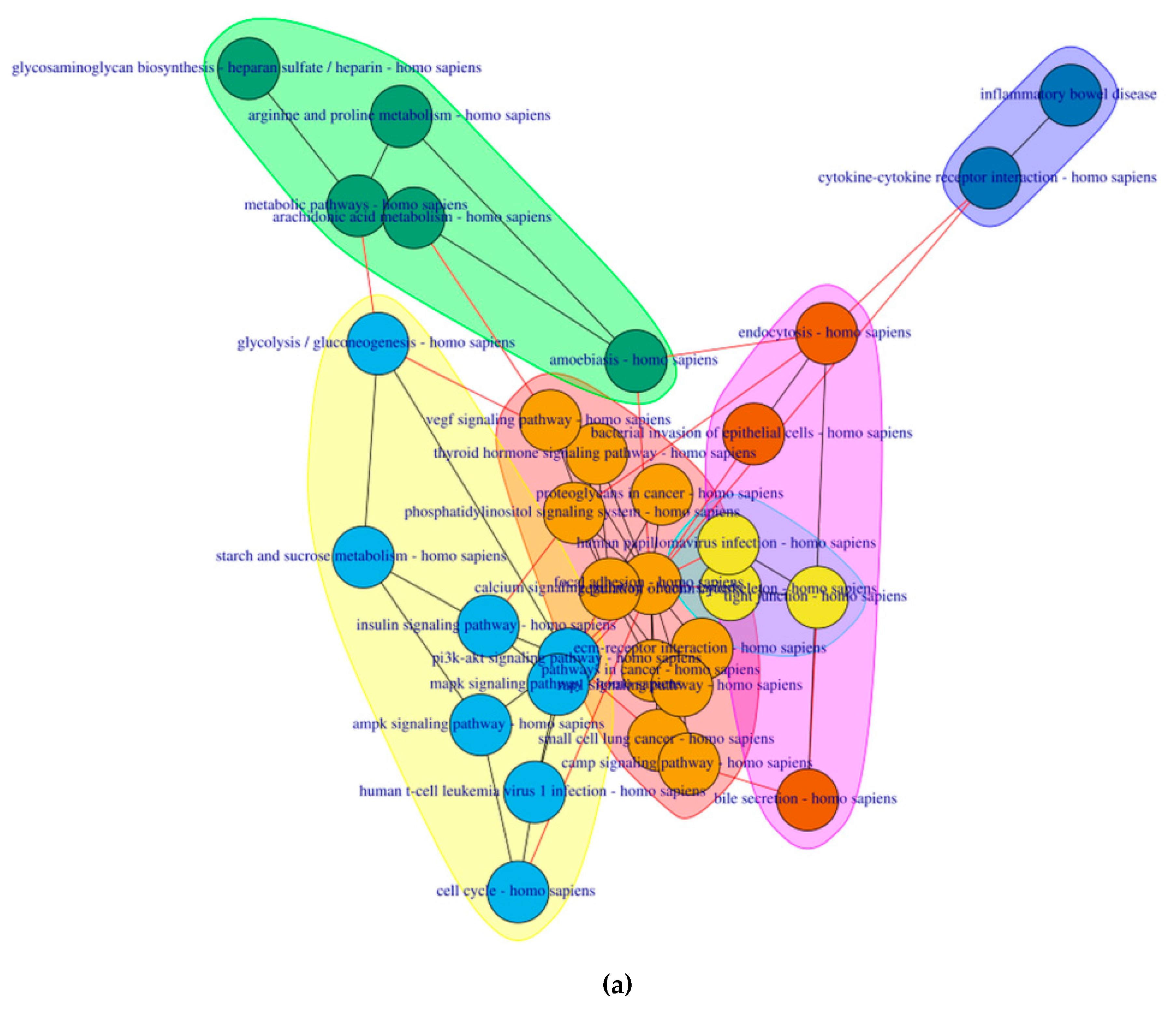
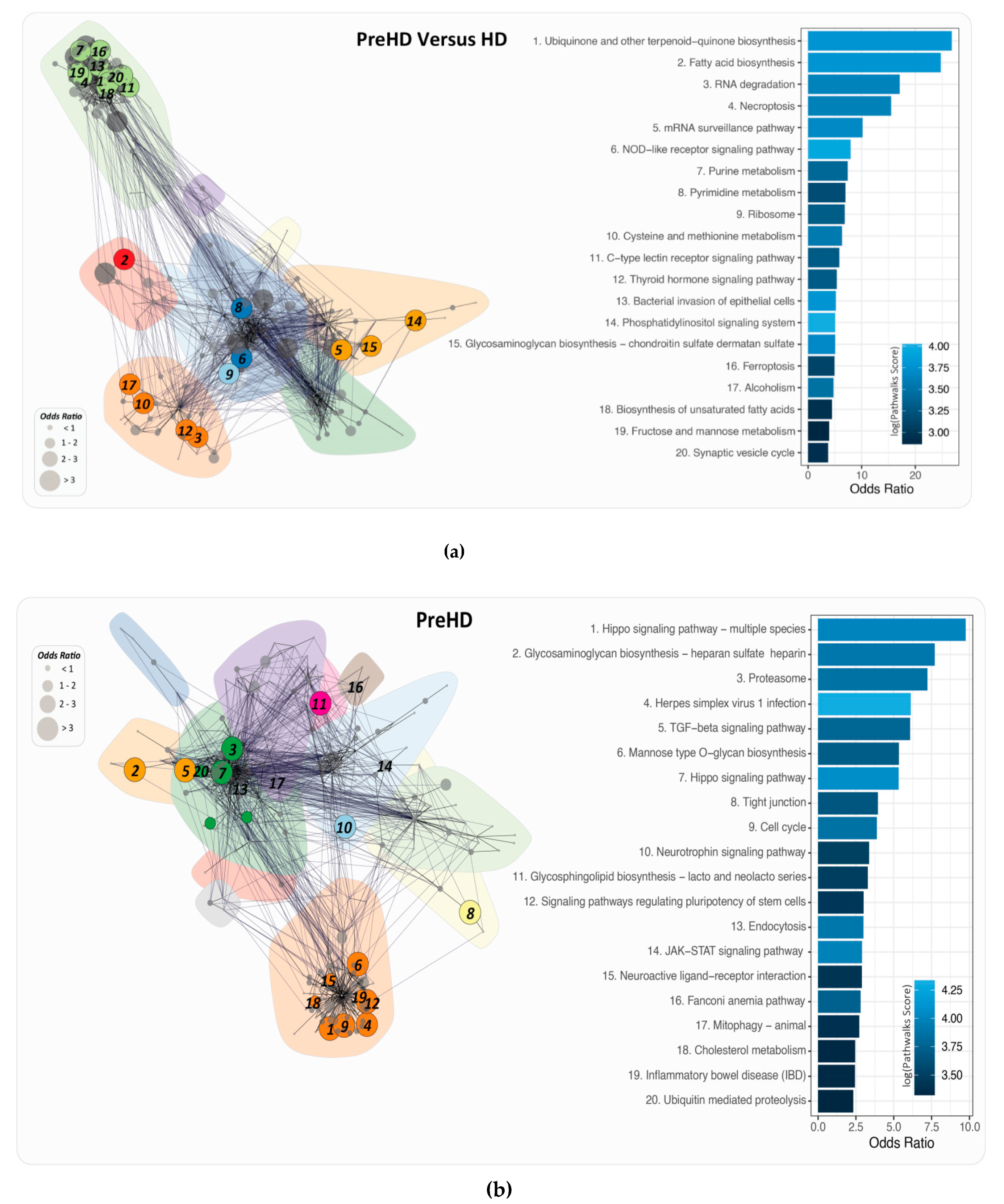
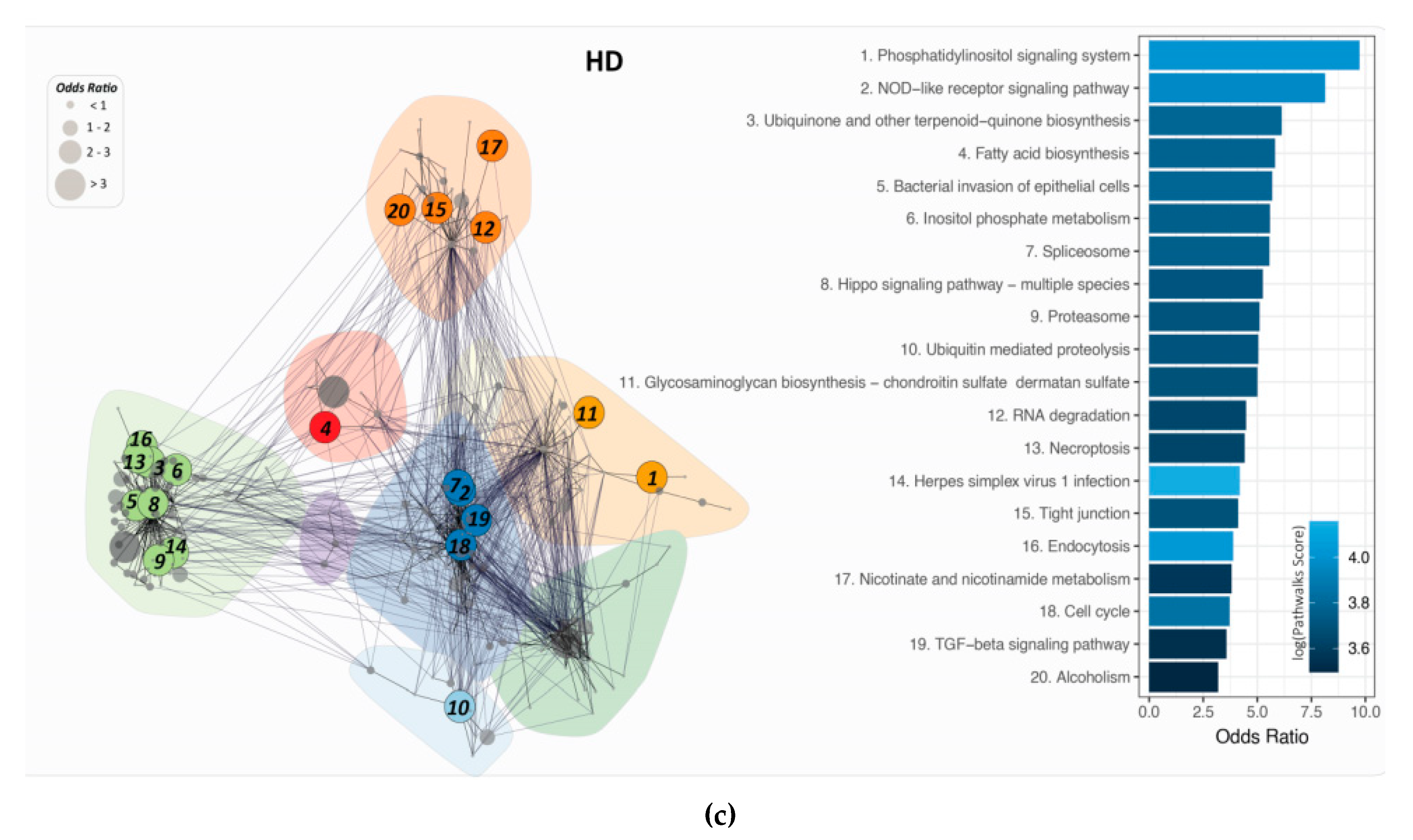
2.4. PathwayConnector Clustering of Pathways Identifed for Pre-Symptomatic and Symptomatic HD Patients
2.5. GeneTrail3 for the Identification of Pathways by Analysing the DEGs for Pre-Symptomatic and Symptomatic HD Patients
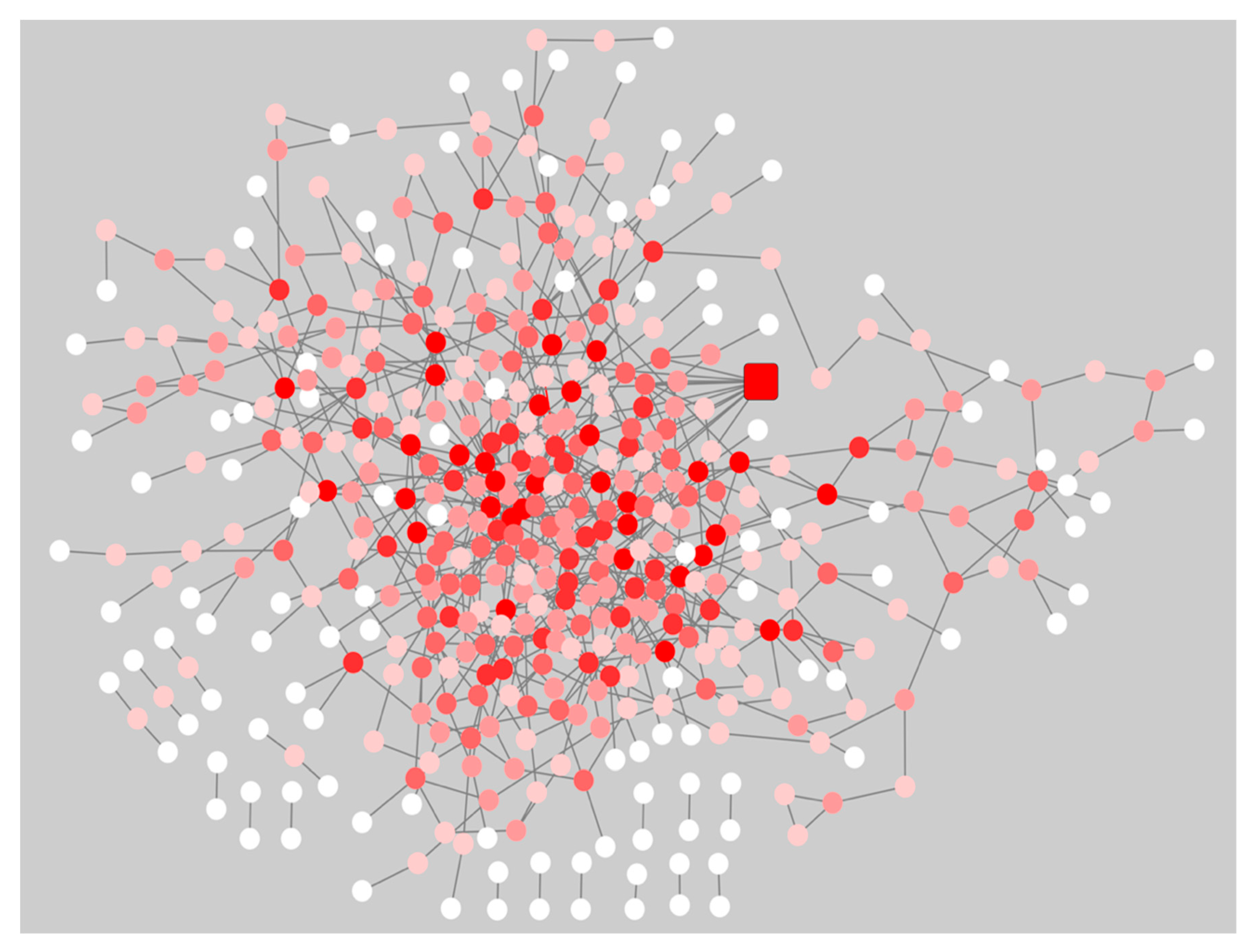
2.6. PathWalks for the Analysis of Over and Under Expressed Genes for Pre-Symptomatic and Symptomatic HD Patients
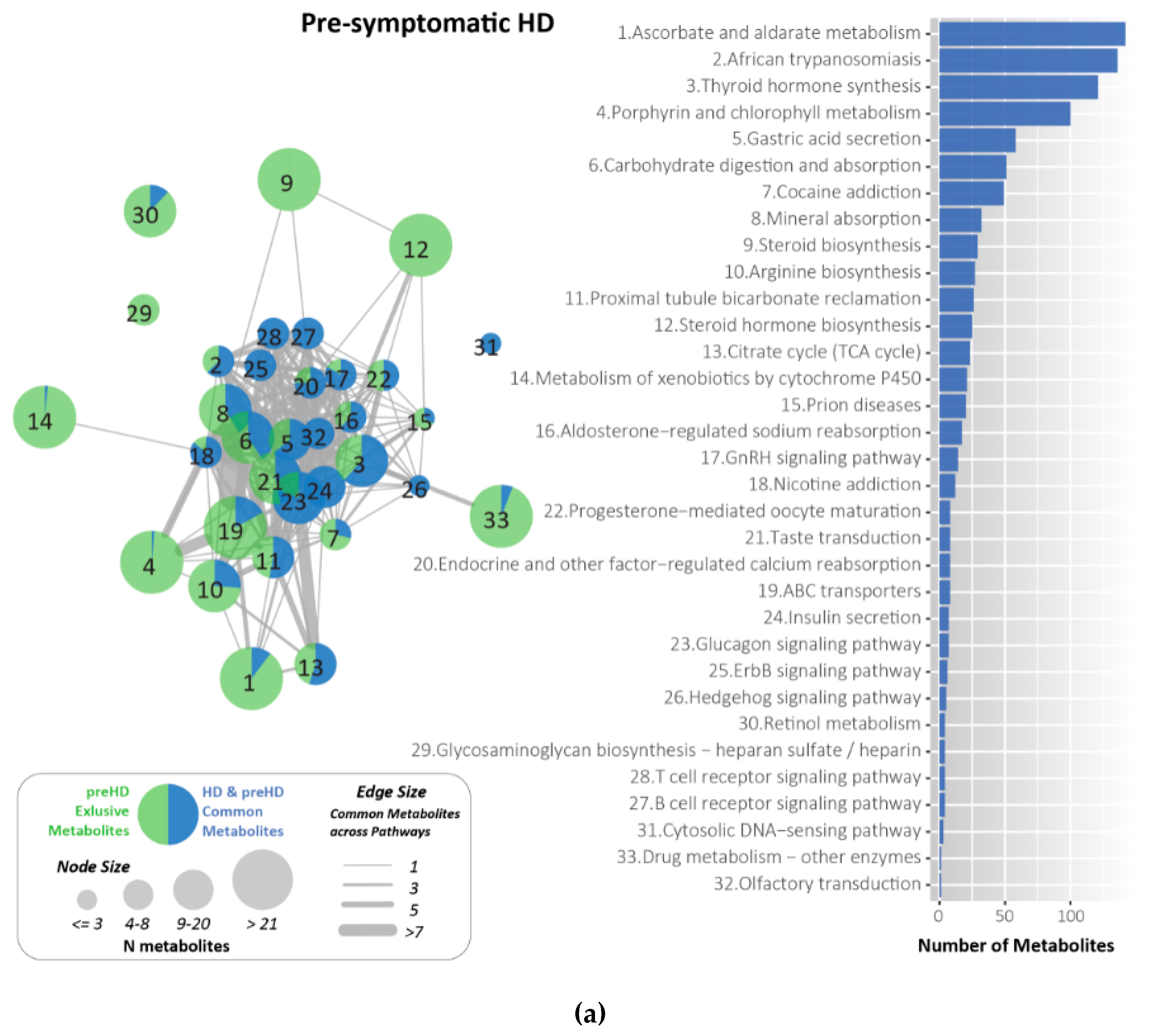
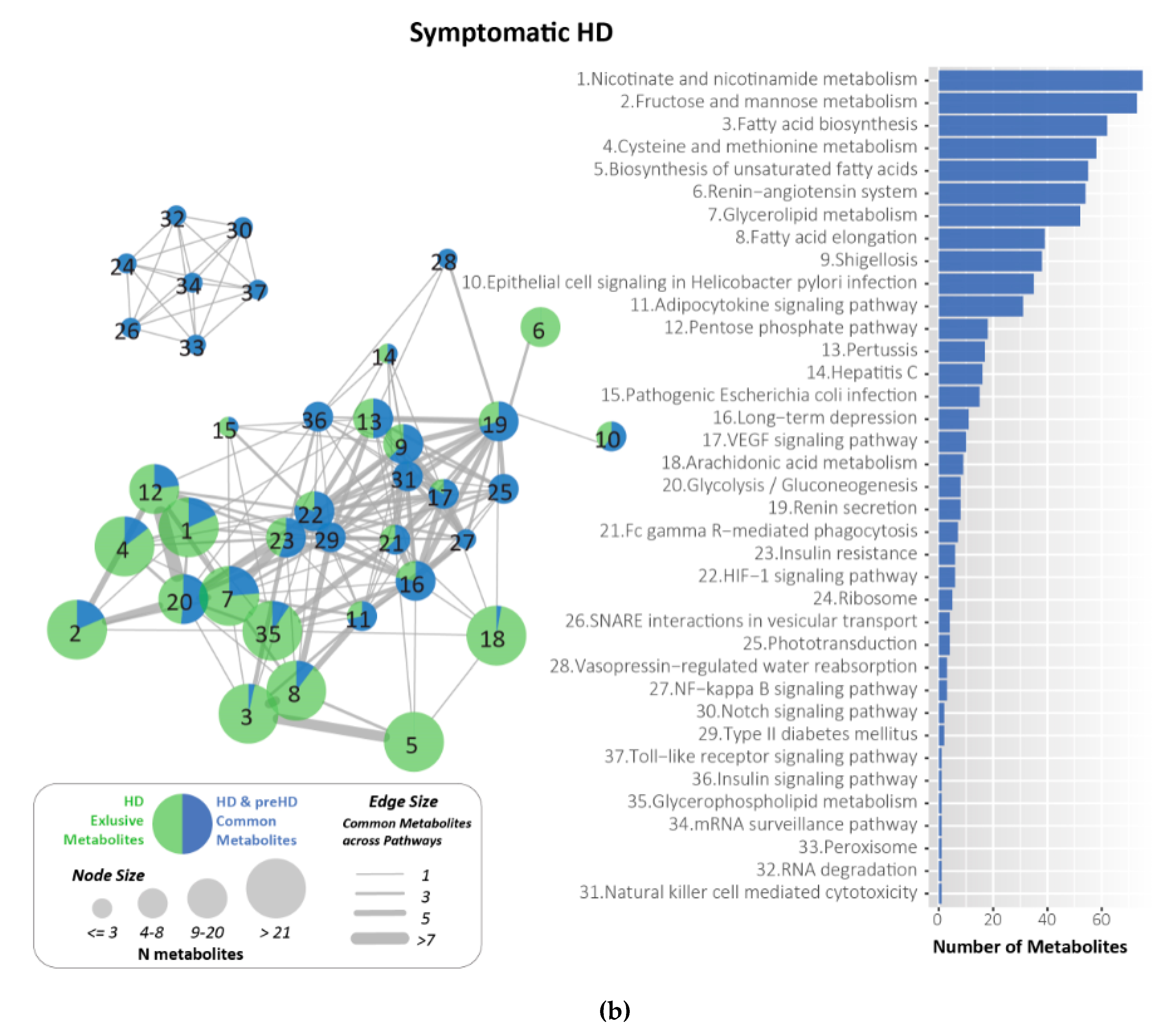
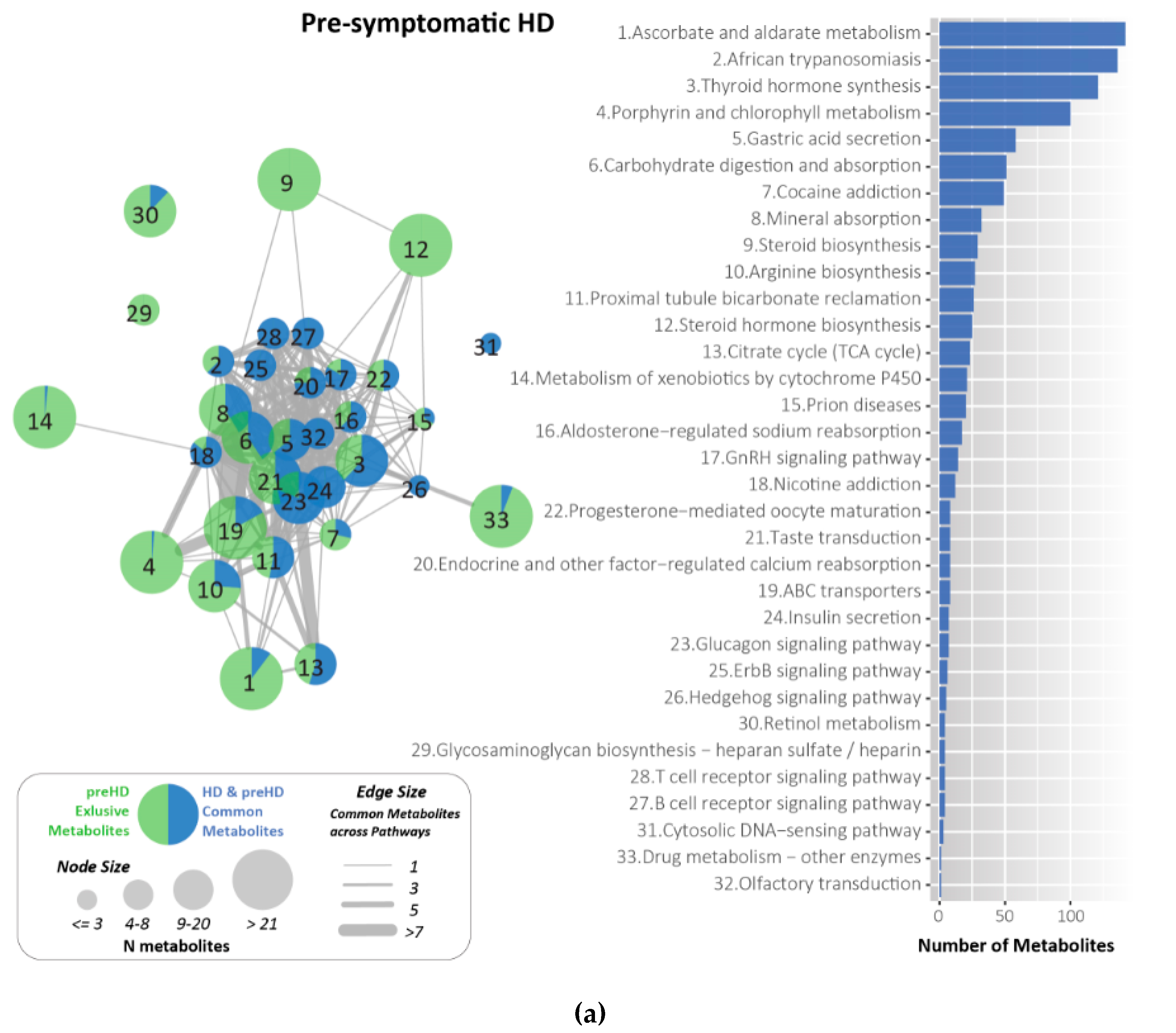
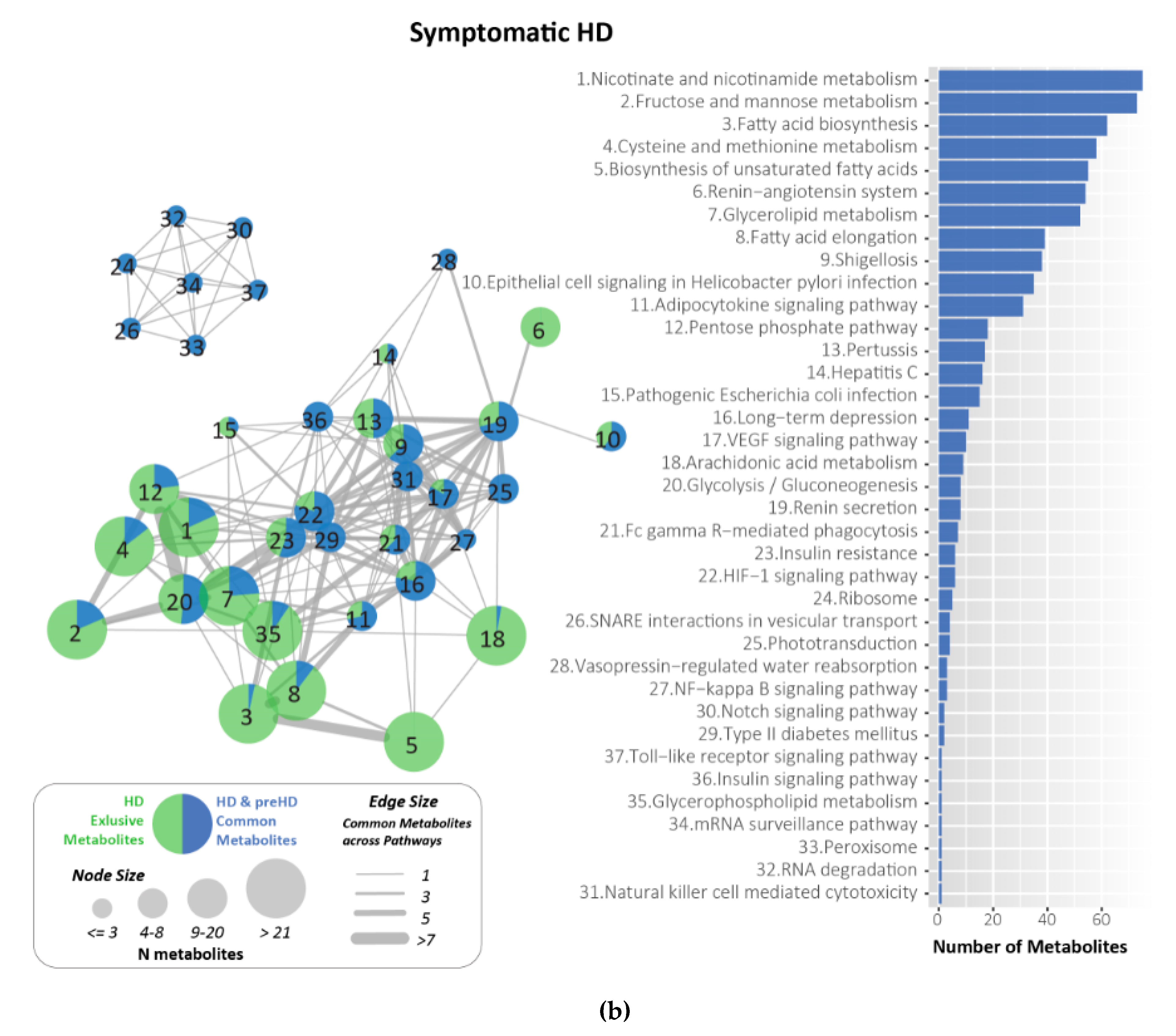
2.7. Metabolites Identified and Related to HD Using KEGG
3. Discussion
4. Materials and Methods
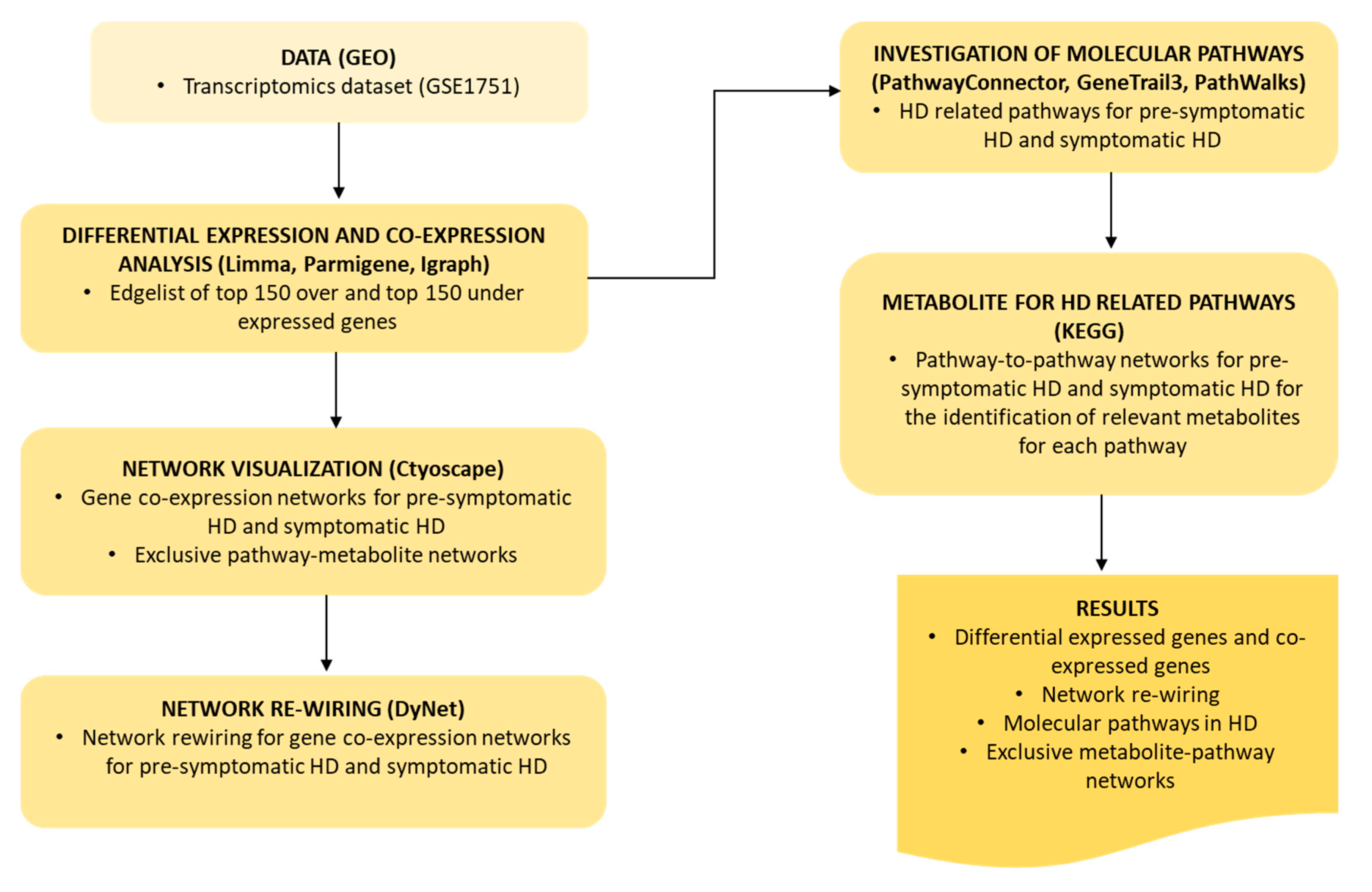
4.1. Data
4.2. Differential Expression and Gene Co-Expression Analysis
4.3. Network Visualization and Analysis
4.4. Network Re-Wiring
4.5. Investigation of Molecular Pathways Related to HD
4.5.1. PathwayConnector for Complementary Pathway-To-Pathway Networks
4.5.2. GeneTrial3 for the Identification of Biological Processes and Pathways
4.5.3. PathWalks Highlighting Pathway Communities
4.6. Metabolites for HD Related Pathways
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACLY | ATP citrate lyase |
| AD | Alzheimer’s Disease |
| ADORA2A | Adenosine A2a receptor |
| BCL-2 | B-cell lymphoma |
| BAG5 | BAG Co-chaperone 5 |
| Ca2+ | Calcium cation |
| CACNA1E | Calcium voltage-gated channel subunit alpha 1S subunit |
| CAG | Cytosine-adenine-guanine |
| CASP | Caspases |
| CBP | Calcium-binding protein |
| CSF | Cerebrospinal Fluid |
| COX7B | Cytochrome c oxidase subunit 7B |
| CNS | Central Nervous System |
| CREB | cAMP response element binding protein |
| DA | Dopamine |
| DEGs | Differentially expressed genes |
| DNAJ | DnaJ heat shock protein family |
| FDR | False Discovery Rate |
| FOXO3 | Forkhead Box O3 |
| GEO | Gene Expression Omnibus |
| GO | Gene Ontology |
| HD | Huntington’s disease |
| HIP3K | Huntingtin Interacting Protein |
| HLADRB4 | Major Histocompatibility Complex, Class II, DR Beta 4 |
| HTT | Huntingtin |
| HVA | Homovanillate |
| InsP3RI | Inositol Trisphosphate Receptor |
| IL | Interleukin |
| iPSCs | Induced Pluripotent Stem Cells |
| ITGA1 | Integrin subunit Alpha 1 |
| KCND2 | Potassium Voltage-Gated Channel Subfamily D Member 2 |
| KEGG | Kyoto Encyclopedia of genes and genomes |
| MAP2 | Microtubule associated protein 2 |
| MAPK | Mitogen-Activated Protein Kinase |
| mtDNA | Mitochondrial DNA |
| MTFRI | Mitochondrial Fission Regulator 1 |
| mHTT | mutant huntingtin |
| NPCs | Neural Progenitors Cells |
| OR | Odds Ratio |
| OXR1 | Oxidation resistance 1 |
| PD | Parkinson’s Disease |
| PET | Positron Emission Tomography |
| PIP4K | Phosphatidylinositol-5-Phosphate 4-Kinase |
| PIP4K2C | Phosphatidylinositol-5-Phosphate 4-Kinase Type 2 Gamma |
| PolyQ | Polyglutamine |
| RYRI | Ryanodine receptor |
| ROS | Reactive Oxygen Species |
| SB | Systems bioinformatics |
| TFC | Total Functional Capacity |
| TGF-β | Transforming Growth Factor Beta |
| TRAF2 | TNF receptor-associated factor 2 |
| UHDRS | Unified Huntington’s Disease Rating Score |
| UPS | Ubiquitin-Proteasome system |
| VGCC | Voltage-gated calcium channels |
References
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Beal, M.F. Antioxidants in Huntington’s disease. Biochim. Biophys. Acta—Mol. Basis Dis. 2012, 1822, 664–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoharan, S.; Guillemin, G.J.; Abiramasundari, R.S.; Essa, M.M.; Akbar, M.; Akbar, M.D. The Role of Reactive Oxygen Species in the Pathogenesis of Alzheimer’s Disease, Parkinson’s Disease, and Huntington’s Disease: A Mini Review. Oxid. Med. Cell. Longev. 2016, 2016, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.O. Huntington’s disease. Semin. Neurol. 2007, 27, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Roos, R.A.C. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, S.J.; Langbehn, D.R.; Leavitt, B.R.; Roos, R.A.C.; Durr, A.; Craufurd, D.; Kennard, C.; Hicks, S.L.; Fox, N.C.; Scahill, R.I.; et al. Biological and clinical manifestations of Huntington’s disease in the longitudinal TRACK-HD study: Cross-sectional analysis of baseline data. Lancet Neurol. 2013, 8, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Oulas, A.; Minadakis, G.; Zachariou, M.; Sokratous, K.; Bourdakou, M.M.; Spyrou, G.M. Systems Bioinformatics: Increasing precision of computational diagnostics and therapeutics through network-based approaches. Brief. Bioinform. 2019, 20, 806–824. [Google Scholar] [CrossRef]
- Fiscon, G.; Conte, F.; Farina, L.; Paci, P. Network-based approaches to explore complex biological systems towards network medicine. Genes 2018, 9, 437. [Google Scholar] [CrossRef] [Green Version]
- Nalbantoglu, S.; Abu-Asab, M.; Suy, S.; Collins, S.; Amri, H. Metabolomics-Based Biosignatures of Prostate Cancer in Patients Following Radiotherapy. OMICS. A J. Integr. Biol. 2019, 23, 214–223. [Google Scholar] [CrossRef]
- Alonso, A.; Marsal, S.; Julià, A. Analytical Methods in Untargeted Metabolomics: State of the Art in 2015. Front. Bioeng. Biotechnol. 2015, 5, 23. [Google Scholar] [CrossRef] [Green Version]
- Goenawan, I.H.; Bryan, K.; Lynn, D.J. DyNet: Visualization and analysis of dynamic molecular interaction networks. Bioinformatics 2016, 32, 2713–2715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safran, M.; Dalah, I.; Alexander, J.; Rosen, N.; Iny Stein, T.; Shmoish, M.; Nativ, N.; Bahir, I.; Doniger, T.; Krug, H.; et al. GeneCards Version 3: The human gene integrator. Database (Oxford) 2010, 2010, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Borovecki, F.; Lovrecic, L.; Zhou, J.; Jeong, H.; Then, F.; Rosas, H.D.; Hersch, S.M.; Hogarth, P.; Bouzou, B.; Jensen, R.V.; et al. Genome-wide expression profiling of human blood reveals biomarkers for Huntington’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 11023–11028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minadakis, G.; Zachariou, M.; Oulas, A.; Spyrou, G.M. PathwayConnector: Finding complementary pathways to enhance functional analysis. Bioinformatics 2019, 35, 889–891. [Google Scholar] [CrossRef] [Green Version]
- Zachariou, M.; Minadakis, G.; Oulas, A.; Afxenti, S.; Spyrou, G.M. Integrating multi-source information on a single network to detect disease-related clusters of molecular mechanisms. J. Proteom. 2018, 188, 15–29. [Google Scholar] [CrossRef]
- Backes, C.; Keller, A.; Kuentzer, J.; Kneissl, B.; Comtesse, N.; Elnakady, Y.A.; Müller, R.; Meese, E.; Lenhof, H.P. GeneTrail-advanced gene set enrichment analysis. Nucleic Acids Res. 2007, 35, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Karatzas, E.; Zachariou, M.; Bourdakou, M.M.; Minadakis, G.; Oulas, A.; Kolios, G.; Delis, A.; Spyrou, G.M. PathWalks: Identifying pathway communities using a disease-related map of integrated information. Bioinformatics 2020. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2009. [Google Scholar] [CrossRef] [Green Version]
- Kashima, R.; Hata, A. The role of TGF-β superfamily signaling in neurological disorders. Acta Biochim. Biophys. Sin. (Shanghai) 2018, 50, 106–120. [Google Scholar] [CrossRef] [Green Version]
- Demaurex, N.; Nunes, P. The role of STIM and ORAI proteins in phagocytic immune cells. Am. J. Physiol.—Cell Physiol. 2016, 310, C496–C508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolobkova, Y.; Vigont, V.; AShakygin, V.; Kaznacheyeva, E. Huntington’s Disease: Calcium Dyshomeostasis and Pathology Models. Acta Nat. 2017, 9, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Calì, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.; Croft, B.; Teng, C.; Awasthi, V. The ubiquitin-proteasome system in neurodegenerative disorders. J. Drug Metab. Toxicol. 2015, 6, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.; Brundin, P.; Li, J.Y. Synaptic dysfunction in Huntington’s disease: A new perspective. Cell. Mol. Life Sci. 2005, 62, 1901–1912. [Google Scholar] [CrossRef]
- Capra, M.; Nuciforo, P.G.; Confalonieri, S.; Quarto, M.; Bianchi, M.; Nebuloni, M.; Boldorini, R.; Pallotti, F.; Viale, G.; Gishizky, M.L.; et al. Frequent alterations in the expression of serine/threonine kinases in human cancers. Cancer Res. 2006, 66, 8147–8154. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Fu, Y.; Liang, Y.; Song, H.; Yao, Y.; Wu, P.; Yao, Y.; Pan, Y.; Wen, X.; Ma, L.; et al. Suppression of MAPK11 or HIPK3 reduces mutant Huntingtin levels in Huntington’s disease models. Cell Res. 2017, 27, 1441–1465. [Google Scholar] [CrossRef]
- Raghu, P.; Joseph, A.; Krishnan, H.; Singh, P.; Saha, S. Phosphoinositides: Regulators of Nervous System Function in Health and Disease. Front. Mol. Neurosci. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Xu, J.; Lazarovici, P.; Quirion, R.; Zheng, W. cAMP Response Element-Binding Protein (CREB): A Possible Signaling Molecule Link in the Pathophysiology of Schizophrenia. Front. Mol. Neurosci. 2018, 11, 1–14. [Google Scholar] [CrossRef]
- Obrietan, K.; Hoyt, K.R. CRE-Mediated Transcription Is Increased in Huntington’s Disease Transgenic Mice. J. Neurosci. 2004, 24, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Wang, E.A.; Cepeda, C.; Levine, M.S. Dopamine imbalance in Huntington’s disease: A mechanism for the lack of behavioral flexibility. Front. Neurosci. 2013, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, G.W.; Eisenhofer, G.; Jennings, G.L.; Esler, M.D. Regional homovanillic acid production in humans. Life Sci. 1993, 53, 63–75. [Google Scholar] [CrossRef]
- Marcelis, M.; Suckling, J.; Hofman, P.; Woodruff, P.; Bullmore, E.; van Os, J. Evidence that brain tissue volumes are associated with HVA reactivity to metabolic stress in schizophrenia. Schizophr. Res. 2006, 86, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Kurlan, R.; Goldblatt, D.; Zaczek, R.; Jeffries, K.; Irvine, C.; Coyle, J.; Shoulson, I. Cerebrospinal fluid homovanillic acid and parkinsonism in Huntington’s disease. Ann. Neurol. 1988, 24, 282–284. [Google Scholar] [CrossRef] [PubMed]
- Markianos, M.; Panas, M.; Kalfakis, N.; Vassilopoulos, D. Plasma homovanillic acid and prolactin in huntington’s disease. Neurochem. Res. 2009, 34, 917–922. [Google Scholar] [CrossRef]
- Reynolds, G.P.; Garrett, N.J. Striatal dopamine and homovanillic acid in Huntington’s Disease. J. Neural Transm. 1986, 65, 151–155. [Google Scholar] [CrossRef]
- Kish, S.J.; Shannak, K.; Hornykiewicz, O. Elevated serotonin and reduced dopamine in subregionally divided Huntington’s disease striatum. Ann. Neurol. 1987, 22, 386–389. [Google Scholar] [CrossRef]
- Saucedo, L.J.; Edgar, B.A. Filling out the Hippo pathway. Nat. Rev. Mol. Cell Biol. 2007, 8, 613–621. [Google Scholar] [CrossRef]
- Mueller, K.A.; Glajch, K.E.; Huizenga, M.N.; Wilson, R.A.; Granucci, E.J.; Dios, A.M.; Tousley, A.R.; Iuliano, M.; Weisman, E.; LaQuaglia, M.J.; et al. Hippo Signaling Pathway Dysregulation in Human Huntington’s Disease Brain and Neuronal Stem Cells. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Majerus, P.W. Phosphatidylinositol signalling reactions. Semin. Cell Dev. Biol. 1998, 9, 153–160. [Google Scholar] [CrossRef]
- Kakouri, A.C.; Christodoulou, C.C.; Zachariou, M.; Oulas, A.; Minadakis, G.; Demetriou, C.A.; Votsi, C.; Zamba-Papanicolaou, E.; Christodoulou, K.; Spyrou, G.M. Revealing clusters of connected pathways through multisource data integration in huntington’s disease and spastic ataxia. IEEE J. Biomed. Heal. Inform. 2019, 23, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The Gene Expression Omnibus database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Sales, G.; Romualdi, C. Parmigene-a parallel R package for mutual information estimation and gene network reconstruction. Bioinformatics 2011, 27, 1876–1877. [Google Scholar] [CrossRef] [PubMed]
- Csárdi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Stuart, J.M.; Segal, E.; Koller, D.; Kim, S.K. A gene-coexpression network for global discovery of conserved genetic modules. Science (80-) 2003, 302, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Fabregat, A.; Sidiropoulos, K.; Viteri, G.; Forner, O.; Marin-Garcia, P.; Arnau, V.; D’Eustachio, P.; Stein, L.; Hermjakob, H. Reactome pathway analysis: A high-performance in-memory approach. BMC Bioinform. 2017, 18. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Pathway Name | p-Value |
|---|---|---|
| 1 | Transforming growth factor-beta (TGF)-beta signaling | 0.00235 |
| 2 | Codeine and morphine metabolism | 0.00878 |
| 3 | Focal adhesion | 0.00878 |
| 4 | PI3K-Akt signaling | 0.00878 |
| 5 | Small cell lung cancer | 0.01354 |
| 6 | Methylene tetrahydrofolate reductase (MTHFR) deficiency | 0.02864 |
| 7 | Chromosomal and microsatellite instability in colorectal cancer | 0.03138 |
| 8 | Development and heterogeneity of the innate lymphoid cell (ILC) family | 0.03138 |
| 9 | Oligodendrocyte specification and differentiation(including remyelination), leading to myelin components for central nervous system (CNS) | 0.03138 |
| 10 | Pregnane X receptor pathway | 0.03138 |
| 11 | Ciliary landscape | 0.03437 |
| 12 | Ectoderm differentiation | 0.03437 |
| 13 | Sleep regulation | 0.03667 |
| 14 | 22q11.2 deletion syndrome | 0.03806 |
| 15 | Mesodermal commitment pathway | 0.03806 |
| Rank | Pathway Name | p-Value |
|---|---|---|
| 1 | Small cell lung cancer | 0.00028 |
| 2 | Adipogenesis | 0.00100 |
| 3 | Pregnane X receptor pathway | 0.00368 |
| 4 | Spinal cord injury | 0.00374 |
| 5 | Aryl hydrocarbon receptor netpath | 0.00993 |
| 6 | Integrated breast cancer pathway | 0.00993 |
| 7 | Phosphodiesterases in neuronal function | 0.00993 |
| 8 | Sudden infant death syndrome (SIDS) susceptibility pathways | 0.00993 |
| 9 | Hippo–Yap signaling | 0.01068 |
| 10 | Nuclear receptors meta-pathway | 0.01068 |
| 11 | Pathways affected in adenoid cystic carcinoma | 0.01565 |
| 12 | Non-small cell lung cancer | 0.02004 |
| 13 | Chromosomal and microsatellite instability in colorectal cancer | 0.02053 |
| 14 | Circadian rhythm-related genes | 0.02117 |
| 15 | Ciliary landscape | 0.02573 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christodoulou, C.C.; Zachariou, M.; Tomazou, M.; Karatzas, E.; Demetriou, C.A.; Zamba-Papanicolaou, E.; Spyrou, G.M. Investigating the Transition of Pre-Symptomatic to Symptomatic Huntington’s Disease Status Based on Omics Data. Int. J. Mol. Sci. 2020, 21, 7414. https://doi.org/10.3390/ijms21197414
Christodoulou CC, Zachariou M, Tomazou M, Karatzas E, Demetriou CA, Zamba-Papanicolaou E, Spyrou GM. Investigating the Transition of Pre-Symptomatic to Symptomatic Huntington’s Disease Status Based on Omics Data. International Journal of Molecular Sciences. 2020; 21(19):7414. https://doi.org/10.3390/ijms21197414
Chicago/Turabian StyleChristodoulou, Christiana C., Margarita Zachariou, Marios Tomazou, Evangelos Karatzas, Christiana A. Demetriou, Eleni Zamba-Papanicolaou, and George M. Spyrou. 2020. "Investigating the Transition of Pre-Symptomatic to Symptomatic Huntington’s Disease Status Based on Omics Data" International Journal of Molecular Sciences 21, no. 19: 7414. https://doi.org/10.3390/ijms21197414
APA StyleChristodoulou, C. C., Zachariou, M., Tomazou, M., Karatzas, E., Demetriou, C. A., Zamba-Papanicolaou, E., & Spyrou, G. M. (2020). Investigating the Transition of Pre-Symptomatic to Symptomatic Huntington’s Disease Status Based on Omics Data. International Journal of Molecular Sciences, 21(19), 7414. https://doi.org/10.3390/ijms21197414