Combined Transcriptome and Proteome Analysis of Masson Pine (Pinus massoniana Lamb.) Seedling Root in Response to Nitrate and Ammonium Supplementations

Abstract
:1. Introduction
2. Results
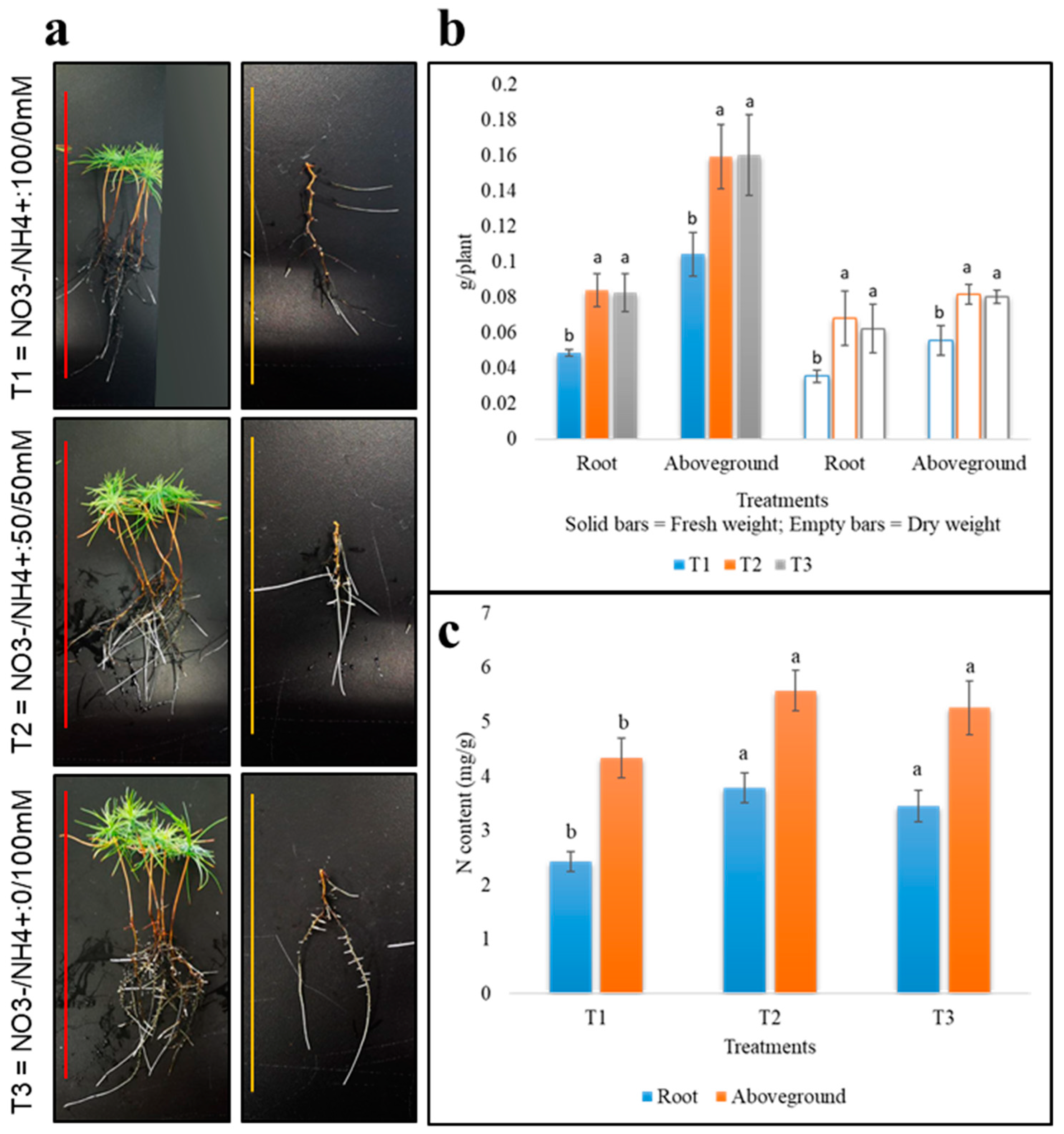
2.1. Morphological Response of Pine Roots under Different N-Nutrition
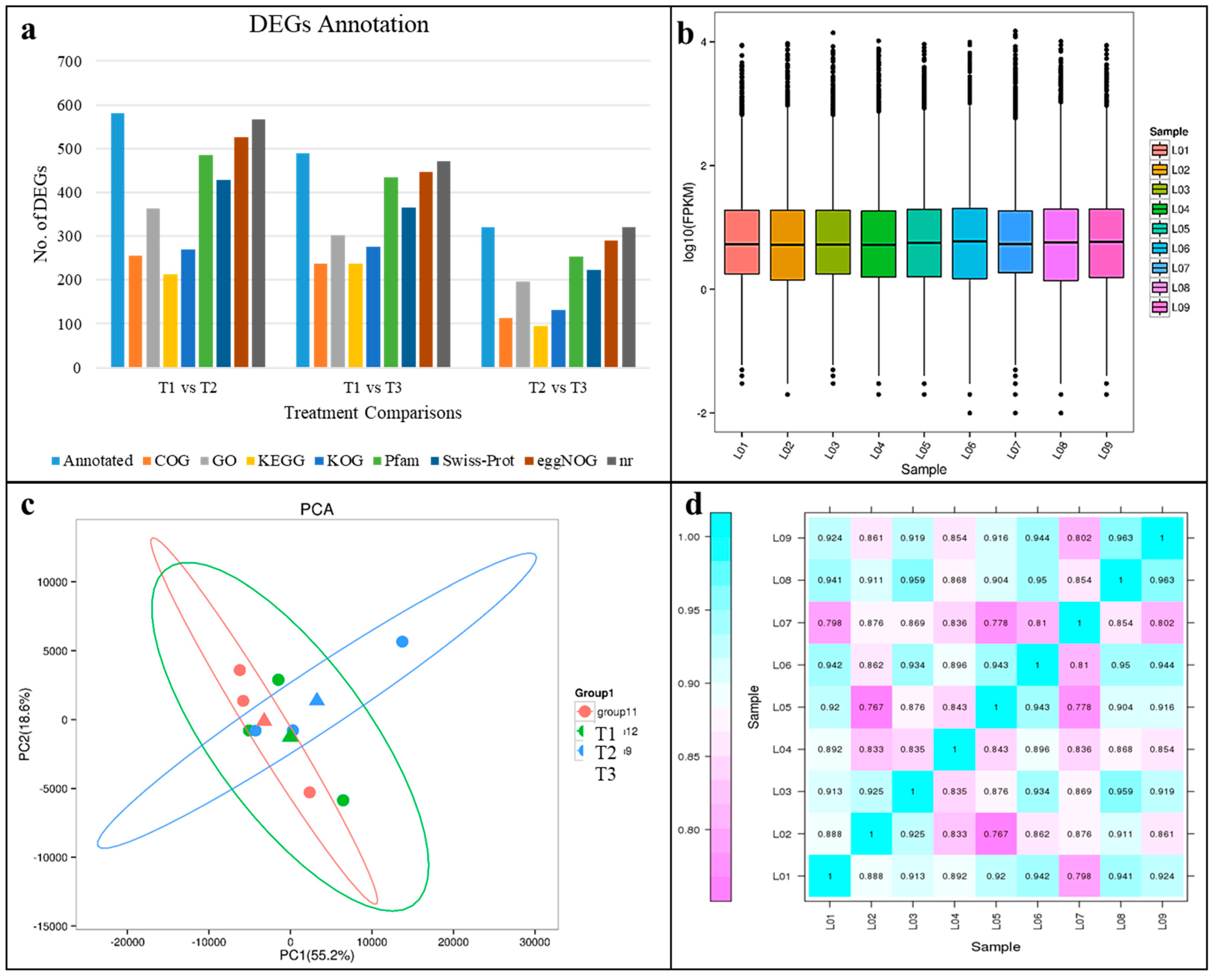
2.2. RNA Sequencing and Transcript Annotation
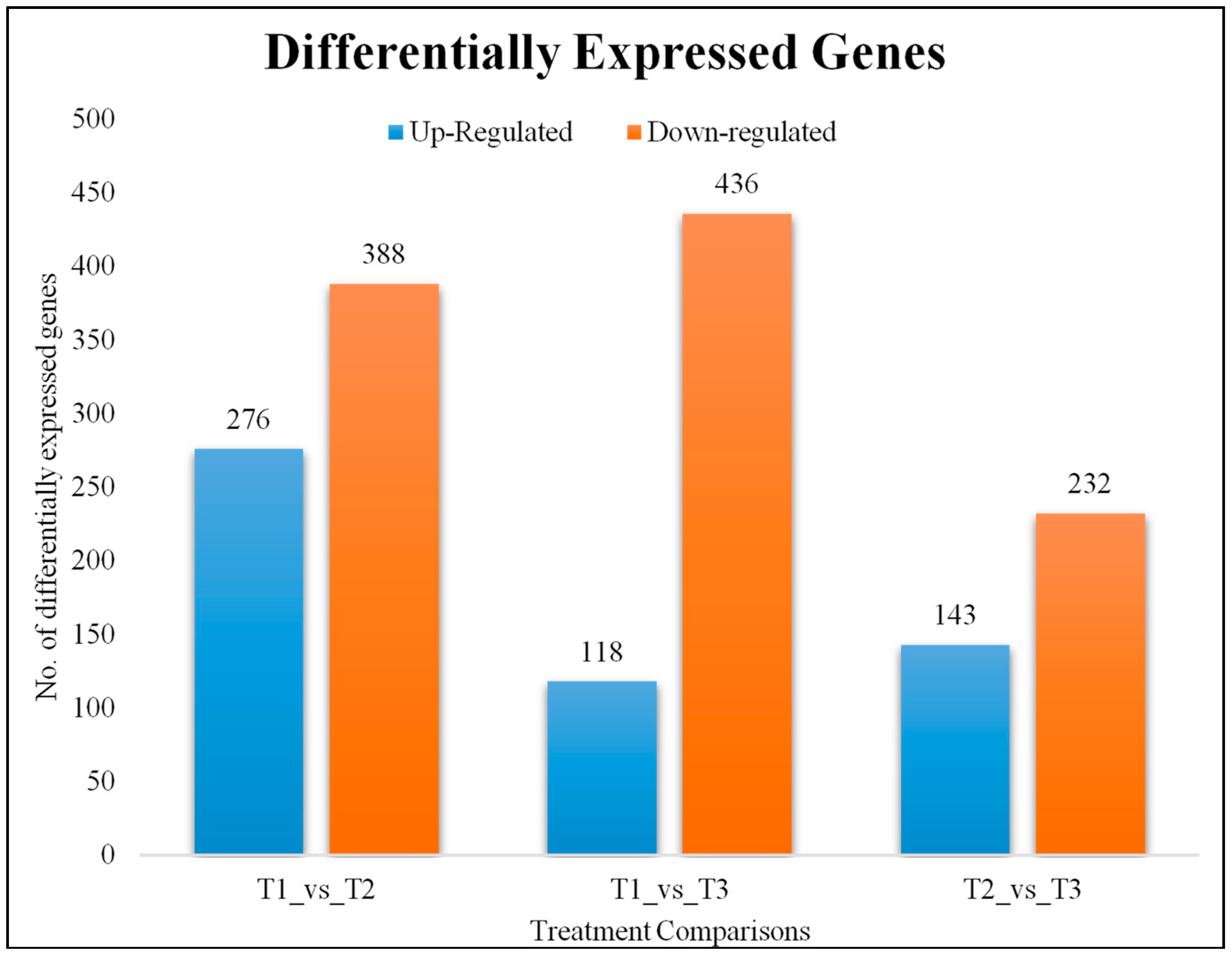
2.3. Differential Gene Expression Analysis
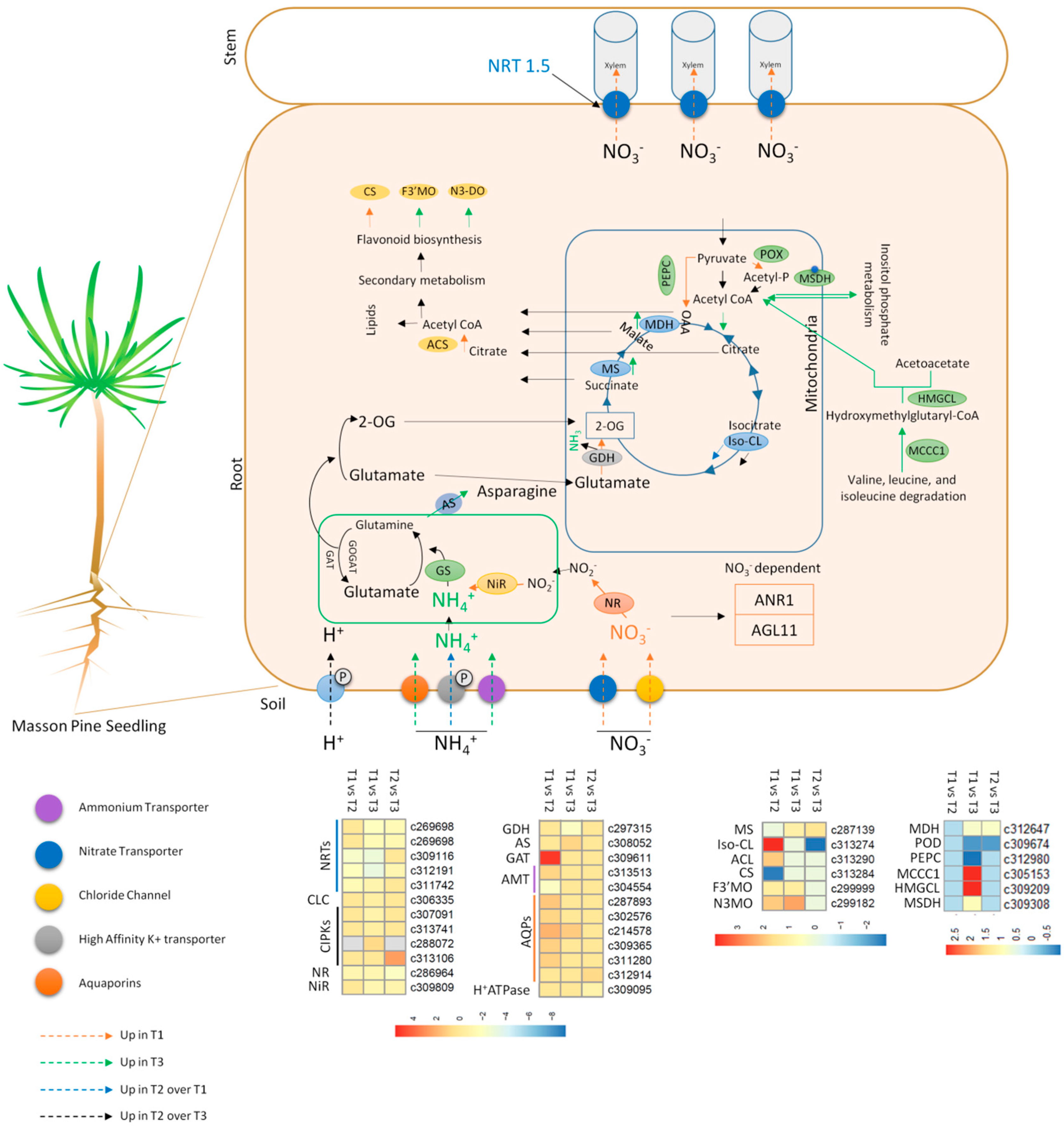
2.4. DEGs Related to N Uptake and Transport
2.5. DEGs Related to Nitrate Reduction
2.6. DEGs Related to Carbon/Nitrogen Metabolism and Secondary Metabolism
2.7. Differentially Expressed TFs
2.8. DEGs Related to Phytohormone Regulation
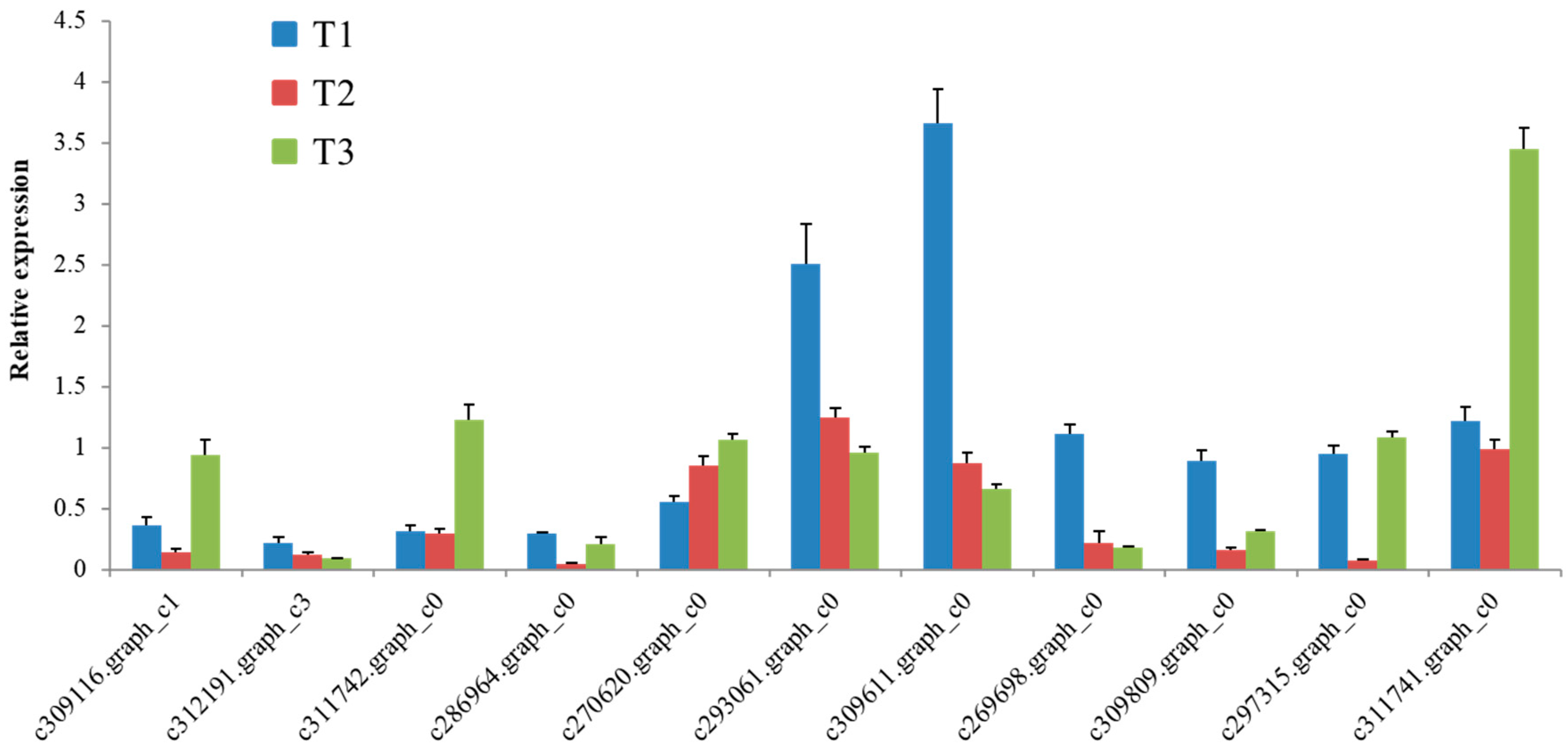
2.9. qRT-PCR Analyses of Selected Genes
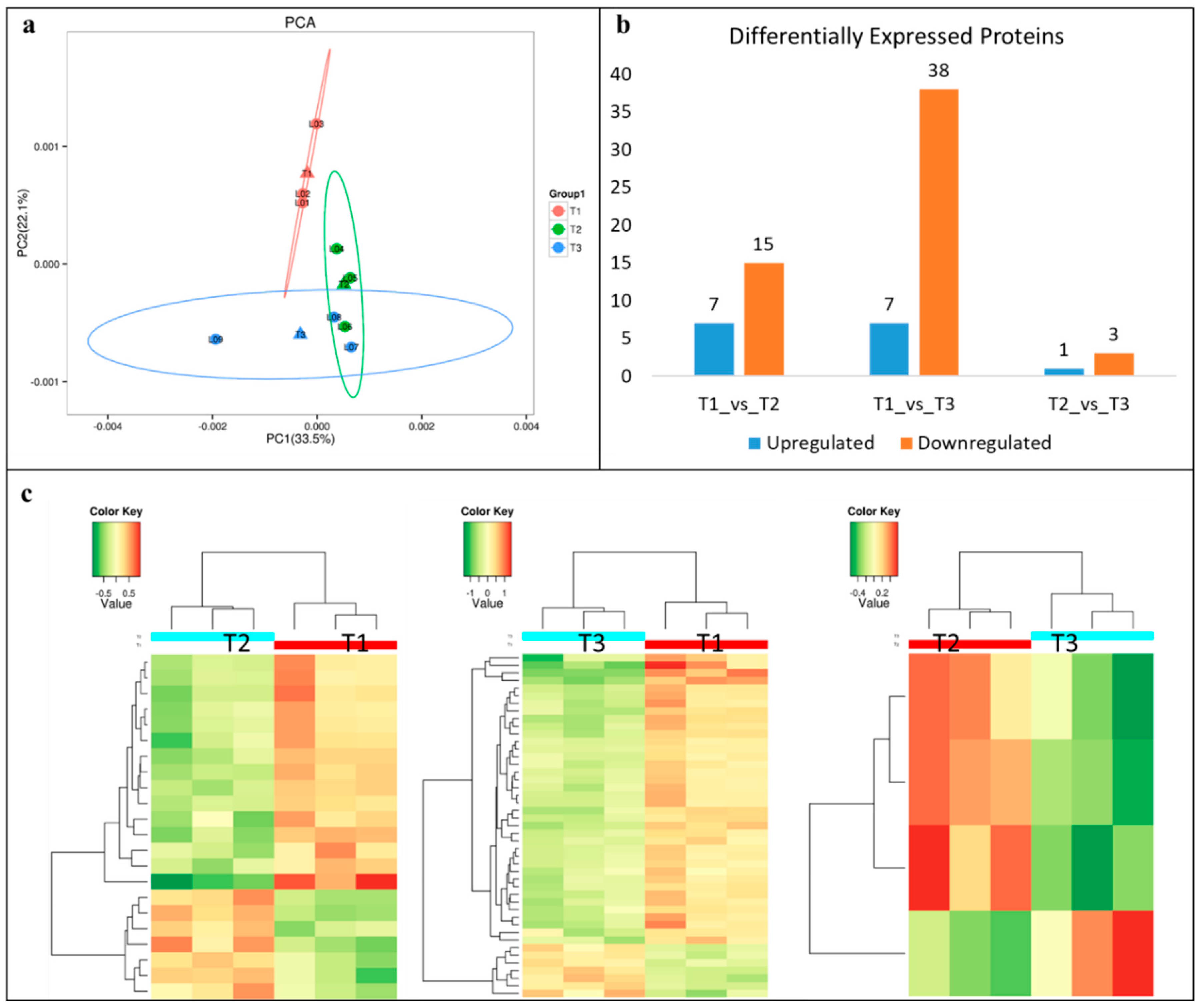
2.10. Proteome Analysis and Identification of DEPs
3. Discussion
3.1. N Uptake and Root Responses to Ammonium
3.2. Nitrogen Assimilation—The GS/GOGAT Cycle
3.3. Carbon/Nitrogen Metabolism Modulation in Response to NH4+ and the Role of TCA Cycle
3.4. Managing Increased NH4+ Accumulation in Masson Pine Roots
4. Material and Methods
4.1. Plant Material and Treatments
4.2. Morphological Evaluation and N-Content Measurements
4.3. Library Preparation for Transcriptome Sequencing
4.4. Clustering and Sequencing
4.5. Data Analysis
4.6. Transcriptome Assembly
4.7. Gene Functional Annotation and Differential Expression Analysis
4.8. Proteome Sequencing
4.8.1. Sample Preparation, Protein Quantification, and SDS Page
4.8.2. Enzymatic Hydrolysis
4.8.3. Classification of Peptide Markers and LC-MS/MS Analysis
4.9. Gene Expression Data Validation by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Torres-Olivar, V.; Villegas-Torres, O.G.; Domínguez-Patiño, M.L.; Sotelo-Nava, H.; Rodríguez-Martínez, A.; Melgoza-Alemán, R.M.; Valdez-Aguilar, L.A.; Alia-Tejacal, I. Role of nitrogen and nutrients in crop nutrition. J. Agric. Sci. Technol. B 2014, 4, 29. [Google Scholar]
- Nacry, P.; Bouguyon, E.; Gojon, A. Nitrogen acquisition by roots: Physiological and developmental mechanisms ensuring plant adaptation to a fluctuating resource. Plant Soil 2013, 370, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Yao, X.; Rehbein, P.J.; Lee, C.J.; Evans, G.J.; Corbin, J.; Jeong, C.-H. A study on the extent of neutralization of sulphate aerosol through laboratory and field experiments using an ATOFMS and a GPIC. Atmos. Environ. 2011, 45, 6251–6256. [Google Scholar] [CrossRef]
- Xu, G.; Fan, X.; Miller, A.J. Plant nitrogen assimilation and use efficiency. Annu. Rev. Plant Biol. 2012, 63, 153–182. [Google Scholar] [CrossRef] [Green Version]
- Britto, D.T.; Kronzucker, H.J. NH4+ toxicity in higher plants: A critical review. J. Plant Physiol. 2002, 159, 567–584. [Google Scholar] [CrossRef] [Green Version]
- Lewis, M.A. The effects of mixtures and other environmental modifying factors on the toxicities of surfactants to freshwater and marine life. Water Res. 1992, 26, 1013–1023. [Google Scholar] [CrossRef]
- Thomas, R.C.; Mead, D.J. Uptake of nitrogen by Pinus radiata and retention within the soil after applying 15N-labelled urea at different frequencies. 2. Seasonal trends and processes. For. Ecol. Manag. 1992, 53, 153–174. [Google Scholar] [CrossRef]
- Kronzucker, H.J.; Siddiqi, M.Y.; Glass, A.D. Conifer root discrimination against soil nitrate and the ecology of forest succession. Nature 1997, 385, 59–61. [Google Scholar] [CrossRef]
- Dechorgnat, J.; Nguyen, C.T.; Armengaud, P.; Jossier, M.; Diatloff, E.; Filleur, S.; Daniel-Vedele, F. From the soil to the seeds: The long journey of nitrate in plants. J. Exp. Bot. 2011, 62, 1349–1359. [Google Scholar] [CrossRef] [Green Version]
- Nour-Eldin, H.H.; Andersen, T.G.; Burow, M.; Madsen, S.R.; Jørgensen, M.E.; Olsen, C.E.; Dreyer, I.; Hedrich, R.; Geiger, D.; Halkier, B.A. NRT/PTR transporters are essential for translocation of glucosinolate defence compounds to seeds. Nature 2012, 488, 531–534. [Google Scholar] [CrossRef]
- De Angeli, A.; Monachello, D.; Ephritikhine, G.; Frachisse, J.; Thomine, S.; Gambale, F.; Barbier-Brygoo, H. The nitrate/proton antiporter AtCLCa mediates nitrate accumulation in plant vacuoles. Nature 2006, 442, 939–942. [Google Scholar] [CrossRef] [PubMed]
- Howitt, S.M.; Udvardi, M.K. Structure, function and regulation of ammonium transporters in plants. Biochim. Biophys. Acta Biomembr. 2000, 1465, 152–170. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Ramirez, C.; Mora, S.I.; Trejo, J.; Pantoja, O. PvAMT1; 1, a highly selective ammonium transporter that functions as H+/NH4+ symporter. J. Biol. Chem. 2011, 286, 31113–31122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Apuzzo, E.; Rogato, A.; Simon-Rosin, U.; El Alaoui, H.; Barbulova, A.; Betti, M.; Dimou, M.; Katinakis, P.; Marquez, A.; Marini, A.-M. Characterization of three functional high-affinity ammonium transporters in Lotus japonicus with differential transcriptional regulation and spatial expression. Plant Physiol. 2004, 134, 1763–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, F.; Giehl, R.F.; Geldner, N.; Salt, D.E.; von Wirén, N. Root zone–specific localization of AMTs determines ammonium transport pathways and nitrogen allocation to shoots. PLoS Biol. 2018, 16, e2006024. [Google Scholar] [CrossRef]
- Hoopen, F.t.; Cuin, T.A.; Pedas, P.; Hegelund, J.N.; Shabala, S.; Schjoerring, J.K.; Jahn, T.P. Competition between uptake of ammonium and potassium in barley and Arabidopsis roots: Molecular mechanisms and physiological consequences. J. Exp. Bot. 2010, 61, 2303–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, A.D.; Britto, D.T.; Kaiser, B.N.; Kinghorn, J.R.; Kronzucker, H.J.; Kumar, A.; Okamoto, M.; Rawat, S.; Siddiqi, M.; Unkles, S.E. The regulation of nitrate and ammonium transport systems in plants. J. Exp. Bot. 2002, 53, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Hachiya, T.; Sakakibara, H. Interactions between nitrate and ammonium in their uptake, allocation, assimilation, and signaling in plants. J. Exp. Bot. 2017, 68, 2501–2512. [Google Scholar] [CrossRef]
- Coleto, I.; Vega-Mas, I.; Glauser, G.; González-Moro, M.B.; Marino, D.; Ariz, I. New insights on Arabidopsis thaliana root adaption to ammonium nutrition by the use of a quantitative proteomic approach. Int. J. Mol. Sci. 2019, 20, 814. [Google Scholar] [CrossRef] [Green Version]
- Falkengren-Grerup, U. Interspecies differences in the preference of ammonium and nitrate in vascular plants. Oecologia 1995, 102, 305–311. [Google Scholar] [CrossRef]
- Britto, D.T.; Kronzucker, H.J. Ecological significance and complexity of N-source preference in plants. Ann. Bot. 2013, 112, 957–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menz, J.; Li, Z.; Schulze, W.X.; Ludewig, U. Early nitrogen-deprivation responses in Arabidopsis roots reveal distinct differences on transcriptome and (phospho-) proteome levels between nitrate and ammonium nutrition. Plant J. 2016, 88, 717–734. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.; Cakmak, T.; Cooper, A.; Lager, I.; Rasmusson, A.G.; Escobar, M.A. Distinct signalling pathways and transcriptome response signatures differentiate ammonium-and nitrate-supplied plants. Plantcell Environ. 2010, 33, 1486–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, D.; Moran, J.F. Can ammonium stress be positive for plant performance? Front. Plant Sci. 2019, 10, 1103. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; Tang, S. Modeling compatible single-tree aboveground biomass equations for masson pine (Pinus massoniana) in southern China. J. For. Res. 2012, 23, 593–598. [Google Scholar] [CrossRef]
- Zhu, F.; Dai, L.; Hobbie, E.A.; Koba, K.; Liu, X.; Gurmesa, G.A.; Huang, S.; Li, S.; Li, Y.; Fang, Y. Uptake patterns of glycine, ammonium, and nitrate differ among four common tree species of northeast China. Front. Plant Sci. 2019, 10, 799. [Google Scholar] [CrossRef]
- Masclaux-Daubresse, C.; Daniel-Vedele, F.; Dechorgnat, J.; Chardon, F.; Gaufichon, L.; Suzuki, A. Nitrogen uptake, assimilation and remobilization in plants: Challenges for sustainable and productive agriculture. Ann. Bot. 2010, 105, 1141–1157. [Google Scholar] [CrossRef] [Green Version]
- Kiba, T.; Krapp, A. Plant nitrogen acquisition under low availability: Regulation of uptake and root architecture. Plant Cell Physiol. 2016, 57, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Coskun, D.; Britto, D.T.; Li, M.; Becker, A.; Kronzucker, H.J. Rapid ammonia gas transport accounts for futile transmembrane cycling under NH3/NH4+ toxicity in plant roots. Plant Physiol. 2013, 163, 1859–1867. [Google Scholar] [CrossRef] [Green Version]
- Bisseling, T.; Scheres, B. Nutrient computation for root architecture. Science 2014, 346, 300–301. [Google Scholar] [CrossRef]
- Yu, C.; Liu, Y.; Zhang, A.; Su, S.; Yan, A.; Huang, L.; Ali, I.; Liu, Y.; Forde, B.G.; Gan, Y. MADS-box transcription factor OsMADS25 regulates root development through affection of nitrate accumulation in rice. PLoS ONE 2015, 10, e0135196. [Google Scholar] [CrossRef] [PubMed]
- Canales, J.; Contreras-López, O.; Álvarez, J.M.; Gutiérrez, R.A. Nitrate induction of root hair density is mediated by TGA 1/TGA 4 and CPC transcription factors in Arabidopsis thaliana. Plant J. 2017, 92, 305–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.Y.; Jang, Y.J.; Park, O.K. AP2/ERF family transcription factors ORA59 and RAP2. 3 interact in the nucleus and function together in ethylene responses. Front. Plant Sci. 2018, 9, 1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najafi, S.; Sorkheh, K.; Nasernakhaei, F. Characterization of the APETALA2/Ethylene-responsive factor (AP2/ERF) transcription factor family in sunflower. Sci. Rep. 2018, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- van der Graaff, E.; Dulk-Ras, A.; Hooykaas, P.; Keller, B. Activation tagging of the LEAFY PETIOLE gene affects leaf petiole development in Arabidopsis thaliana. Development 2000, 127, 4971–4980. [Google Scholar]
- Van Der Graaff, E.; Hooykaas, P.J.; Keller, B. Activation tagging of the two closely linked genes LEP and VAS independently affects vascular cell number. Plant J. 2002, 32, 819–830. [Google Scholar] [CrossRef]
- Ward, J.M.; Smith, A.M.; Shah, P.K.; Galanti, S.E.; Yi, H.; Demianski, A.J.; van der Graaff, E.; Keller, B.; Neff, M.M. A new role for the Arabidopsis AP2 transcription factor, LEAFY PETIOLE, in gibberellin-induced germination is revealed by the misexpression of a homologous gene, SOB2/DRN-LIKE. Plant Cell 2006, 18, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Ravazzolo, L.; Trevisan, S.; Forestan, C.; Varotto, S.; Sut, S.; Dall’Acqua, S.; Malagoli, M.; Quaggiotti, S. Nitrate and Ammonium Affect the Overall Maize Response to Nitrogen Availability by Triggering Specific and Common Transcriptional Signatures in Roots. Int. J. Mol. Sci. 2020, 21, 686. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Yoon, J.; Choi, H.; Fan, Y.; Chen, R.; An, G. Transcriptome analysis of nitrogen-starvation-responsive genes in rice. BMC Plant Biol. 2015, 15, 31. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Zhang, L.; Lewis, R.S.; Bovet, L.; Goepfert, S.; Jack, A.M.; Crutchfield, J.D.; Ji, H.; Dewey, R.E. Expression of a constitutively active nitrate reductase variant in tobacco reduces tobacco-specific nitrosamine accumulation in cured leaves and cigarette smoke. Plant Biotechnol. J. 2016, 14, 1500–1510. [Google Scholar] [CrossRef] [Green Version]
- Miflin, B.J.; Habash, D.Z. The role of glutamine synthetase and glutamate dehydrogenase in nitrogen assimilation and possibilities for improvement in the nitrogen utilization of crops. J. Exp. Bot. 2002, 53, 979–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiaochuang, C.; Meiyan, W.; Chunquan, Z.; Chu, Z.; Junhua, Z.; Lianfeng, Z.; Lianghuan, W.; Qianyu, J. Glutamate dehydrogenase mediated amino acid metabolism after ammonium uptake enhances rice growth under aeration condition. Plant Cell Rep. 2020, 39, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Causin, H.; Barneix, A. Regulation of NH4+ uptake in wheat plants: Effect of root ammonium concentration and amino acids. Plant Soil 1993, 151, 211–218. [Google Scholar] [CrossRef]
- Nunes-Nesi, A.; Fernie, A.R.; Stitt, M. Metabolic and signaling aspects underpinning the regulation of plant carbon nitrogen interactions. Mol. Plant 2010, 3, 973–996. [Google Scholar] [CrossRef]
- Vega-Mas, I.; Cukier, C.; Coleto, I.; González-Murua, C.; Limami, A.M.; González-Moro, M.B.; Marino, D. Isotopic labelling reveals the efficient adaptation of wheat root TCA cycle flux modes to match carbon demand under ammonium nutrition. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Prinsi, B.; Espen, L. Mineral nitrogen sources differently affect root glutamine synthetase isoforms and amino acid balance among organs in maize. BMC Plant Biol. 2015, 15, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Lea, P.J.; Sodek, L.; Parry, M.A.; Shewry, P.R.; Halford, N.G. Asparagine in plants. Ann. Appl. Biol. 2007, 150, 1–26. [Google Scholar] [CrossRef]
- Sarkissian, G.; Fowler, M. Interrelationship between nitrate assimilation and carbohydrate metabolism in plant roots. Planta 1974, 119, 335–349. [Google Scholar] [CrossRef]
- Schomburg, D.; Stephan, D. Galactinol-sucrose galactosyltransferase. In Enzyme Handbook 12; Springer: Berlin, Germany, 1996; pp. 431–433. [Google Scholar]
- Prabha, T.; Bhagyalakshmi, N. Carbohydrate metabolism in ripening banana fruit. Phytochemistry 1998, 48, 915–919. [Google Scholar] [CrossRef]
- Tadege, M.; Bucher, M.; Stähli, W.; Suter, M.; Dupuis, I.; Kuhlemeier, C. Activation of plant defense responses and sugar efflux by expression of pyruvate decarboxylase in potato leaves. Plant J. 1998, 16, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Soriano, M.; Diaz, P.; Pastor, F.I.J. Pectate lyase C from Bacillus subtilis: A novel endo-cleaving enzyme with activity on highly methylated pectin. Microbiology 2006, 152, 617–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oraby, H.; Venkatesh, B.; Dale, B.; Ahmad, R.; Ransom, C.; Oehmke, J.; Sticklen, M. Enhanced conversion of plant biomass into glucose using transgenic rice-produced endoglucanase for cellulosic ethanol. Transgenic Res. 2007, 16, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Zhang, B.; Dai, Y.; Zhang, L.; Shang-Guan, K.; Peng, Y.; Zhou, Y.; Zhu, Z. Brittle culm15 encodes a membrane-associated chitinase-like protein required for cellulose biosynthesis in rice. Plant Physiol. 2012, 159, 1440–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igarashi, D.; Ishizaki, T.; Totsuka, K.; Ohsumi, C. ASN2 is a key enzyme in asparagine biosynthesis under ammonium sufficient conditions. Plant Biotechnol. 2009, 26, 153–159. [Google Scholar] [CrossRef]
- Wong, H.-K.; Chan, H.-K.; Coruzzi, G.M.; Lam, H.-M. Correlation of ASN2 gene expression with ammonium metabolism in Arabidopsis. Plant Physiol. 2004, 134, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Crespo, E.; Gómez-Pastor, R.; Scalschi, L.; Llorens, E.; Camanes, G.; García-Agustín, P. NH4+ induces antioxidant cellular machinery and provides resistance to salt stress in citrus plants. Trees 2014, 28, 1693–1704. [Google Scholar] [CrossRef]
- Yang, S.; Hao, D.; Jin, M.; Li, Y.; Liu, Z.; Huang, Y.; Chen, T.; Su, Y. Internal ammonium excess induces ROS-mediated reactions and causes carbon scarcity in rice. BMC Plant Biol. 2020, 20, 143. [Google Scholar] [CrossRef]
- Kumar, S.; Trivedi, P.K. Glutathione S-transferases: Role in combating abiotic stresses including arsenic detoxification in plants. Front. Plant Sci. 2018, 9, 751. [Google Scholar] [CrossRef] [Green Version]
- Asim, M.; Ullah, Z.; Oluwaseun, A.; Wang, Q.; Liu, H. Signalling Overlaps between Nitrate and Auxin in Regulation of The Root System Architecture: Insights from the Arabidopsis thaliana. Int. J. Mol. Sci. 2020, 21, 2880. [Google Scholar] [CrossRef] [Green Version]
- Lowther, J. Use of a single sulphuric acid-hydrogen peroxide digest for the analysis of Pinus radiata needles. Commun. Soil Sci. Plant Anal. 1980, 11, 175–188. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apweiler, R. Functional information in SWISS-PROT: The basis for large-scale characterisation of protein sequences. Brief. Bioinf. 2001, 2, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.; Birney, E.; Cerruti, L.; Durbin, R.; Etwiller, L.; Eddy, S.R.; Griffiths-Jones, S.; Howe, K.L.; Marshall, M.; Sonnhammer, E.L. The Pfam protein families database. Nucleic Acids Res. 2002, 30, 276–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Wilke, A.; Harrison, T.; Wilkening, J.; Field, D.; Glass, E.M.; Kyrpides, N.; Mavrommatis, K.; Meyer, F. The M5nr: A novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinf. 2012, 13, 141. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.-Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average No. of Root Tips | Primary Root Length (cm) | Total Root Length (cm) | Root Surface Area (cm2) | Root Volume (cm3) | |
|---|---|---|---|---|---|
| T1 | 9.2 ± 2.1 c | 9.06 ± 1.71 b | 33.12 ± 2.15 b | 4.42 ± 0.49 b | 0.065 ± 0.013 b |
| T2 | 12.5 ± 1.8 b | 10.6 ± 1.39 ab | 38.1 ± 1.99 a | 5.44 ± 0.54 a | 0.097 ± 0.013 ab |
| T3 | 18 ± 3.0 a | 12.4 ± 1.62 a | 39.67 ± 1.95 a | 5.3 ± 0.98 a | 0.077 ± 0.02 a |
| Gene ID | log2FC | KEGG_Annotation | ||
|---|---|---|---|---|
| T1 vs. T2 | T1 vs. T3 | T2 vs. T3 | ||
| c269432 | 0.731 | --- | --- | abscisic acid receptor PYR/PYL family |
| c309091 | −1.261 | −1.104 | --- | pathogenesis-related protein 1 |
| c304520 | 1.059 | --- | --- | abscisic acid receptor PYR/PYL family |
| c312413 | 1.222 | --- | --- | auxin-responsive protein SAUR32-like |
| c305519 | 1.301 | --- | --- | bZIP; transcription factor TGA2 |
| c299206 | 1.431 | --- | --- | F-box protein GID2 |
| c306724 | 1.574 | --- | --- | SAUR family protein |
| c301864 | −0.768 | --- | --- | protein phosphatase 2C |
| c272209 | −2.702 | --- | --- | jasmonate ZIM domain-containing protein (TIFY 9) |
| c294828 | 2.119 | --- | --- | SAUR-like auxin-responsive family protein |
| c309011 | --- | −1.981 | --- | SAUR family protein |
| c305774 | --- | --- | −1.163 | jasmonate ZIM domain-containing protein (TIFY 10B-like) |
| c295030 | --- | --- | −0.831 | transcription factor HBP-1b (c38)-like |
| c313439 | --- | --- | −2.311 | jasmonate ZIM domain-containing protein |
| T1 | T2 | T3 | |
|---|---|---|---|
| NO3−/NH4+:100/0 mM | NO3−/NH4+:50/50 mM | NO3−/NH4+:0/100 mM | |
| KNO3 | 0.6 | 0.1 | 0 |
| Ca(NO3)2·H2O | 0.2 | 0.2 | 0 |
| CaCl2 | 0.425 | 0.425 | 0.625 |
| (NH4)2SO4 | 0 | 0.25 | 0.5 |
| KH2PO4 | 0.125 | 0.125 | 0.125 |
| MgSO4·7H2O | 0.25 | 0.25 | 0.25 |
| KCl | 0.15 | 0.65 | 0.75 |
| K2SO4 | 0 | 0 | 0 |
| DCD | 0.007 µM/L |
| Gene ID | Gene Description | Forward Primer Sequence | Reverse Primer Sequence |
|---|---|---|---|
| c309116 | high affinity nitrate transporter 2.4-like (HA-NRT2.4-Like) | GCGTTGCCTATGTCCT | TAACTGATTTCGGCTTTG |
| c312191 | High-affinity nitrate transporter 3.2 (HA-NRT3.2) | CCATTGTATGCCTCTT | GCCTTGCTCTGATTTA |
| c311742 | Nitrate transporter 1.5 (NRT1.5) | GATCGCTTCTACTTGTTATT | TGAGCCAGTTCTTCGT |
| c286964 | nitrate reductase (NADH] (NR) | GACTTGATCGTCTTTCG | CAACACCGTGCCTACT |
| c270620 | vacuolar amino acid transporter 1 (AVT1C) | TGGGACTAAGGGTCGA | TAAATCTTTCAGGCATACAG |
| c293061 | Protein GLUTAMINE DUMPER 2 (GDU2) | ACAACCCTATGTCTCG | CAGCCATTTATCACTAT |
| c309611 | Putative glutamine amidotransferase (GAT1_2.1) | CATGCGTTCAGGGTGG | CTATCATCATCAGGGCGTAA |
| c269698 | High-affinity nitrate transporter 3.1 (HA-NRT3.1) | TAGCCACAGAATCCTATCAA | GGGCAGAGCACCAACA |
| c309809 | ferredoxin-nitrite reductase (NiR) | GCAAGAGCCCTGAAAA | ACCGAAGGCAGTAGCA |
| c297315 | glutamate dehydrogenase (NAD(P)+) (GDH) | AAACAGATGCGGGATA | AAGGTCGGATACAACG |
| c311741 | peptide-N4-(N-acetyl-beta-glucosaminyl) asparagine amidase (PNGase A) | AGACCTCGCAGGCAGT | GATTCATCCGCAAACAC |
| tuublin β-TUB | Internal standard | GTCGTGAATCATGGCATGGC | GCCTCACTATCGGTTTCCCA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, Q.; Zhou, Y.; Zhou, X. Combined Transcriptome and Proteome Analysis of Masson Pine (Pinus massoniana Lamb.) Seedling Root in Response to Nitrate and Ammonium Supplementations. Int. J. Mol. Sci. 2020, 21, 7548. https://doi.org/10.3390/ijms21207548
Ren Q, Zhou Y, Zhou X. Combined Transcriptome and Proteome Analysis of Masson Pine (Pinus massoniana Lamb.) Seedling Root in Response to Nitrate and Ammonium Supplementations. International Journal of Molecular Sciences. 2020; 21(20):7548. https://doi.org/10.3390/ijms21207548
Chicago/Turabian StyleRen, Qifei, Yunchao Zhou, and Xinwei Zhou. 2020. "Combined Transcriptome and Proteome Analysis of Masson Pine (Pinus massoniana Lamb.) Seedling Root in Response to Nitrate and Ammonium Supplementations" International Journal of Molecular Sciences 21, no. 20: 7548. https://doi.org/10.3390/ijms21207548