Functional Expression and One-Step Protein Purification of Manganese Peroxidase 1 (rMnP1) from Phanerochaete chrysosporium Using the E. coli-Expression System

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
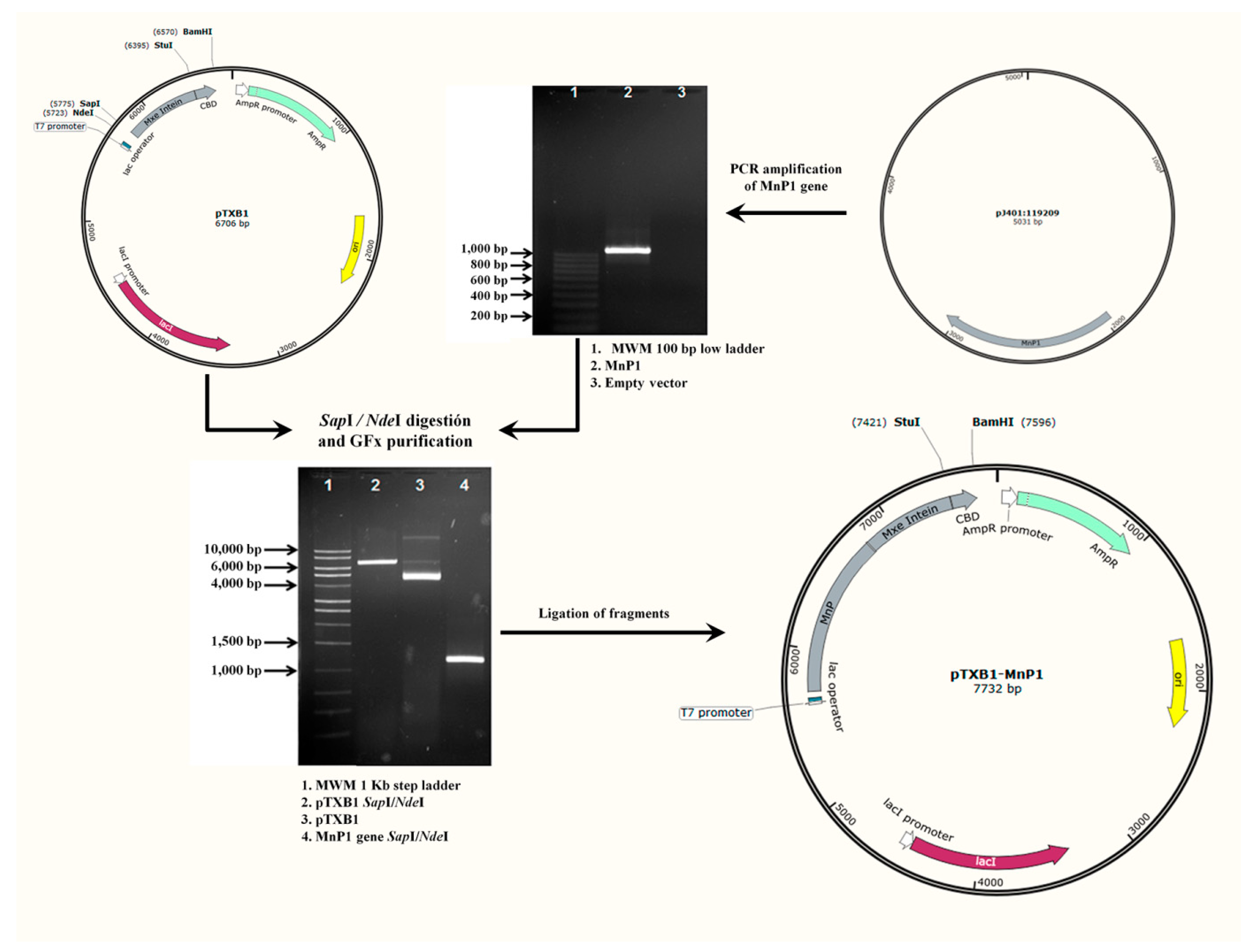
2.1. Construction of Recombinant Expression Vector pTXB1-MnP1
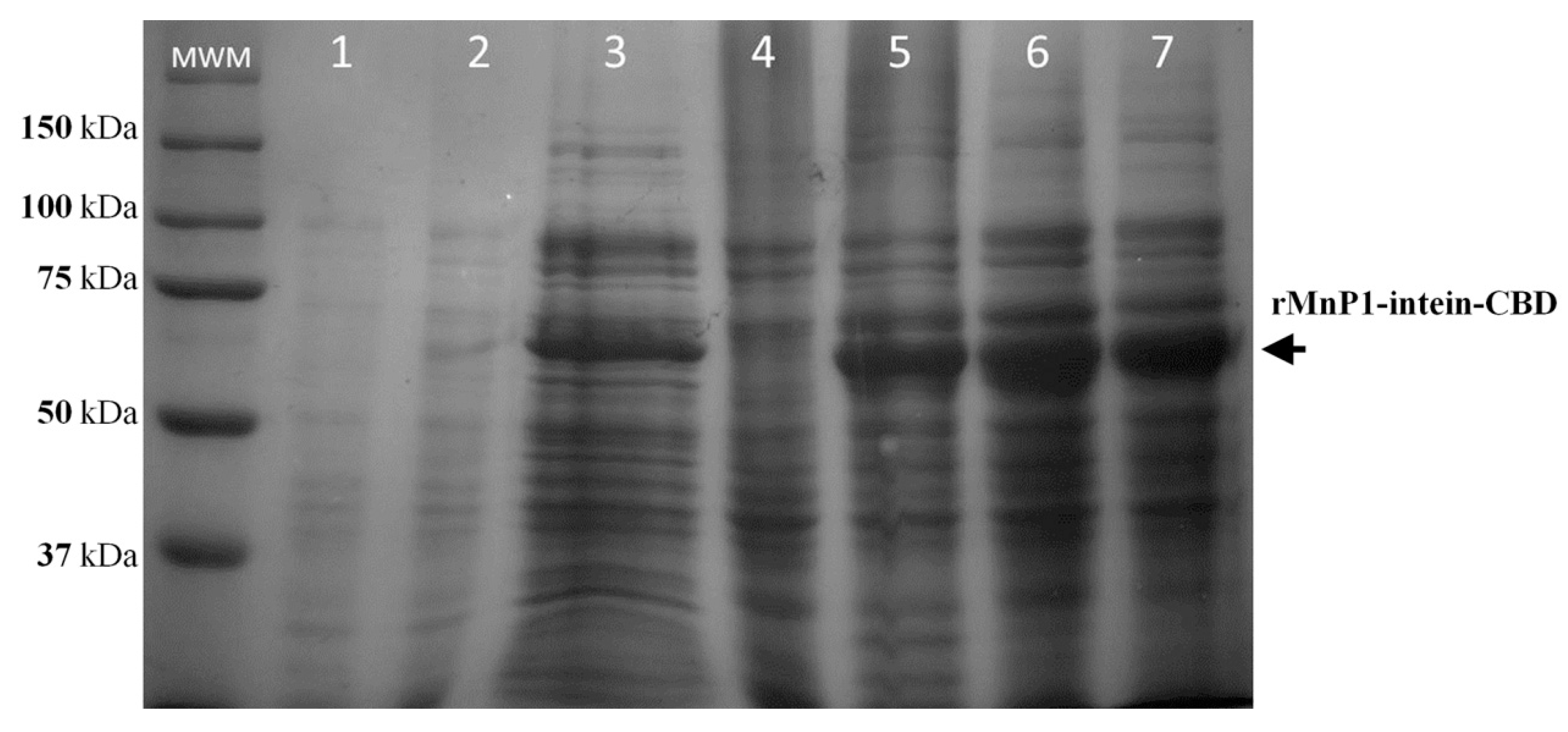
2.2. Expression of Fusion Protein rMnP1−Intein−CBD
2.3. Solubilisation and Purification of rMnP1
2.4. Quantification and Protein Sequencing of rMnP1
2.5. Enzyme Activity
3. Discussion
4. Materials and Methods
4.1. Synthetic MnP1 Gene
4.2. Strains, Vector, Chemicals, Media, Culture Conditions
4.3. PCR Conditions, Cloning, Transformation, and DNA Sequencing
4.4. Expression of Fusion Protein rMnP1−Intein−CBD in E. coli
4.5. Solubilisation of Fusion Protein rMnP1-Intein-CBD from Inclusion Bodies
4.6. Purification of rMnP1 by One-Step Affinity Chromatography and Intein Self-Cleavage
4.7. Quantification and Protein Sequencing of rMnP1
4.8. Enzyme Assays
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wariishi, H.; Valli, K.; Gold, M.H. Manganese(II) oxidation by manganese peroxidase from the basidiomycete Phanerochaete chrysosporium. Kinetic mechanism and role of chelators. J. Biol. Chem. 1992, 267, 23688–23695. [Google Scholar] [PubMed]
- Glenn, J.K.; Gold, M.H. Purification and characterization of an extracellular Mn(II)-dependent peroxidase from the lignin-degrading basidiomycete, Phanerochaete chrysosporium. Arch. Biochem. Biophys. 1985, 242, 329–341. [Google Scholar] [CrossRef]
- Paszczyński, A.; Huynh, V.-B.; Crawford, R. Enzymatic activities of an extracellular, manganese-dependent peroxidase from Phanerochaete chrysosporium. FEMS Microbiol. Lett. 1985, 29, 37–41. [Google Scholar] [CrossRef]
- Glenn, J.K.; Akileswaran, L.; Gold, M.H. Mn(II) oxidation is the principal function of the extracellular Mn-peroxidase from Phanerochaete chrysosporium. Arch. Biochem. Biophys. 1986, 251, 688–696. [Google Scholar] [CrossRef]
- Hatakka, A. Biodegradation of Lignin. In Biopolymers Online; Steinbüchel, A., Ed.; John Wiley and Sons: New York, NY, USA, 2005. [Google Scholar] [CrossRef]
- Higuchi, T. Look back over the studies of lignin biochemistry. J. Wood Sci. 2006, 52, 2–8. [Google Scholar] [CrossRef]
- Martinez, A.T. Molecular biology and structure-function of lignin-degrading heme peroxidases. Enzym. Microb. Technol. 2002, 30, 425–444. [Google Scholar] [CrossRef]
- Conesa, A.; Punt, P.J.; van den Hondel, C.A. Fungal peroxidases: Molecular aspects and applications. J. Biotechnol. 2002, 93, 143–158. [Google Scholar] [CrossRef]
- Miyazaki, C.; Takahashi, H. Engineering of the H2O2-binding pocket region of a recombinant manganese peroxidase to be resistant to H2O2. FEBS Lett. 2001, 509, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Reading, N.S.; Aust, S.D. Engineering a disulfide bond in recombinant manganese peroxidase results in increased thermostability. Biotechnol. Prog. 2000, 16, 326–333. [Google Scholar] [CrossRef]
- Ziegenhagen, D.; Hofrichter, M. A simple and rapid method to gain high amounts of manganese peroxidase with immobilized mycelium of the agaric white-rot fungus Clitocybula dusenii. Appl. Microbiol. Biotechnol. 2000, 53, 553–557. [Google Scholar] [CrossRef]
- Urek, R.O.; Pazarlioglu, N.K. Enhanced production of manganese peroxidase by Phanerochaete chrysosporium. Braz. Arch. Biol. Technol. 2007, 50, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Buswell, J.A.; Mollet, B.; Odier, E. Ligninolytic enzyme production by Phanerochaete chrysosporium under conditions of nitrogen sufficiency. FEMS Microbiol. Lett. 1984, 25, 295–299. [Google Scholar] [CrossRef]
- Bono, J.-J.; Goulas, P.; Boe, J.-F.; Portet, N.; Seris, J.-L. Effect of Mn(II) on reactions catalyzed by lignin peroxidase from Phanerochaete chrysosporium. Eur. J. Biochem. 1990, 192, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Fujian, X.; Hongzhang, C.; Zuohu, L. Solid-state production of lignin peroxidase (LiP) and manganese peroxidase (MnP) by Phanerochaete chrysosporium using steam-exploded straw as substrate. Bioresour. Technol. 2001, 80, 149–151. [Google Scholar] [CrossRef]
- Whitwam, R.E.; Gazarian, I.G.; Tien, M. Expression of fungal Mn peroxidase in E. coli and refolding to yield active enzyme. Biochem. Biophys. Res. Commun. 1995, 216, 1013–1017. [Google Scholar] [CrossRef]
- Whitwam, R.; Tien, M. Heterologous expression and reconstitution of fungal Mn peroxidase. Arch. Biochem. Biophys. 1996, 333, 439–446. [Google Scholar] [CrossRef]
- Wang, N.; Ren, K.; Jia, R.; Chen, W.; Sun, R. Expression of a fungal manganese peroxidase in Escherichia coli: A comparison between the soluble and refolded enzymes. BMC Biotechnol. 2016, 16, 87. [Google Scholar] [CrossRef] [Green Version]
- Pease, E.A.; Aust, S.D.; Tien, M. Heterologous expression of active manganese peroxidase from Phanerochaete chrysosporium using the baculovirus expression system. Biochem. Biophys. Res. Commun. 1991, 179, 897–903. [Google Scholar] [CrossRef]
- Jiang, F.; Kongsaeree, P.; Schilke, K.; Lajoie, C.; Kelly, C. Effects of pH and temperature on recombinant manganese peroxidase production and stability. Appl. Biochem. Biotechnol. 2008, 146, 15–27. [Google Scholar] [CrossRef]
- Xu, H.; Guo, M.-Y.; Gao, Y.-H.; Bai, X.-H.; Zhou, X.-W. Expression and characteristics of manganese peroxidase from Ganoderma lucidum in Pichia pastoris and its application in the degradation of four dyes and phenol. BMC Biotechnol. 2017, 17, 19. [Google Scholar] [CrossRef] [Green Version]
- Hilden, K.; Makela, M.R.; Lundell, T.; Kuuskeri, J.; Chernykh, A.; Golovleva, L.; Archer, D.B.; Hatakka, A. Heterologous expression and structural characterization of two low pH laccases from a biopulping white-rot fungus Physisporinus rivulosus. Appl. Microbiol. Biotechnol. 2013, 97, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Lajoie, C.; Kelly, C. Expression of a Phanerochaete chrysosporium manganese peroxidase gene in the yeast Pichia pastoris. Biotechnol. Prog. 2003, 19, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Kongsaeree, P.; Charron, R.; Lajoie, C.; Xu, H.; Scott, G.; Kelly, C. Production and separation of manganese peroxidase from heme amended yeast cultures. Biotechnol. Bioeng. 2008, 99, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.; Whitwam, R.E.; Kersten, P.J.; Cullen, D.; Tien, M. Efficient expression of a Phanerochaete chrysosporium manganese peroxidase gene in Aspergillus oryzae. Appl. Environ. Microbiol. 1996, 62, 860–864. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Espinosa, D.V.; Absalon, A.E.; Sanchez, N.; Loera, O.; Rodriguez-Vazquez, R.; Fernandez, F.J. Heterologous expression of manganese peroxidase in Aspergillus niger and its effect on phenanthrene removal from soil. J. Mol. Microbiol. Biotechnol. 2011, 21, 120–129. [Google Scholar] [CrossRef]
- Nakano, H.; Yamane, T. Novel techniques using PCR and cell-free protein synthesis systems for combinatorial bioengineering. In Cell-Free Protein Synthesis: Methods and Protocols; Spirin, A.S., Swartz, J.R., Eds.; Wiley-VCH: Weinheim, Germany, 2008; pp. 179–189. [Google Scholar] [CrossRef]
- Ninomiya, R.; Zhu, B.; Kojima, T.; Iwasaki, Y.; Nakano, H. Role of disulfide bond isomerase DsbC, calcium ions, and hemin in cell-free protein synthesis of active manganese peroxidase isolated from Phanerochaete chrysosporium. J. Biosci. Bioeng. 2014, 117, 652–657. [Google Scholar] [CrossRef]
- Perler, F.B.; Davis, E.O.; Dean, G.E.; Gimble, F.S.; Jack, W.E.; Neff, N.; Noren, C.J.; Thorner, J.; Belfort, M. Protein splicing elements: Inteins and exteins. A definition of terms and recommended nomenclature. Nucleic Acids Res. 1994, 22, 1125–1127. [Google Scholar] [CrossRef]
- Singleton, S.F.; Simonette, R.A.; Sharma, N.C.; Roca, A.I. Intein-mediated affinity-fusion purification of the Escherichia coli RecA protein. Protein Expr. Purif. 2002, 26, 476–488. [Google Scholar] [CrossRef]
- Guo, C.; Li, Z.; Shi, Y.; Xu, M.; Wise, J.G.; Trommer, W.E.; Yuan, J. Intein-mediated fusion expression, high efficient refolding, and one-step purification of gelonin toxin. Protein Expr. Purif. 2004, 37, 361–367. [Google Scholar] [CrossRef]
- Wu, W.-Y.; Miller, K.D.; Coolbaugh, M.; Wood, D.W. Intein-mediated one-step purification of Escherichia coli secreted human antibody fragments. Protein Expr. Purif. 2011, 76, 221–228. [Google Scholar] [CrossRef]
- Carrillo-Campos, J. Expression and Purification of the Manganese Peroxidase 1 from Phanerochaete chrysosporium (Expresión y Purificación de la Manganeso Peroxidasa 1 de Phanerochaete chrysosporium). Master’s Thesis, Autonomous University of Chihuahua, Chihuahua, Mexico, 2014. Unpublished. [Google Scholar]
- Wariishi, H.; Akileswaran, L.; Gold, M.H. Manganese peroxidase from the basidiomycete Phanerochaete chrysosporium: Spectral characterization of the oxidized states and the catalytic cycle. Biochemistry 1988, 27, 5365–5370. [Google Scholar] [CrossRef] [PubMed]
- Alfi, A.; Zhu, B.; Damnjanovic, J.; Kojima, T.; Iwasaki, Y.; Nakano, H. Production of active manganese peroxidase in Escherichia coli by co-expression of chaperones and in vitro maturation by ATP-dependent chaperone release. J. Biosci. Bioeng. 2019, 128, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; van den Hondel, C.A.M.J.J.; Punt, P.J. Studies on the production of fungal peroxidases in Aspergillus niger. Appl. Environ. Microbiol. 2000, 66, 3016–3023. [Google Scholar] [CrossRef] [Green Version]
- Paszczyński, A.; Crawford, R.L.; Huynh, V.-B. Manganese peroxidase of Phanerochaete chrysosporium: Purification. In Methods in Enzymology; Academic Press: New York, NY, USA, 1988; Volume 161, pp. 264–270. [Google Scholar]
- Kuan, I.C.; Johnson, K.A.; Tien, M. Kinetic analysis of manganese peroxidase. The reaction with manganese complexes. J. Biol. Chem. 1993, 268, 20064–20070. [Google Scholar] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pech-Canul, A.D.L.C.; Carrillo-Campos, J.; Ballinas-Casarrubias, M.d.L.; Solis-Oviedo, R.L.; Hernández-Rascón, S.K.; Hernández-Ochoa, L.R.; Gutiérrez-Méndez, N.; García-Triana, A. Functional Expression and One-Step Protein Purification of Manganese Peroxidase 1 (rMnP1) from Phanerochaete chrysosporium Using the E. coli-Expression System. Int. J. Mol. Sci. 2020, 21, 416. https://doi.org/10.3390/ijms21020416
Pech-Canul ADLC, Carrillo-Campos J, Ballinas-Casarrubias MdL, Solis-Oviedo RL, Hernández-Rascón SK, Hernández-Ochoa LR, Gutiérrez-Méndez N, García-Triana A. Functional Expression and One-Step Protein Purification of Manganese Peroxidase 1 (rMnP1) from Phanerochaete chrysosporium Using the E. coli-Expression System. International Journal of Molecular Sciences. 2020; 21(2):416. https://doi.org/10.3390/ijms21020416
Chicago/Turabian StylePech-Canul, Angel De La Cruz, Javier Carrillo-Campos, María de Lourdes Ballinas-Casarrubias, Rosa Lidia Solis-Oviedo, Selena Karina Hernández-Rascón, León Raúl Hernández-Ochoa, Néstor Gutiérrez-Méndez, and Antonio García-Triana. 2020. "Functional Expression and One-Step Protein Purification of Manganese Peroxidase 1 (rMnP1) from Phanerochaete chrysosporium Using the E. coli-Expression System" International Journal of Molecular Sciences 21, no. 2: 416. https://doi.org/10.3390/ijms21020416