Allosteric Binding Sites On Nuclear Receptors: Focus On Drug Efficacy and Selectivity



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
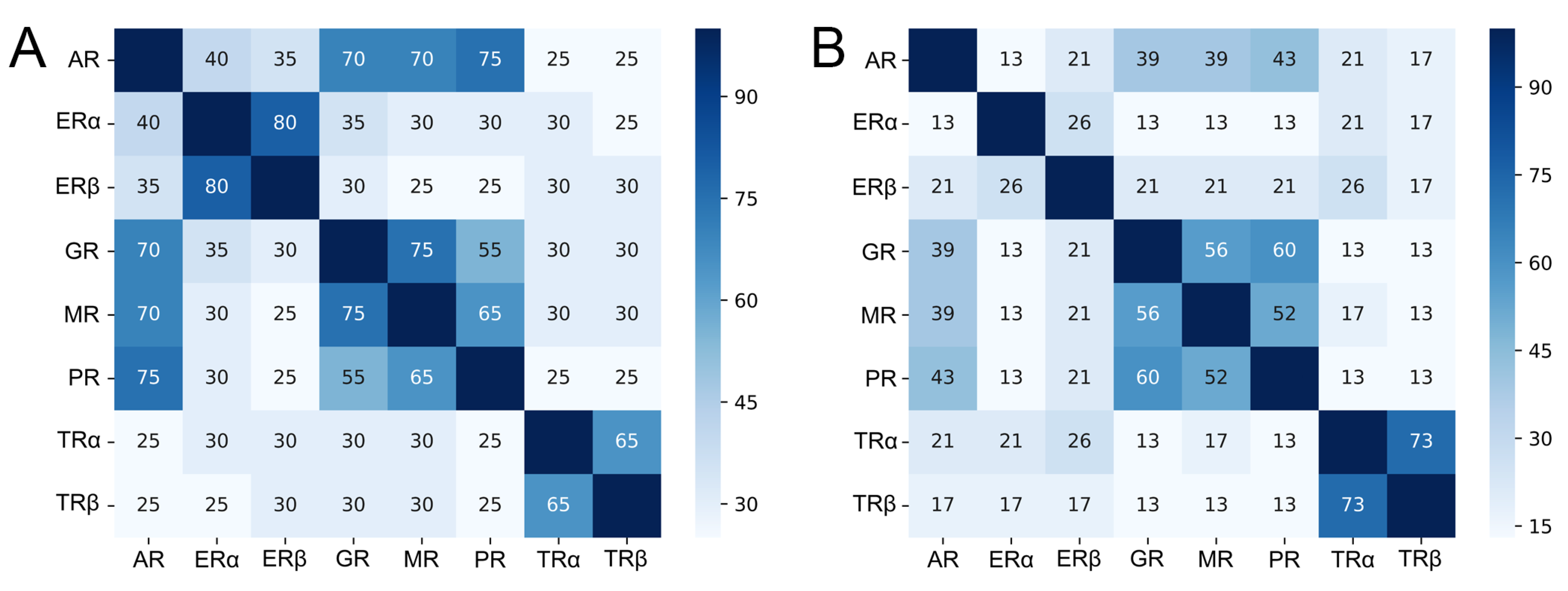
2.1. Sequence Similarity Among Hormonal NRs
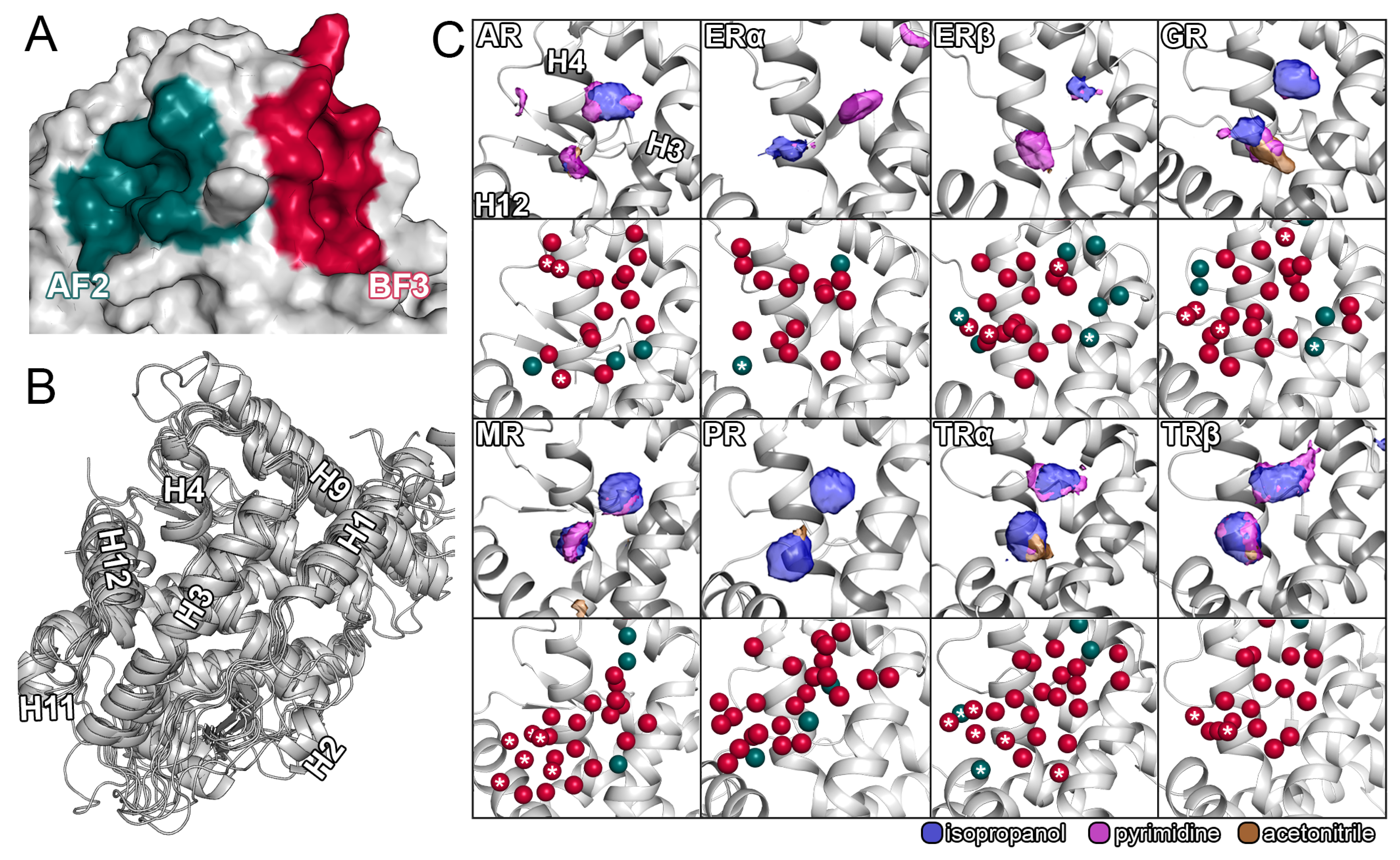
2.2. Distinct Pharmacophores of the Allosteric Sites
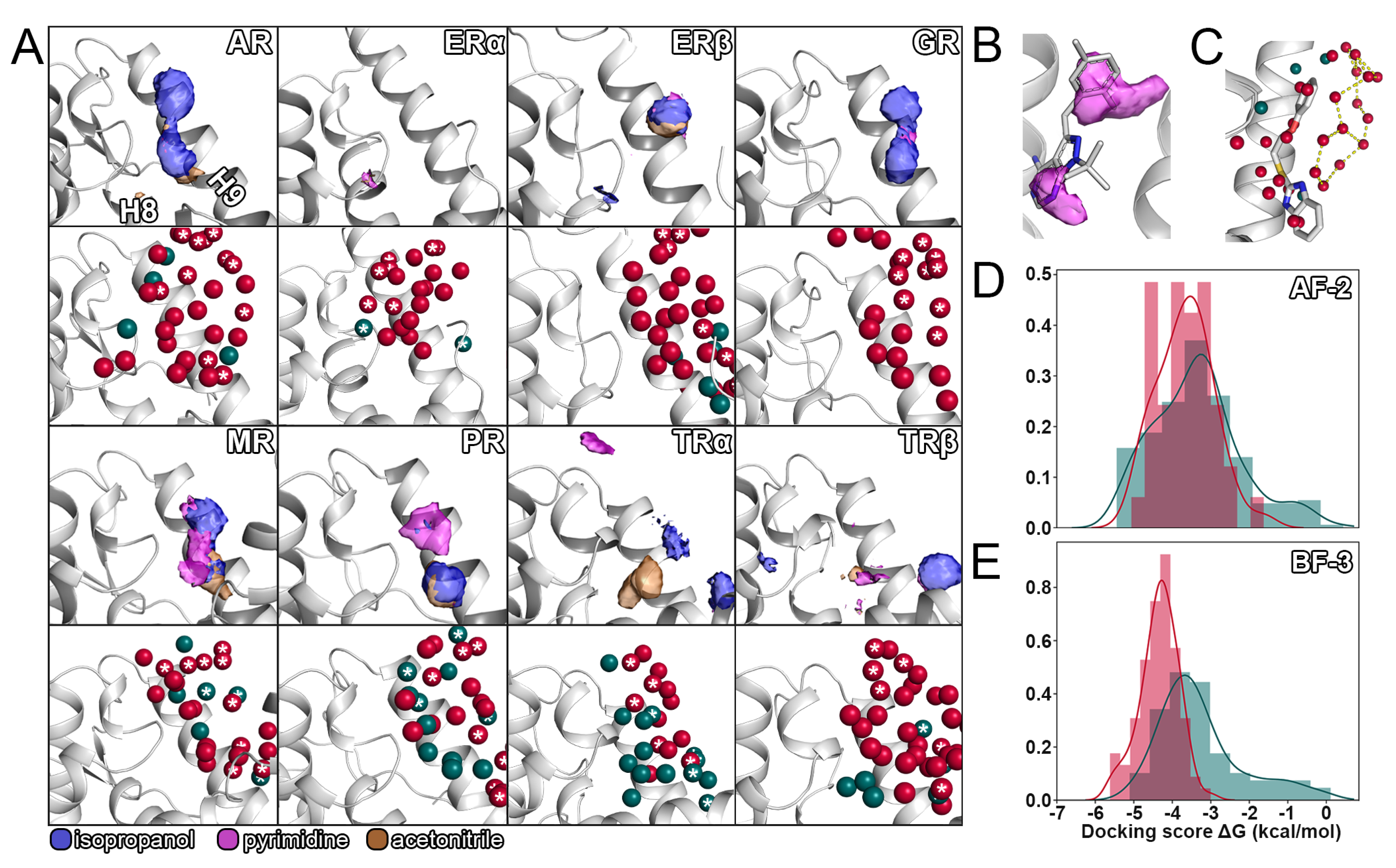
2.3. Conformational Changes of the Allosteric Sites
2.4. Displacement of Water Molecules from the Allosteric Sites
2.5. Selectivity of Allosteric Inhibitors Explored by Molecular Docking
3. Materials and Methods
3.1. Sequence Alignment and Analysis
3.2. Ligand Preparation
3.3. Protein Preparation
3.4. MD Simulations and Evaluation
3.5. Crystal Structure Analysis
3.6. Molecular Docking
3.7. Data Availability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pawlak, M.; Lefebvre, P.; Staels, B. General molecular biology and architecture of nuclear receptors. Curr. Topics Med. Chem. 2012, 12, 486–504. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Ariza, G.; Hulme, C. Recent advances in allosteric androgen receptor inhibitors for the potential treatment of castration-resistant prostate cancer. Pharm. Patent Analyst 2015, 4, 387–402. [Google Scholar] [CrossRef]
- Gao, W.; Bohl, C.E.; Dalton, J.T. Chemistry and structural biology of androgen receptor. Chem. Rev. 2005, 105, 3352–3370. [Google Scholar] [CrossRef] [Green Version]
- Ban, F.; Leblanc, E.; Li, H.; Munuganti, R.S.N.; Frewin, K.; Rennie, P.S.; Cherkasov, A. Discovery of 1 H-indole-2-carboxamides as novel inhibitors of the androgen receptor binding function 3 (BF3). J. Med. Chem. 2014, 57, 6867–6872. [Google Scholar] [CrossRef]
- Gunther, J.R.; Moore, T.W.; Collins, M.L.; Katzenellenbogen, J.A. Amphipathic benzenes are designed inhibitors of the estrogen receptor alpha/steroid receptor coactivator interaction. ACS Chem. Biol. 2008, 3, 282–286. [Google Scholar] [CrossRef] [Green Version]
- Estebanez-Perpina, E.; Arnold, L.A.; Nguyen, P.; Rodrigues, E.D.; Mar, E.; Bateman, R.; Pallai, P.; Shokat, K.M.; Baxter, J.D.; Guy, R.K.; et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc. Nat. Acad. Sci. USA 2007, 104, 16074–16079. [Google Scholar] [CrossRef] [Green Version]
- Caboni, L.; Kinsella, G.K.; Blanco, F.; Fayne, D.; Jagoe, W.N.; Carr, M.; Williams, D.C.; Meegan, M.J.; Lloyd, D.G. “True” antiandrogens-selective non-ligand-binding pocket disruptors of androgen receptor-coactivator interactions: Novel tools for prostate cancer. J. Med. Chem. 2012, 55, 1635–1644. [Google Scholar] [CrossRef]
- Munuganti, R.; Hassona, M.; Leblanc, E.; Frewin, K.; Singh, K.; Ma, D.; Ban, F.; Hsing, M.; Adomat, H.; Lallous, N.; et al. Identification of a Potent Antiandrogen that Targets the BF3 Site of the Androgen Receptor and Inhibits Enzalutamide-Resistant Prostate Cancer. Chem. Biol. 2014, 21, 1476–1485. [Google Scholar] [CrossRef] [Green Version]
- Buzón, V.; Carbó, L.R.; Estruch, S.B.; Fletterick, R.J.; Estébanez-Perpiñ, E. A conserved surface on the ligand binding domain of nuclear receptors for allosteric control. Mol. Cell. Endocrinol. 2012, 348, 394–402. [Google Scholar] [CrossRef]
- Biron, E.; Bédard, F. Recent progress in the development of protein-protein interaction inhibitors targeting androgen receptor-coactivator binding in prostate cancer. J. Steroid Biochem. Mol. Biol. 2016, 161, 36–44. [Google Scholar] [CrossRef]
- Axerio-Cilies, P.; Lack, N.A.; Nayana, M.R.S.; Chan, K.H.; Yeung, A.; Leblanc, E.; Guns, E.S.; Rennie, P.S.; Cherkasov, A. Inhibitors of androgen receptor activation function-2 (AF2) site identified through virtual screening. J. Med. Chem. 2011, 54, 6197–6205. [Google Scholar] [CrossRef]
- Lack, N.A.; Axerio-Cilies, P.; Tavassoli, P.; Han, F.Q.; Chan, K.H.; Feau, C.; LeBlanc, E.; Guns, E.T.; Guy, R.K.; Rennie, P.S.; et al. Targeting the binding function 3 (BF3) site of the human androgen receptor through virtual screening. J. Med. Chem. 2011, 54, 8563–8573. [Google Scholar] [CrossRef] [Green Version]
- Munuganti, R.S.N.; Leblanc, E.; Axerio-Cilies, P.; Labriere, C.; Frewin, K.; Singh, K.; Hassona, M.D.H.; Lack, N.A.; Li, H.; Ban, F.; et al. Targeting the binding function 3 (BF3) site of the androgen receptor through virtual screening. 2. Development of 2-((2-phenoxyethyl) thio)-1H-benzimidazole derivatives. J. Med. Chem. 2013, 56, 1136–1148. [Google Scholar] [CrossRef]
- Gunther, J.R.; Parent, A.A.; Katzenellenbogen, J.A. Alternative inhibition of androgen receptor signaling: Peptidomimetic pyrimidines as direct androgen receptor/coactivator disruptors. ACS Chem. Biol. 2009, 4, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wu, M.; Wang, T.; Xie, Y.; Cui, X.; He, L.; He, Y.; Li, X.; Liu, M.; Hu, L.; et al. Structural Based Screening of Antiandrogen Targeting Activation Function-2 Binding Site. Front. Pharmacol. 2018, 9, 1419. [Google Scholar] [CrossRef] [Green Version]
- Ravindranathan, P.; Lee, T.K.; Yang, L.; Centenera, M.M.; Butler, L.; Tilley, W.D.; Hsieh, J.T.; Ahn, J.M.; Raj, G.V. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nat. Commun. 2013, 4, 1923. [Google Scholar] [CrossRef]
- Lallous, N.; Leblanc, E.; Munuganti, R.S.N.; Hassona, M.D.H.; Nakouzi, N.A.; Awrey, S.; Morin, H.; Roshan-Moniri, M.; Singh, K.; Lawn, S.; et al. Targeting Binding Function-3 of the Androgen Receptor Blocks Its Co-Chaperone Interactions, Nuclear Translocation, and Activation. Mol. Cancer Ther. 2016, 15, 2936–2945. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.B.; Collins, M.L.; Gunther, J.R.; Comninos, J.S.; Katzenellenbogen, J.A. Bicyclo[2.2.2]octanes: close structural mimics of the nuclear receptor-binding motif of steroid receptor coactivators. Bioorganic Med. Chem. Lett. 2007, 17, 4118–4122. [Google Scholar] [CrossRef] [Green Version]
- Parent, A.A.; Gunther, J.R.; Katzenellenbogen, J.A. Blocking estrogen signaling after the hormone: pyrimidine-core inhibitors of estrogen receptor-coactivator binding. J. Med. Chem. 2008, 51, 6512–6530. [Google Scholar] [CrossRef]
- Rodriguez, A.L.; Tamrazi, A.; Collins, M.L.; Katzenellenbogen, J.A. Design, Synthesis, and in Vitro Biological Evaluation of Small Molecule Inhibitors of Estrogen Receptor α Coactivator Binding. J. Med. Chem. 2004, 47, 600–611. [Google Scholar] [CrossRef]
- Singh, K.; Munuganti, R.S.N.; Leblanc, E.; Lin, Y.L.; Leung, E.; Lallous, N.; Butler, M.; Cherkasov, A.; Rennie, P.S. In silico discovery and validation of potent small-molecule inhibitors targeting the activation function 2 site of human oestrogen receptor alpha. Breast Cancer Res. BCR 2015, 17, 27. [Google Scholar] [CrossRef] [Green Version]
- Sun, A.; Moore, T.W.; Gunther, J.R.; Kim, M.S.; Rhoden, E.; Du, Y.; Fu, H.; Snyder, J.P.; Katzenellenbogen, J.A. Discovering small-molecule estrogen receptor α/coactivator binding inhibitors: high-throughput screening, ligand development, and models for enhanced potency. ChemMedChem 2011, 6, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Berrodin, T.J.; Manas, E.; Hauze, D.; Powers, R.; Bapat, A.; Gonder, D.; Winneker, R.C.; Frail, D.E. Identification of novel estrogen receptor α antagonists. J. Steroid Biochem. Mol. Biol. 2004, 88, 351–360. [Google Scholar] [CrossRef]
- Singh, K.; Munuganti, R.S.; Lallous, N.; Dalal, K.; Yoon, J.S.; Sharma, A.; Yamazaki, T.; Cherkasov, A.; Rennie, P.S. Benzothiophenone derivatives targeting mutant forms of estrogen receptor-α in hormone-resistant breast cancers. Int. J. Mol. Sci. 2018, 19, 579. [Google Scholar] [CrossRef] [Green Version]
- Arnold, L.A.; Estebanez-Perpina, E.; Togashi, M.; Jouravel, N.; Shelat, A.; McReynolds, A.C.; Mar, E.; Nguyen, P.; Baxter, J.D.; Fletterick, R.J.; et al. Discovery of small molecule inhibitors of the interaction of the thyroid hormone receptor with transcriptional coregulators. J. Biol. Chem. 2005, 280, 43048–43055. [Google Scholar] [CrossRef] [Green Version]
- Jong, Y.H.; Arnold, L.A.; Zhu, F.; Kosinski, A.; Mangano, T.J.; Setola, V.; Roth, B.L.; Guy, R.K. Improvement of pharmacological properties of irreversible thyroid receptor coactivator binding inhibitors. J. Med. Chem. 2009, 52, 3892–3901. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.Y.; Huang, W.; Arnold, L.A.; Huang, R.; Attia, R.R.; Connelly, M.; Wichterman, J.; Zhu, F.; Augustinaite, I.; Austin, C.P.; et al. Methylsulfonylnitrobenzoates, a new class of irreversible inhibitors of the interaction of the thyroid hormone receptor and its obligate coactivators that functionally antagonizes thyroid hormone. J. Biol. Chem. 2011, 286, 11895–11908. [Google Scholar] [CrossRef] [Green Version]
- Lallous, N.; Dalal, K.; Cherkasov, A.; Rennie, P.S. Targeting alternative sites on the androgen receptor to treat Castration-Resistant Prostate Cancer. Int. J. Mol. Sci. 2013, 14, 12496–12519. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, S.; Lonard, D.M.; O’Malley, B.W. Nuclear Receptor Coactivators: Master Regulators of Human Health and Disease. Ann. Rev. Med. 2014, 65, 279–292. [Google Scholar] [CrossRef] [Green Version]
- Thangudu, R.R.; Bryant, S.H.; Panchenko, A.R.; Madej, T. Modulating protein-protein interactions with small molecules: The importance of binding hotspots. J. Mol. Biol. 2012, 415, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.S.; Spring, D.R.; Abell, C.; Verma, C.S. The Application of Ligand-Mapping Molecular Dynamics Simulations to the Rational Design of Peptidic Modulators of Protein-Protein Interactions. J. Chem. Theory Comput. 2015, 11, 3199–3210. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Lakkaraju, S.K.; Raman, E.P.; Fang, L.; Mackerell, A.D. Pharmacophore modeling using site-identification by ligand competitive saturation (SILCS) with multiple probe molecules. J. Chem. Inf. Model. 2015, 55, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Lakkaraju, S.K.; Yu, W.; Raman, E.P.; Hershfeld, A.V.; Fang, L.; Deshpande, D.A.; MacKerell, A.D. Mapping Functional Group Free Energy Patterns at Protein Occluded Sites: Nuclear Receptors and G-Protein Coupled Receptors. J. Chem. Inf. Model. 2015, 55, 700–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.Y.; Wang, S. Hydrophobic Binding Hot Spots of Bcl-xL Protein-Protein Interfaces by Cosolvent Molecular Dynamics Simulation. ACS Med. Chem. Lett. 2011, 2, 280–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uehara, S.; Tanaka, S. Cosolvent-Based Molecular Dynamics for Ensemble Docking: Practical Method for Generating Druggable Protein Conformations. J. Chem. Inf. Model. 2017, 57, 742–756. [Google Scholar] [CrossRef]
- Mahmoud, A.H.; Yang, Y.; Lill, M.A. Improving Atom-Type Diversity and Sampling in Cosolvent Simulations Using λ-Dynamics. J. Chem. Theory Comput. 2019, 15, 3272–3287. [Google Scholar] [CrossRef]
- Ghanakota, P.; Carlson, H.A. Moving beyond Active-Site Detection: MixMD Applied to Allosteric Systems. J. Phys. Chem. B 2016, 120, 8685–8695. [Google Scholar] [CrossRef] [Green Version]
- Seco, J.; Luque, F.J.; Barril, X. Binding site detection and druggability index from first principles. J. Med. Chem. 2009, 52, 2363–2371. [Google Scholar] [CrossRef]
- Graham, S.E.; Smith, R.D.; Carlson, H.A. Predicting Displaceable Water Sites Using Mixed-Solvent Molecular Dynamics. J. Chem. Inf. Model. 2018, 58, 305–314. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Hu, B.; Lill, M.A. WATsite2.0 with PyMOL Plugin: Hydration Site Prediction and Visualization BT —Protein Function Prediction: Methods and Protocols; Springer: New York, NY, USA, 2017; pp. 123–134. [Google Scholar] [CrossRef]
- Hu, B.; Lill, M.A. WATsite: Hydration site prediction program with PyMOL interface. J. Comput. Chem. 2014, 35, 1255–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lill, M.; Yang, Y.; Mahmoud, A.; Masters, M.; ChemRxiv Preprint, A. Elucidating the Multiple Roles of Hydration in Protein-Ligand Binding via Layerwise Relevance Propagation and Big Data Analytics: Elucidating the Multiple Roles of Hydration in Protein-Ligand Binding via Layerwise Relevance Propagation and Big Data. Chemrxiv 2019, 1–17. [Google Scholar] [CrossRef]
- Tan, M.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallastegui, N.; Estébanez-Perpiñá, E. Thinking Outside the Box: Alternative Binding Sites in the Ligand Binding Domain of Nuclear Receptors BT - Nuclear Receptors: From Structure to the Clinic; Springer International Publishing: Cham, Switzerland, 2015; pp. 179–203. [Google Scholar] [CrossRef]
- Bleach, R.; McIlroy, M. The divergent function of androgen receptor in breast cancer; analysis of steroid mediators and tumor intracrinology. Front. Endocrinol. 2018, 9, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ghanakota, P.; DasGupta, D.; Carlson, H.A. Free Energies and Entropies of Binding Sites Identified by MixMD Cosolvent Simulations. J. Chem. Inf. Model. 2019, 59, 2035–2045. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Smiesko, M.; Smieško, M. Ligand Pathways in Nuclear Receptors. J. Chem. Inf. Model. 2019, 59, 3100–3109. [Google Scholar] [CrossRef]
- Ghanakota, P.; Carlson, H.A. Driving Structure-Based Drug Discovery through Cosolvent Molecular Dynamics. J. Med. Chem. 2016, 59, 10383–10399. [Google Scholar] [CrossRef] [Green Version]
- Adhireksan, Z.; Palermo, G.; Riedel, T.; Ma, Z.; Muhammad, R.; Rothlisberger, U.; Dyson, P.J.; Davey, C.A. Allosteric cross-talk in chromatin can mediate drug-drug synergy. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2015, 8, 38022–38043. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.R.; Hu, H.P.; Ruvinsky, A.M.; Sherman, W.; Favia, A.D. Deciphering Cryptic Binding Sites on Proteins by Mixed-Solvent Molecular Dynamics. J. Chem. Inf. Model. 2017, 57, 1388–1401. [Google Scholar] [CrossRef]
- Geistlinger, T.R.; Guy, R.K. Novel selective inhibitors of the interaction of individual nuclear hormone receptors with a mutually shared steroid receptor coactivator 2. J. Am. Chem. Soc. 2003, 125, 6852–6853. [Google Scholar] [CrossRef] [PubMed]
- Wahl, J.; Smieško, M. Thermodynamic Insight into the Effects of Water Displacement and Rearrangement upon Ligand Modifications using Molecular Dynamics Simulations. ChemMedChem 2018, 13, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Vedani, A.; Dobler, M.; Hu, Z.; Smieško, M. OpenVirtualToxLab-A platform for generating and exchanging in silico toxicity data. Toxicol. Lett. 2015, 232, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Xie, L.; Bourne, P.E. Structure-based systems biology for analyzing off-target binding. Curr. Opin. Struct. Biol. 2011, 21, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Repasky, M.P.; Murphy, R.B.; Banks, J.L.; Greenwood, J.R.; Tubert-Brohman, I.; Bhat, S.; Friesner, R.A. Docking performance of the glide program as evaluated on the Astex and DUD datasets: A complete set of glide SP results and selected results for a new scoring function integrating WaterMap and glide. J. Comput.-Aided Mol. Des. 2012, 26, 787–799. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Varlamov, A.; Vaskin, Y.; Efremov, I.; German Grehov, O.G.; Kandrov, D.; Rasputin, K.; Syabro, M.; et al. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [Green Version]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger LCC. LigPrep 2019-3. 2019. Available online: https://www.schrodinger.com/ligprep (accessed on 30 April 2019).
- Schrödinger LCC. Maestro Small-Molecule Drug Discovery Suite 2019-3. 2019. Available online: https://www.schrodinger.com/jp/suites/small-molecule-drug-discovery-suite (accessed on 30 April 2019).
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comp.-Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comp.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Alpi, E.; Antunes, R.; Bely, B.; Bingley, M.; Bonilla, C.; Britto, R.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Bowers, K.; Chow, E.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossvary, I.; Moraes, M.; Sacerdoti, F.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the ACM/IEEE SC 2006 Conference (SC’06), Tampa, FL, USA, 11–17 November 2006; p. 43. [Google Scholar] [CrossRef] [Green Version]
- Shaw, D.E.; Grossman, J.P.; Bank, J.A.; Batson, B.; Butts, J.A.; Chao, J.C.; Deneroff, M.M.; Dror, R.O.; Even, A.; Fenton, C.H.; et al. Anton 2: Raising the Bar for Performance and Programmability in a Special-Purpose Molecular Dynamics Supercomputer. In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, SC Louisana, NO, USA, 16–21 November 2014; pp. 41–53. [Google Scholar] [CrossRef]
- Defelipe, L.A.; Arcon, J.P.; Modenutti, C.P.; Marti, M.A.; Turjanski, A.G.; Barril, X. Solvents to fragments to drugs: MD applications in drug design. Molecules 2018, 23, 3269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, A.R.; Strauss, C.E.M.; Olmea, O. MAMMOTH (Matching molecular models obtained from theory): An automated method for model comparison. Protein Sci. 2002, 11, 2606–2621. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrodinger LLC: New York, NY, USA, 2015. [Google Scholar]
- Jukič, M.; Konc, J.; Gobec, S.; Janežič, D. Identification of Conserved Water Sites in Protein Structures for Drug Design. J. Chem. Inf. Model. 2017, 57, 3094–3103. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Wahl, J.; Smieško, M. Endocrine disruption at the androgen receptor: Employing molecular dynamics and docking for improved virtual screening and toxicity prediction. Int. J. Mol. Sci. 2018, 19, 1784. [Google Scholar] [CrossRef] [Green Version]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fischer, A.; Smieško, M. Allosteric Binding Sites On Nuclear Receptors: Focus On Drug Efficacy and Selectivity. Int. J. Mol. Sci. 2020, 21, 534. https://doi.org/10.3390/ijms21020534
Fischer A, Smieško M. Allosteric Binding Sites On Nuclear Receptors: Focus On Drug Efficacy and Selectivity. International Journal of Molecular Sciences. 2020; 21(2):534. https://doi.org/10.3390/ijms21020534
Chicago/Turabian StyleFischer, André, and Martin Smieško. 2020. "Allosteric Binding Sites On Nuclear Receptors: Focus On Drug Efficacy and Selectivity" International Journal of Molecular Sciences 21, no. 2: 534. https://doi.org/10.3390/ijms21020534