Role of Antimicrobial Peptides in Skin Barrier Repair in Individuals with Atopic Dermatitis


 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
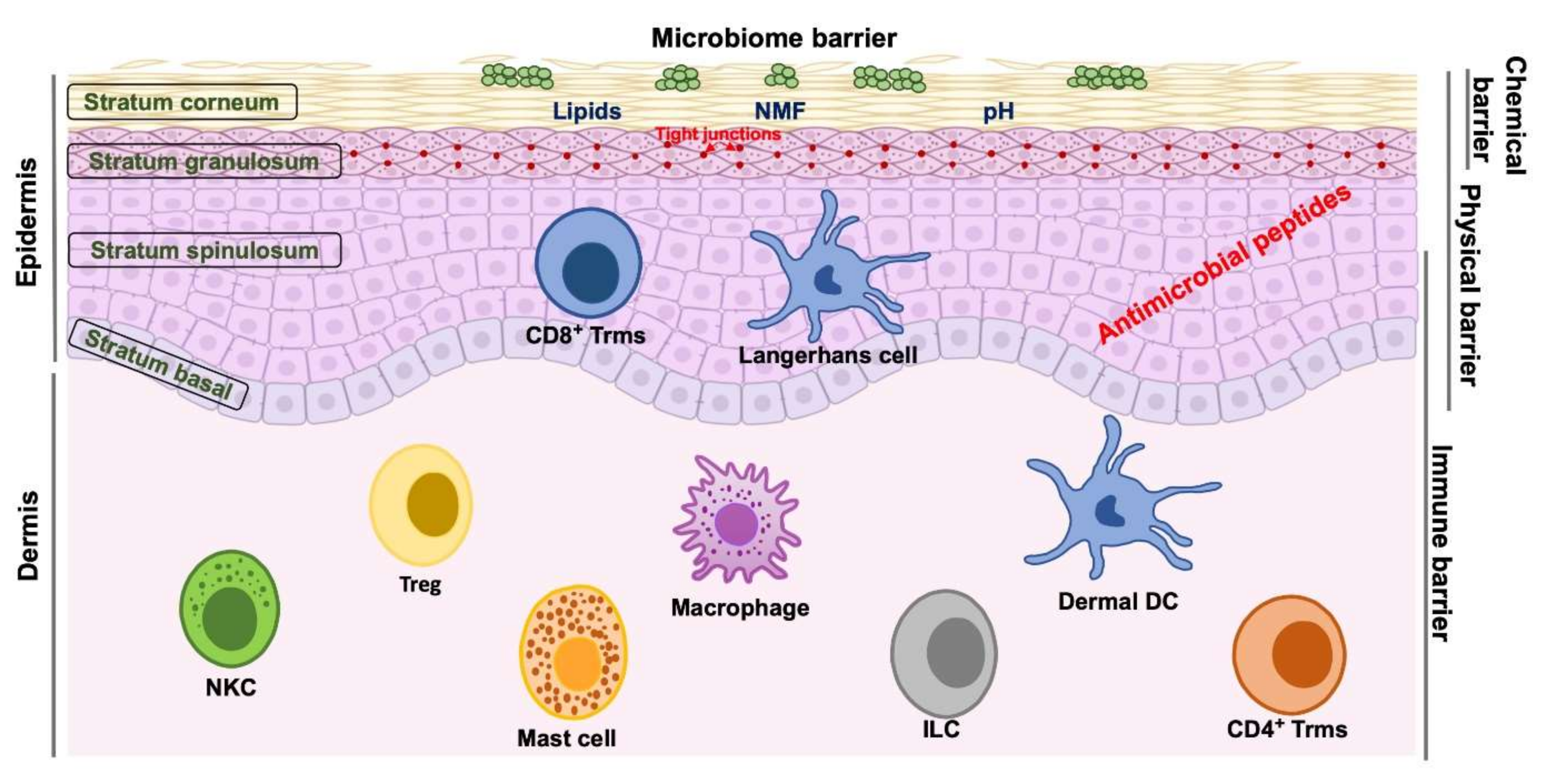
2. Skin Barrier
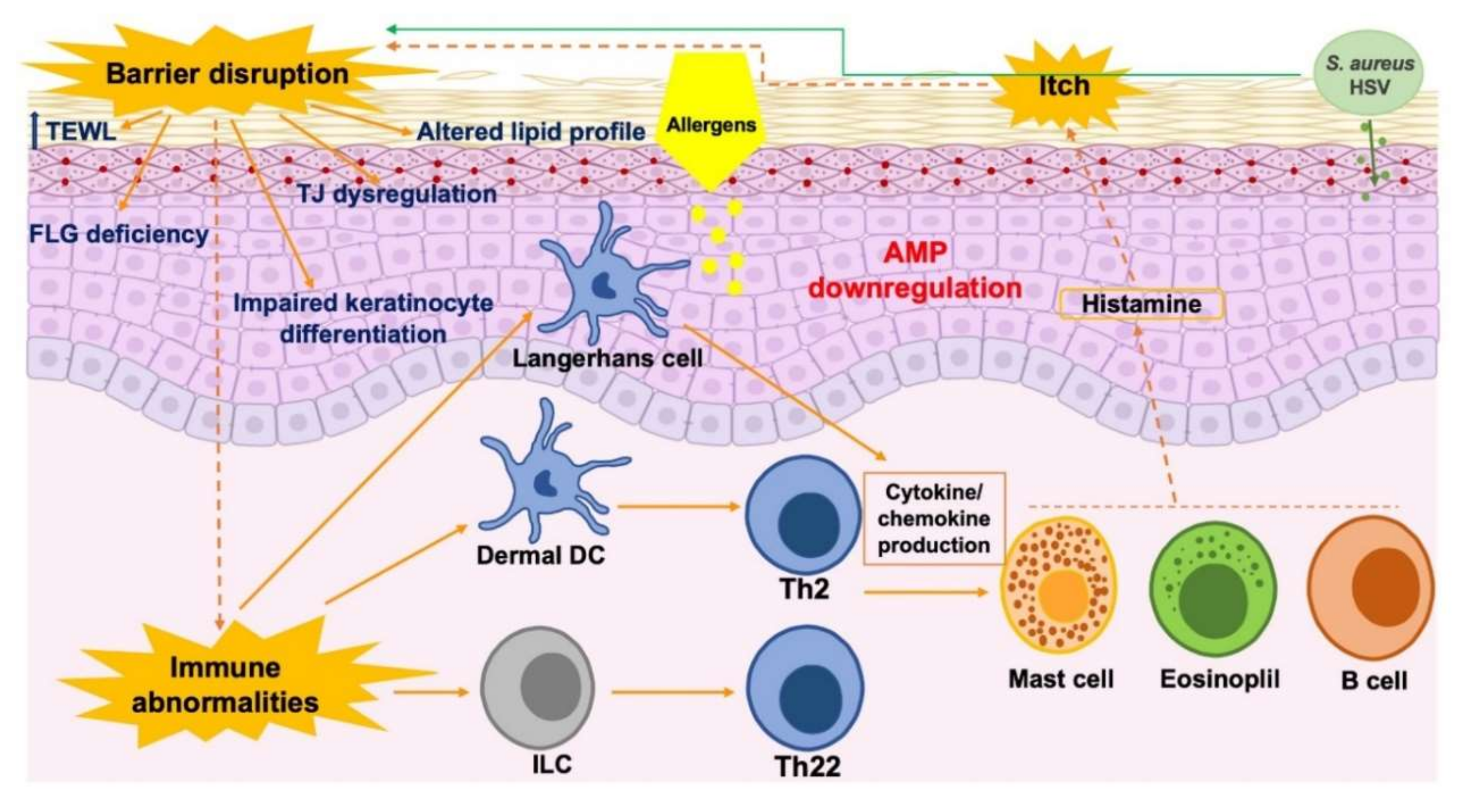
3. Skin Barrier Dysfunction in Individuals with AD
4. Skin Barrier Dysfunction-Related Infections in AD
5. Roles of AMPs in AD
6. Influence of AD Treatments on AMPs
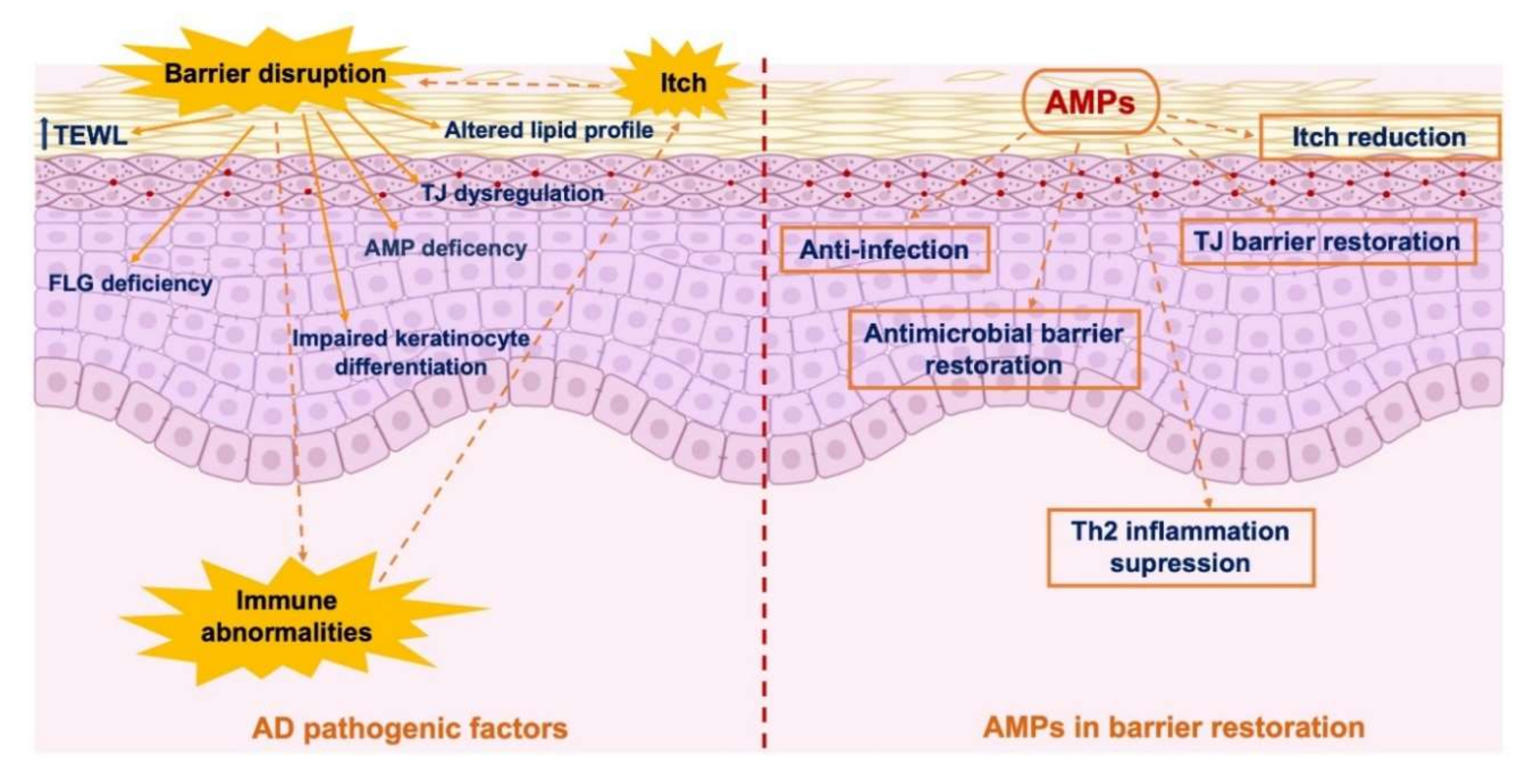
7. AMPs in Skin Barrier Repair: An Option for AD Treatment?
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | atopic dermatitis |
| AMP | antimicrobial peptide |
| DC | dendritic cell |
| dsRNA | double-stranded RNA |
| hBD | human b-defensin |
| HDPs | host defense peptides |
| FDA | US Food and Drug Administration |
| FLG | filaggrin |
| HSV | Herpes simplex virus |
| IL | interleukin |
| LC | Langerhans cell |
| NKC | natural killer cell |
| NMF | natural moisturizing factor |
| S. aureus | Staphylococcus aureus |
| SC | stratum corneum |
| S. epidermidis | Staphylococcus epidermidis |
| SG | stratum granulosum |
| TCI | topical calcineurin inhibitor |
| TCS | topical corticosteroids |
| TEWL | transepidermal water loss |
| Th | helper T-cell |
| TJ | tight junction |
| UV | ultraviolet |
References
- Kraft, M.T.; Prince, B.T. Atopic dermatitis is a barrier issue, not an allergy issue. Immunol. Allergy Clin. N. Am. 2019, 39, 507–519. [Google Scholar] [CrossRef]
- Fujii, M. Current understanding of pathophysiological mechanisms of atopic dermatitis: Interactions among skin barrier dysfunction, immune abnormalities and pruritus. Biol. Pharm. Bull. 2020, 43, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Schneider, L.; Tilles, S.; Lio, P.; Boguniewicz, M.; Beck, L.; LeBovidge, J.; Novak, N.; Bernstein, D.; Blessing-Moore, J.; Khan, D.; et al. Atopic dermatitis: A practice parameter update 2012. J. Allergy Clin. Immunol. 2013, 131, 295–299.e27. [Google Scholar] [CrossRef]
- Palmer, C.N.A.; Irvine, A.D.; Terron-Kwiatkowski, A.; Zhao, Y.; Liao, H.; Lee, S.P.; Goudie, D.R.; Sandilands, A.; Campbell, L.E.; Smith, F.J.D.; et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat. Genet. 2006, 38, 441–446. [Google Scholar] [CrossRef]
- Enomoto, H.; Hirata, K.; Otsuka, K.; Kawai, T.; Takahashi, T.; Hirota, T.; Suzuki, Y.; Tamari, M.; Otsuka, F.; Fujieda, S.; et al. Filaggrin null mutations are associated with atopic dermatitis and elevated levels of IgE in the Japanese population: A family and case–control study. J. Hum. Genet. 2008, 53, 615–621. [Google Scholar] [CrossRef] [Green Version]
- Barker, J.N.; Palmer, C.N.; Zhao, Y.; Liao, H.; Hull, P.R.; Lee, S.P.; Allen, M.H.; Meggitt, S.J.; Reynolds, N.J.; Trembath, R.C.; et al. Null mutations in the filaggrin gene (FLG) determine major susceptibility to early-onset atopic dermatitis that persists into adulthood. J. Invest. Dermatol. 2007, 127, 564–567. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.J.; Sandilands, A.; Zhao, Y.; Liao, H.; Relton, C.L.; Meggitt, S.J.; Trembath, R.C.; Barker, J.N.; Reynolds, N.J.; Cordell, H.J.; et al. Prevalent and low-frequency null mutations in the filaggrin gene are associated with early-onset and persistent atopic eczema. J. Invest. Dermatol. 2008, 128, 1591–1594. [Google Scholar] [CrossRef] [Green Version]
- Carson, C.G.; Rasmussen, M.A.; Thyssen, J.P.; Menné, T.; Bisgaard, H. Clinical presentation of atopic dermatitis by filaggrin gene mutation status during the first 7 years of life in a prospective cohort study. PLoS ONE 2012, 7, e48678. [Google Scholar] [CrossRef] [Green Version]
- Eyerich, S.; Eyerich, K.; Traidl-Hoffmann, C.; Biedermann, T. Cutaneous barriers and skin immunity: Differentiating a connected network. Trends Immunol. 2018, 39, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Boguniewicz, M.; Leung, D.Y. Atopic dermatitis: A disease of altered skin barrier and immune dysregulation. Immunol. Rev. 2011, 242, 233–246. [Google Scholar] [CrossRef]
- De Benedetto, A.; Georas, S.N.; Cheadle, C.; Berger, A.E. Tight junction defects in atopic dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786. [Google Scholar] [CrossRef] [Green Version]
- Yuki, T.; Tobiishi, M.; Kusaka-Kikushima, A.; Ota, Y.; Tokura, Y. Impaired tight junctions in atopic dermatitis skin and in a skin-equivalent model treated with interleukin-17. PLoS ONE 2016, 11, e0161759. [Google Scholar] [CrossRef]
- Niyonsaba, F.; Nagaoka, I.; Ogawa, H.; Okumura, K. Multifunctional antimicrobial proteins and peptides: Natural activators of immune systems. Curr. Pharm. Des. 2009, 15, 2393–2413. [Google Scholar] [CrossRef]
- Niyonsaba, F.; Kiatsurayanon, C.; Chieosilapatham, P.; Ogawa, H. Friends or Foes? Host defense (antimicrobial) peptides and proteins in human skin diseases. Exp. Dermatol. 2017, 26, 989–998. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, T.; Niyonsaba, F.; Kiatsurayanon, C.; Nguyen, T.T.; Ushio, H.; Fujimura, T.; Ueno, T.; Okumura, K.; Ogawa, H.; Ikeda, S. The human cathelicidin LL-37 host defense peptide upregulates tight junction-related proteins and increases human epidermal keratinocyte barrier function. J. Innate Immun. 2014, 6, 739–753. [Google Scholar] [CrossRef]
- Goto, H.; Hongo, M.; Ohshima, H.; Kurasawa, M.; Hirakawa, S.; Kitajima, Y. Human beta defensin-1 regulates the development of tight junctions in cultured human epidermal keratinocytes. J. Dermatol. Sci. 2013, 71, 145–148. [Google Scholar] [CrossRef]
- Kiatsurayanon, C.; Niyonsaba, F.; Smithrithee, R.; Akiyama, T.; Ushio, H.; Hara, M.; Okumura, K.; Ikeda, S.; Ogawa, H. Host defense (antimicrobial) peptide, human β-defensin-3, improves the function of the epithelial tight-junction barrier in human keratinocytes. J. Invest. Dermatol. 2014, 134, 2163–2173. [Google Scholar] [CrossRef] [Green Version]
- Hattori, F.; Kiatsurayanon, C.; Okumura, K.; Ogawa, H.; Ikeda, S.; Okamoto, K.; Niyonsaba, F. The antimicrobial protein S100A7/psoriasin enhances the expression of keratinocyte differentiation markers and strengthens the skin’s tight junction barrier. Cutan. Biol. 2014, 171, 742–753. [Google Scholar] [CrossRef]
- Sanford, J.A.; Gallo, R.L. Functions of the skin microbiota in health and disease. Semin. Immunol. 2013, 25, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Iwase, T.; Uehara, Y.; Shinji, H.; Tajima, A.; Seo, H.; Takada, K.; Agata, T.; Mizunoe, Y. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nat. Cell Biol. 2010, 465, 346–349. [Google Scholar] [CrossRef]
- Lai, Y.; Di Nardo, A.; Nakatsuji, T.; Leichtle, A.; Yang, Y.; Cogen, A.L.; Wu, Z.-R.; Hooper, L.V.; Von Aulock, S.; Radek, K.A.; et al. Commensal bacteria regulate TLR3-dependent inflammation following skin injury. Nat. Med. 2009, 15, 1377–1382. [Google Scholar] [CrossRef]
- Kezic, S.; O’Regan, G.M.; Lutter, R.; Jakasa, I.; Koster, E.S.; Saunders, S.; Caspers, P.; Kemperman, P.M.J.H.; Puppels, G.J.; Sandilands, A.; et al. Filaggrin loss-of-function mutations are associated with enhanced expression of IL-1 cytokines in the stratum corneum of patients with atopic dermatitis and in a murine model of filaggrin deficiency. J. Allergy Clin. Immunol. 2012, 129, 1031–1039.e1. [Google Scholar] [CrossRef] [Green Version]
- Verdier-Sévrain, S.; Bonté, F. Skin hydration: A review on its molecular mechanisms. J. Cosmet. Dermatol. 2007, 6, 75–82. [Google Scholar] [CrossRef]
- Marekov, L.N.; Steinert, P.M. Ceramides are bound to structural proteins of the human foreskin epidermal cornified cell envelope. J. Biol. Chem. 1998, 273, 17763–17770. [Google Scholar] [CrossRef] [Green Version]
- Swartzendruber, D.C.; Wertz, P.W.; Madison, K.C.; Downing, D.T. Evidence that the corneocyte has a chemically bound lipid envelope. J. Invest. Dermatol. 1987, 88, 709–713. [Google Scholar] [CrossRef] [Green Version]
- Lazo, N.D.; Meine, J.G.; Downing, D.T. Lipids are covalently attached to rigid corneocyte protein envelopes existing predominantly as β-sheets: A solid-state nuclear magnetic resonance study. J. Invest. Dermatol. 1995, 105, 296–300. [Google Scholar] [CrossRef] [Green Version]
- Eckert, R.L.; Rorke, E.A. Molecular biology of keratinocyte differentiation. Environ. Health Perspect. 1989, 80, 109–116. [Google Scholar] [CrossRef]
- Matsui, T.; Amagai, M. Dissecting the formation, structure and barrier function of the stratum corneum. Int. Immunol. 2015, 27, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Clark, R.A.; Liu, L.; Wagers, A.J.; Fuhlbrigge, R.C.; Kupper, T.S. Skin infection generates non-migratory memory CD8+ TRM cells providing global skin immunity. Nat. Cell Biol. 2012, 483, 227–231. [Google Scholar] [CrossRef]
- Gebhardt, T.; Wakim, L.M.; Eidsmo, L.; Reading, P.C.; Heath, W.R.; Carbone, F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009, 10, 524–530. [Google Scholar] [CrossRef]
- Proksch, E.; Brasch, J. Abnormal epidermal barrier in the pathogenesis of contact dermatitis. Clin. Dermatol. 2012, 30, 335–344. [Google Scholar] [CrossRef]
- Smith, F.J.D.; Irvine, A.D.; Terron-Kwiatkowski, A.; Sandilands, A.; Campbell, L.E.; Zhao, Y.; Liao, H.; Evans, A.T.; Goudie, D.R.; Lewis-Jones, S.; et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat. Genet. 2006, 38, 337–342. [Google Scholar] [CrossRef]
- Vávrová, K.; Henkes, D.; Strüver, K.; Sochorová, M.; Školová, B.; Witting, M.Y.; Friess, W.; Schreml, S.; Meier, R.J.; Schäfer-Korting, M.; et al. Filaggrin deficiency leads to impaired lipid profile and altered acidification pathways in a 3D skin construct. J. Invest. Dermatol. 2014, 134, 746–753. [Google Scholar] [CrossRef] [Green Version]
- Gruber, R.; Elias, P.M.; Crumrine, D.; Lin, T.-K.; Brandner, J.M.; Hachem, J.-P.; Presland, R.B.; Fleckman, P.; Janecke, A.R.; Sandilands, A.; et al. Filaggrin genotype in ichthyosis vulgaris predicts abnormalities in epidermal structure and function. Am. J. Pathol. 2011, 178, 2252–2263. [Google Scholar] [CrossRef] [Green Version]
- Bosko, C.A. Skin barrier insights: From bricks and mortar to molecules and microbes. J. Drugs Dermatol. 2019, 18, s63–s67. [Google Scholar]
- Gao, P.-S.; Rafaels, N.M.; Hand, T.; Murray, T.; Boguniewicz, M.; Hata, T.; Schneider, L.; Hanifin, J.M.; Gallo, R.L.; Gao, L.; et al. Filaggrin mutations that confer risk of atopic dermatitis confer greater risk for eczema herpeticum. J. Allergy Clin. Immunol. 2009, 124, 507–513.e7. [Google Scholar] [CrossRef] [Green Version]
- Kezic, S.; Kemperman, P.; Koster, E.; De Jongh, C.; Thio, H.; Campbell, L.; Irvine, A.; McLean, I.; Puppels, G.; Caspers, P. Loss-of-function mutations in the filaggrin gene lead to reduced level of natural moisturizing factor in the stratum corneum. J. Invest. Dermatol. 2008, 128, 2117–2119. [Google Scholar] [CrossRef] [Green Version]
- Tsakok, T.; Woolf, R.; Smith, C.H.; Weidinger, S.; Flohr, C. Atopic dermatitis: The skin barrier and beyond. Br. J. Dermatol. 2018, 180, 464–474. [Google Scholar] [CrossRef]
- Henderson, J.; Northstone, K.; Lee, S.P.; Liao, H.; Zhao, Y.; Pembrey, M.; Mukhopadhyay, S.; Smith, G.D.; Palmer, C.N.; McLean, W.H.I.; et al. The burden of disease associated with filaggrin mutations: A population-based, longitudinal birth cohort study. J. Allergy Clin. Immunol. 2008, 121, 872–877.e9. [Google Scholar] [CrossRef]
- Tsukita, S.; Furuse, M. Claudin-based barrier in simple and stratified cellular sheets. Curr. Opin. Cell Biol. 2002, 14, 531–536. [Google Scholar] [CrossRef]
- Brandner, J.M. Importance of tight junctions in relation to skin barrier function. Curr. Probl. Dermatol. 2016, 49, 27–37. [Google Scholar]
- Gruber, R.; Börnchen, C.; Rose, K.; Daubmann, A.; Volksdorf, T.; Wladykowski, E.; Vidal-y-Sy, S.; Peters, E.M.; Danso, M.; Bouwstra, J.; et al. Diverse regulation of claudin-1 and claudin-4 in atopic dermatitis. Am. J. Pathol. 2015, 185, 2777–2789. [Google Scholar] [CrossRef]
- Yuki, T.; Komiya, A.; Kusaka, A.; Kuze, T.; Sugiyama, Y.; Inoue, S. Impaired tight junctions obstruct stratum corneum formation by altering polar lipid and profilaggrin processing. J. Dermatol. Sci. 2013, 69, 148–158. [Google Scholar] [CrossRef]
- Furuse, M.; Hata, M.; Furuse, K.; Yoshida, Y.; Haratake, A.; Sugitani, Y.; Noda, T.; Kubo, A.; Tsukita, S. Claudin-based tight junctions are crucial for the mammalian epidermal barrier. J. Cell Biol. 2002, 156, 1099–1111. [Google Scholar] [CrossRef]
- Batista, D.I.S.; Perez, L.; Orfali, R.L.; Zaniboni, M.C.; Samorano, L.P.; Pereira, N.V.; Sotto, M.N.; Ishizaki, A.S.; Oliveira, L.M.S.; Sato, M.N.; et al. Profile of skin barrier proteins (filaggrin, claudins 1 and 4) and Th1/Th2/Th17 cytokines in adults with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1091–1095. [Google Scholar] [CrossRef]
- De Benedetto, A.; Slifka, M.K.; Rafaels, N.M.; Kuo, I.-H.; Georas, S.N.; Boguniewicz, M.; Hata, T.; Schneider, L.C.; Hanifin, J.M.; Gallo, R.L.; et al. Reductions in claudin-1 may enhance susceptibility to herpes simplex virus 1 infections in atopic dermatitis. J. Allergy Clin. Immunol. 2011, 128, 242–246.e5. [Google Scholar] [CrossRef] [Green Version]
- Dębińska, A.; Danielewicz, H.; Drabik-Chamerska, A.; Kalita, D.; Boznański, A. Filaggrin loss-of-function mutations as a predictor for atopic eczema, allergic sensitization and eczema-associated asthma in Polish children population. Adv. Clin. Exp. Med. 2017, 26, 991–998. [Google Scholar] [CrossRef] [Green Version]
- Cubero, J.L.; Isidoro-García, M.; Segura, N.; Pescador, D.B.; Sanz, C.; Lorente, F.; Davila, I.; Colás, C. Filaggrin gene mutations and new SNPs in asthmatic patients: A cross-sectional study in a Spanish population. Allergy Asthma Clin. Immunol. 2016, 12, 31. [Google Scholar] [CrossRef] [Green Version]
- Kubo, A.; Nagao, K.; Amagai, M.; Kubo, A.; Nagao, K.; Amagai, M. Epidermal barrier dysfunction and cutaneous sensitization in atopic diseases. J. Clin. Invest. 2012, 122, 440–447. [Google Scholar] [CrossRef] [Green Version]
- Lack, G. Epidemiologic risks for food allergy. J. Allergy Clin. Immunol. 2008, 121, 1331–1336. [Google Scholar] [CrossRef]
- Tsakok, T.; Marrs, T.; Mohsin, M.; Baron, S.; Du Toit, G.; Till, S.; Flohr, C. Does atopic dermatitis cause food allergy? a systematic review. J. Allergy Clin. Immunol. 2016, 137, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.J.; Asai, Y.; Cordell, H.J.; Campbell, L.E.; Zhao, Y.; Liao, H.; Northstone, K.; Henderson, J.; Alizadehfar, R.; Ben-Shoshan, M.; et al. Loss-of-function variants in the filaggrin gene are a significant risk factor for peanut allergy. J. Allergy Clin. Immunol. 2011, 127, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Takai, T.; Ikeda, S. Barrier dysfunction caused by environmental proteases in the pathogenesis of allergic diseases. Allergol. Int. 2011, 60, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Dickel, H.; Gambichler, T.; Kamphowe, J.; Altmeyer, P.; Skrygan, M. Standardized tape stripping prior to patch testing induces upregulation of Hsp90, Hsp70, IL-33, TNF-α and IL-8/CXCL8 mRNA: New insights into the involvement of “alarmins”. Contact Dermatitis 2010, 63, 215–222. [Google Scholar] [CrossRef]
- Leyva-Castillo, J.; Hener, P.; Michea, P.; Karasuyama, H.; Chan, S.; Soumelis, V.; Li, M. Skin thymic stromal lymphopoietin initiates Th2 responses through an orchestrated immune cascade. Nat. Commun. 2013, 4, 2847. [Google Scholar] [CrossRef]
- Black, A.P.B.; Ardern-Jones, M.R.; Kasprowicz, V.; Bowness, P.; Jones, A.; Bailey, A.S.; Ogg, G.S. Human keratinocyte induction of rapid effector function in antigen-specific memory CD4+ and CD8+ T cells. Eur. J. Immunol. 2007, 37, 1485–1493. [Google Scholar] [CrossRef]
- Gittler, J.K.; Shemer, A.; Suárez-Fariñas, M.; Fuentes-Duculan, J.; Gulewicz, K.J.; Wang, C.Q.F.; Mitsui, H.; Cardinale, I.; De Guzman Strong, C.; Krueger, J.G.; et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis. J. Allergy Clin. Immunol. 2012, 130, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Suárez-Fariñas, M.; Tintle, S.J.; Shemer, A.; Chiricozzi, A.; Nograles, K.; Cardinale, I.; Duan, S.; Bowcock, A.M.; Krueger, J.G.; Guttman-Yassky, E. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 2011, 127, 954–964.e4. [Google Scholar] [CrossRef] [Green Version]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.-C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.; et al. A role for IL-25 and IL-33–driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef]
- Hijnen, D.; Knol, E.F.; Gent, Y.Y.; Giovannone, B.; Beijn, S.J.; Kupper, T.S.; Bruijnzeel-Koomen, C.A.; Clark, R.A. CD8+ T cells in the lesional skin of atopic dermatitis and psoriasis patients are an important source of IFN-γ, IL-13, IL-17, and IL-22. J. Invest. Dermatol. 2013, 133, 973–979. [Google Scholar] [CrossRef] [Green Version]
- Sonkoly, E.; Muller, A.; Lauerma, A.I.; Pivarcsi, A.; Soto, H.; Kemeny, L.; Alenius, H.; Dieu-Nosjean, M.-C.; Meller, S.; Rieker, J.; et al. IL-31: A new link between T cells and pruritus in atopic skin inflammation. J. Allergy Clin. Immunol. 2006, 117, 411–417. [Google Scholar] [CrossRef]
- Kopfnagel, V.; Harder, J.; Werfel, T. Expression of antimicrobial peptides in atopic dermatitis and possible immunoregulatory functions. Curr. Opin. Allergy Clin. Immunol. 2013, 13, 531–536. [Google Scholar] [CrossRef]
- Howell, M.D.; Kim, B.E.; Gao, P.; Grant, A.V.; Boguniewicz, M.; DeBenedetto, A.; Schneider, L.; Beck, L.A.; Barnes, K.C.; Leung, D.Y. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol. 2007, 120, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Gutowska-Owsiak, D.; Schaupp, A.; Salimi, M.; Taylor, S.; Ogg, G. Interleukin-22 downregulates filaggrin expression and affects expression of profilaggrin processing enzymes. Br. J. Dermatol. 2011, 165, 492–498. [Google Scholar] [CrossRef]
- Aberg, K.M.; Man, M.-Q.; Gallo, R.L.; Ganz, T.; Crumrine, D.; Brown, B.E.; Choi, E.-H.; Kim, D.-K.; Schröder, J.M.; Feingold, K.R.; et al. Co-regulation and interdependence of the mammalian epidermal permeability and antimicrobial barriers. J. Invest. Dermatol. 2008, 128, 917–925. [Google Scholar] [CrossRef] [Green Version]
- Borkowski, A.W.; Gallo, R.L. The coordinated response of the physical and antimicrobial peptide barriers of the skin. J. Invest. Dermatol. 2011, 131, 285–287. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.S. The role of microorganisms in atopic dermatitis. Clin. Exp. Immunol. 2006, 144, 1–9. [Google Scholar] [CrossRef]
- Leung, D.Y.; Harbeck, R.; Bina, P.; Reiser, R.F.; Yang, E.; Norris, D.A.; Hanifin, J.M.; Sampson, H.A. Presence of IgE antibodies to staphylococcal exotoxins on the skin of patients with atopic dermatitis. evidence for a new group of allergens. J. Clin. Invest. 1993, 92, 1374–1380. [Google Scholar] [CrossRef] [Green Version]
- Elias, P.M.; Hatano, Y.; Williams, M.L. Basis for the barrier abnormality in atopic dermatitis: Outside-inside-outside pathogenic mechanisms. J. Allergy Clin. Immunol. 2008, 121, 1337–1343. [Google Scholar] [CrossRef] [Green Version]
- Schlievert, P.M.; Case, L.C.; Strandberg, K.L.; Abrams, B.B.; Leung, D.Y.M. Superantigen profile of staphylococcus aureus isolates from patients with steroid-resistant atopic dermatitis. Clin. Infect. Dis. 2008, 46, 1562–1567. [Google Scholar] [CrossRef] [Green Version]
- Patel, D.; Jahnke, M.N. Serious complications from staphylococcal aureus in atopic dermatitis. Pediatr. Dermatol. 2015, 32, 792–796. [Google Scholar] [CrossRef]
- Hoeger, P.H.; Ganschow, R.; Finger, G. Staphylococcal septicemia in children with atopic dermatitis. Pediatr. Dermatol. 2000, 17, 111–114. [Google Scholar] [CrossRef]
- Mathé, P.J.; Joost, I.; Peyerl-Hoffmann, G.; Schneider, C.; Kern, W.; Rieg, S. Staphylococcus aureus bloodstream infection in patients with atopic dermatitis, or: Think twice before placing a venous catheter into lesional atopic skin. J. Invest. Dermatol. 2020, 140, 1870–1872. [Google Scholar] [CrossRef]
- Narla, S.; Silverberg, J.I. Association between atopic dermatitis and serious cutaneous, multiorgan and systemic infections in US adults. Ann. Allergy Asthma Immunol. 2018, 120, 66–72.e11. [Google Scholar] [CrossRef]
- Oestergaard, L.B.; Schmiegelow, M.D.; Bruun, L.; Skov, R.; Andersen, P.S.; Larsen, A.R.; Gerds, T.A.; Dahl, A.; Petersen, A.; Lauridsen, T.K.; et al. Staphylococcus aureus bacteremia in children aged 5-18 years—risk factors in the new millennium. J. Pediatr. 2018, 203, 108–115.e3. [Google Scholar] [CrossRef]
- Leung, D.Y. Why is eczema herpeticum unexpectedly rare? Antivir. Res. 2013, 98, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Bin, L.; Kim, B.E.; Brauweiler, A.; Goleva, E.; Streib, J.; Ji, Y.; Schlievert, P.M.; Leung, D.Y.M. Staphylococcus aureus α-toxin modulates skin host response to viral infection. J. Allergy Clin. Immunol. 2012, 130, 683–691.e2. [Google Scholar] [CrossRef] [Green Version]
- Scheynius, A.; Johansson, C.; Buentke, E.; Zargari, A.; Linder, M.T. Atopic eczema/dermatitis syndrome and Malassezia. Int. Arch. Allergy Immunol. 2002, 127, 161–169. [Google Scholar] [CrossRef]
- Lange, L.; Alter, N.; Keller, T.; Rietschel, E. Sensitization to malassezia in infants and children with atopic dermatitis: Prevalence and clinical characteristics. Allergy 2008, 63, 486–487. [Google Scholar] [CrossRef]
- Glatz, M.; Buchner, M.; Von Bartenwerffer, W.; Schmid-Grendelmeier, P.; Worm, M.; Hedderich, J.; Fölster-Holst, R. Malassezia spp.-specific immunoglobulin E level is a marker for severity of atopic dermatitis in adults. Acta Derm. Venereol. 2015, 95, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Zhang, E.; Tanaka, T.; Tajima, M.; Tsuboi, R.; Kato, H.; Nishikawa, A.; Sugita, T. Anti-malassezia-specific IgE antibodies production in Japanese patients with head and neck atopic dermatitis: Relationship between the level of specific IgE antibody and the colonization frequency of cutaneous Malassezia species and clinical severity. J. Allergy 2011, 2011, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Buentke, E.; Scheynius, A. Dendritic cells and fungi. APMIS 2003, 111, 789–796. [Google Scholar] [CrossRef]
- Ong, P.Y.; Ohtake, T.; Brandt, C.; Strickland, I.; Boguniewicz, M.; Ganz, T.; Gallo, R.L.; Leung, D.Y.M. Endogenous antimicrobial peptides and skin infections in atopic dermatitis. N. Engl. J. Med. 2002, 347, 1151–1160. [Google Scholar] [CrossRef] [Green Version]
- Rieg, S.; Steffen, H.; Seeber, S.; Humeny, A.; Kalbacher, H.; Dietz, K.; Garbe, C.; Schittek, B. Deficiency of dermcidin-derived antimicrobial peptides in sweat of patients with atopic dermatitis correlates with an impaired innate defense of human skin in vivo. J. Immunol. 2005, 174, 8003–8010. [Google Scholar] [CrossRef] [Green Version]
- Gambichler, T.; Skrygan, M.; Tomi, N.S.; Othlinghaus, N.; Brockmeyer, N.H.; Altmeyer, P.; Kreuter, A. Differential mRNA expression of antimicrobial peptides and proteins in atopic dermatitis as compared to psoriasis vulgaris and healthy skin. Int. Arch. Allergy Immunol. 2008, 147, 17–24. [Google Scholar] [CrossRef]
- Kisich, K.O.; Carspecken, C.W.; Fiéve, S.; Boguniewicz, M.; Leung, D.Y. Defective killing of Staphylococcus aureus in atopic dermatitis is associated with reduced mobilization of human β-defensin-3. J. Allergy Clin. Immunol. 2008, 122, 62–68. [Google Scholar] [CrossRef]
- Nomura, I.; Goleva, E.; Howell, M.D.; Hamid, Q.A.; Ong, P.Y.; Hall, C.F.; Darst, M.A.; Gao, B.; Boguniewicz, M.; Travers, J.B.; et al. Cytokine milieu of atopic dermatitis, as compared to psoriasis, skin prevents induction of innate immune response genes. J. Immunol. 2003, 171, 3262–3269. [Google Scholar] [CrossRef] [Green Version]
- Howell, M.D.; Gallo, R.L.; Boguniewicz, M.; Jones, J.F.; Wong, C.; Streib, J.E.; Leung, D.Y. Cytokine milieu of atopic dermatitis skin subverts the innate immune response to vaccinia virus. Immunity 2006, 24, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Chertov, O.; Bykovskaia, S.N.; Chen, Q.; Buffo, M.J.; Shogan, J.; Anderson, M.; Schröder, J.M.; Wang, J.M.; Howard, O.M.; et al. Beta-defensins: Linking innate and adaptive immunity through dendritic and T cell CCR6. Science 1999, 286, 525–528. [Google Scholar] [CrossRef]
- Röhrl, J.; Yang, D.; Oppenheim, J.J.; Hehlgans, T. Human beta-defensin 2 and 3 and their mouse orthologs induce chemotaxis through interaction with CCR2. J. Immunol. 2010, 184, 6688–6694. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Niyonsaba, F.; Ushio, H.; Nagaoka, I.; Ikeda, S.; Okumura, K.; Ogawa, H. Human cathelicidin LL-37 increases vascular permeability in the skin via mast cell activation, and phosphorylates MAP kinases p38 and ERK in mast cells. J. Dermatol. Sci. 2006, 43, 63–66. [Google Scholar] [CrossRef]
- Subramanian, H.; Gupta, K.; Guo, Q.; Price, R.; Ali, H. Mas-related gene X2 (MrgX2) is a novel G protein-coupled receptor for the antimicrobial peptide LL-37 in human mast cells: Resistance to receptor phosphorylation, desensitization, and internalization. J. Biol. Chem. 2011, 286, 44739–44749. [Google Scholar] [CrossRef] [Green Version]
- Niyonsaba, F.; Ushio, H.; Hara, M.; Yokoi, H.; Tominaga, M.; Takamori, K.; Kajiwara, N.; Saito, H.; Nagaoka, I.; Ogawa, H.; et al. Antimicrobial peptides human β-defensins and cathelicidin LL-37 induce the secretion of a pruritogenic cytokine IL-31 by human mast cells. J. Immunol. 2010, 184, 3526–3534. [Google Scholar] [CrossRef] [Green Version]
- Umehara, Y.; Kamata, Y.; Tominaga, M.; Niyonsaba, F.; Ogawa, H.; Takamori, K. Cathelicidin LL-37 induces semaphorin 3A expression in human epidermal keratinocytes: Implications for possible application to pruritus. J. Invest. Dermatol. 2015, 135, 2887–2890. [Google Scholar] [CrossRef] [Green Version]
- Kanda, N.; Watanabe, S. Increased serum human β-defensin-2 levels in atopic dermatitis: Relationship to IL-22 and oncostatin M. Immunobiol. 2012, 217, 436–445. [Google Scholar] [CrossRef]
- Kanda, N.; Hau, C.S.; Tada, Y.; Sato, S.; Watanabe, S. Decreased serum LL-37 and vitamin D3 levels in atopic dermatitis: Relationship between IL-31 and oncostatin M. Allergy 2012, 67, 804–812. [Google Scholar] [CrossRef]
- Chen, X.; Niyonsaba, F.; Ushio, H.; Hara, M.; Yokoi, H.; Matsumoto, K.; Saito, H.; Nagaoka, I.; Ikeda, S.; Okumura, K.; et al. Antimicrobial peptides human beta-defensin (hBD)-3 and hBD-4 activate mast cells and increase skin vascular permeability. Eur. J. Immunol. 2007, 37, 434–444. [Google Scholar] [CrossRef]
- Chamlin, S.L.; Kao, J.; Frieden, I.J.; Sheu, M.Y.; Fowler, A.J.; Fluhr, J.W.; Williams, M.L.; Elias, P.M. Ceramide-dominant barrier repair lipids alleviate childhood atopic dermatitis: Changes in barrier function provide a sensitive indicator of disease activity. J. Am. Acad. Dermatol. 2002, 47, 198–208. [Google Scholar] [CrossRef]
- Miller, D.W.; Koch, S.B.; Yentzer, B.A.; Clark, A.R.; O’Neill, J.R.; Fountain, J.; Weber, T.M.; Fleischer, A.B., Jr. An over-the-counter moisturizer is as clinically effective as, and more cost-effective than, prescription barrier creams in the treatment of children with mild-to-moderate atopic dermatitis: A randomized, controlled trial. J. Drugs Dermatol. 2011, 10, 531–537. [Google Scholar]
- Bissonnette, R.; Maari, C.; Provost, N.; Bolduc, C.; Nigen, S.; Rougier, A.; Seite, S. A double-blind study of tolerance and efficacy of a new urea-containing moisturizer in patients with atopic dermatitis. J. Cosmet. Dermatol. 2010, 9, 16–21. [Google Scholar] [CrossRef]
- Grimalt, R.; Mengeaud, V.; Cambazard, F. The steroid-sparing effect of an emollient therapy in infants with atopic dermatitis: A randomized controlled study. Dermatology 2006, 214, 61–67. [Google Scholar] [CrossRef]
- Frankel, A.J.; Sohn, A.; Patel, R.V.; Lebwohl, M. Bilateral comparison study of pimecrolimus cream 1% and a ceramide-hyaluronic acid emollient foam in the treatment of patients with atopic dermatitis. J. Drugs Dermatol. 2011, 10, 666–672. [Google Scholar]
- Park, K.Y.; Kim, D.H.; Jeong, M.S.; Li, K.; Seo, S.J. Changes of antimicrobial peptides and transepidermal water loss after topical application of tacrolimus and ceramide-dominant emollient in patients with atopic dermatitis. J. Korean Med Sci. 2010, 25, 766–771. [Google Scholar] [CrossRef]
- Katayama, I.; Aihara, M.; Ohya, Y.; Saeki, H.; Shimojo, N.; Shoji, S.; Taniguchi, M.; Yamada, H. Japanese guidelines for atopic dermatitis 2017. Allergol. Int. 2017, 66, 230–247. [Google Scholar] [CrossRef]
- Ring, J.; AlOmar, A.; Bieber, T.; Deleuran, M.; Fink-Wagner, A.; Gelmetti, C.; Gieler, U.; Lipozencic, J.; Luger, T.; Oranje, A.; et al. Guidelines for treatment of atopic eczema (atopic dermatitis) Part I. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1045–1060. [Google Scholar] [CrossRef]
- Wollenberg, A.; Barbarot, S.; Bieber, T.; Christen-Zaech, S.; Deleuran, M.; Fink-Wagner, A.; Gieler, U.; Girolomoni, G.; Lau, S.; Muraro, A.; et al. Consensus-based European guidelines for treatment of atopic eczema (atopic dermatitis) in adults and children: Part I. Pract. Guidel. 2018, 32, 657–682. [Google Scholar]
- Yawalkar, S.J.; Schwerzmann, L. Double-blind, comparative clinical trials with halobetasol propionate cream in patients with atopic dermatitis. J. Am. Acad. Dermatol. 1991, 25, 1163–1166. [Google Scholar] [CrossRef]
- Eichenfield, L.F.; Basu, S.; Calvarese, B.; Trancik, R.J. Effect of desonide hydrogel 0.05% on the hypothalamic-pituitary-adrenal axis in pediatric subjects with moderate to severe atopic dermatitis. Pediatr. Dermatol. 2007, 24, 289–295. [Google Scholar] [CrossRef]
- Yentzer, B.A.; Ade, R.A.; Fountain, J.M.; Clark, A.R.; Taylor, S.L.; Borgerding, E.; Feldman, S.R. Improvement in treatment adherence with a 3-day course of fluocinonide cream 0.1% for atopic dermatitis. Cutis 2010, 86, 208–213. [Google Scholar]
- Callen, J.; Chamlin, S.; Eichenfield, L.; Ellis, C.; Girardi, M.; Goldfarb, M.; Hanifin, J.; Lee, P.; Margolis, D.; Paller, A.S.; et al. A systematic review of the safety of topical therapies for atopic dermatitis. Br. J. Dermatol. 2007, 156, 203–221. [Google Scholar] [CrossRef] [Green Version]
- Sheu, H.M.; Lee, J.Y.; Chai, C.Y.; Kuo, K.W. Depletion of stratum corneum intercellular lipid lamellae and barrier function abnormalities after long-term topical corticosteroids. Br. J. Dermatol. 1997, 136, 884–890. [Google Scholar] [CrossRef]
- Kao, J.S.; Fluhr, J.W.; Man, M.-Q.; Fowler, A.J.; Hachem, J.-P.; Crumrine, D.; Ahn, S.K.; Brown, B.E.; Elias, P.M.; Feingold, K.R. Short-term glucocorticoid treatment compromises both permeability barrier homeostasis and stratum corneum integrity: Inhibition of epidermal lipid synthesis accounts for functional abnormalities. J. Invest. Dermatol. 2003, 120, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Aberg, K.M.; Radek, K.A.; Choi, E.-H.; Kim, D.-K.; Demerjian, M.; Hupe, M.; Kerbleski, J.; Gallo, R.L.; Ganz, T.; Mauro, T.; et al. Psychological stress downregulates epidermal antimicrobial peptide expression and increases severity of cutaneous infections in mice. J. Clin. Invest. 2007, 117, 3339–3349. [Google Scholar] [CrossRef] [Green Version]
- Eichenfield, L.; Tom, W.; Chamlin, S.; Feldman, S.; Hanifin, J.; Simpson, E.; Berfer, T.; Bergman, J.; Cohen, D.; Cooper, K.; et al. Guidelines of care for the management of atopic dermatitis: Section 1. Diagnosis and assessement of atopic dermatitis. J. Am. Acad. Dermatol. 2014, 70, 338–351. [Google Scholar] [CrossRef] [Green Version]
- Cury-Martins, J.; Martins, C.; Aoki, V.; Góis, A.F.; Ishii, H.; Da Silva, E.M. Topical tacrolimus for atopic dermatitis. Cochrane Database Syst. Rev. 2015, 2015, 009864. [Google Scholar] [CrossRef]
- Chittock, J.; Brown, K.; Cork, M.; Danby, S.G. Comparing the effect of a twice-weekly tacrolimus and betamethasone valerate dose on the subclinical epidermal barrier defect in atopic dermatitis. Acta Derm. Venereol. 2015, 95, 653–658. [Google Scholar] [CrossRef]
- Büchau, A.S.; Schauber, J.; Hultsch, T.; Stuetz, A.; Gallo, R.L. Pimecrolimus enhances TLR2/6-induced expression of antimicrobial peptides in keratinocytes. J. Invest. Dermatol. 2008, 128, 2646–2654. [Google Scholar] [CrossRef] [Green Version]
- Sidbury, R.; Davis, D.M.; Cohen, D.E.; Cordoro, K.M.; Berger, T.G.; Bergman, J.N.; Chamlin, S.L.; Cooper, K.D.; Feldman, S.R.; Hanifin, J.M.; et al. Guidelines of care for the management of atopic dermatitis: Section 3. Management and treatment with phototherapy and systemic agents. Pract. Guideline 2014, 71, 327–349. [Google Scholar]
- Garritsen, F.; Brouwer, M.; Limpens, J.; Spuls, P.I. Photo(chemo)therapy in the management of atopic dermatitis: An updated systematic review with implications for practice and research. Br. J. Dermatol. 2014, 170, 501–513. [Google Scholar] [CrossRef]
- Rubiano, M.O.; Arenas, C.M.; Chalela, J.G. UVA-1 phototherapy for the management of atopic dermatitis: A large retrospective study conducted in a low-middle income country. Int. J. Dermatol. 2018, 57, 799–803. [Google Scholar] [CrossRef]
- Hong, S.P.; Kim, M.J.; Jung, M.-Y.; Jeon, H.; Goo, J.; Ahn, S.K.; Lee, S.H.; Elias, P.M.; Choi, E.H. Biopositive effects of low-dose UVB on epidermis: Coordinate upregulation of antimicrobial peptides and permeability barrier reinforcement. J. Invest. Dermatol. 2008, 128, 2880–2887. [Google Scholar] [CrossRef] [Green Version]
- Gambichler, T.; Skrygan, M.; Tomi, N.; Altmeyer, P.; Kreuter, A. Changes of antimicrobial peptide mRNA expression in atopic eczema following phototherapy. Br. J. Dermatol. 2006, 155, 1275–1278. [Google Scholar] [CrossRef]
- Vähävihu, K.; Ala-Houhala, M.; Perić, M.; Karisola, P.; Kautiainen, H.; Hasan, T.; Snellman, E.; Alenius, H.; Schauber, J.; Reunala, T. Narrowband ultraviolet B treatment improves vitamin D balance and alters antimicrobial peptide expression in skin lesions of psoriasis and atopic dermatitis. Br. J. Dermatol. 2010, 163, 321–328. [Google Scholar] [CrossRef]
- De Jongh, G.J.; Zeeuwen, P.L.; Kucharekova, M.; Pfundt, R.; Van Der Valk, P.G.; Blokx, W.A.; Dogan, A.; Hiemstra, P.; Van De Kerkhof, P.C.; Schalkwijk, J. High expression levels of keratinocyte antimicrobial proteins in psoriasis compared with atopic dermatitis. J. Invest. Dermatol. 2005, 125, 1163–1173. [Google Scholar] [CrossRef]
- Jensen, J.-M.; Ahrens, K.; Meingassner, J.; Scherer, A.; Braeutigam, M.; Stütz, A.; Schwarz, T.; Fölster-Holst, R.; Harder, J.; Gläser, R.; et al. Differential suppression of epidermal antimicrobial protein expression in atopic dermatitis and in EFAD mice by pimecrolimus compared to corticosteroids. Exp. Dermatol. 2011, 20, 783–788. [Google Scholar] [CrossRef]
- Howell, M.D.; Wollenberg, A.; Gallo, R.L.; Flaig, M.; Streib, J.E.; Wong, C.; Pavicic, T.; Boguniewicz, M.; Leung, D.Y. Cathelicidin deficiency predisposes to eczema herpeticum. J. Allergy Clin. Immunol. 2006, 117, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Hazrati, E.; Galen, B.; Lu, W.; Wang, W.; Ouyang, Y.; Keller, M.J.; Lehrer, R.I.; Herold, B.C. Human alpha- and beta-defensins block multiple steps in herpes simplex virus infection. J. Immunol. 2006, 177, 8658–8666. [Google Scholar] [CrossRef] [Green Version]
- Harder, J.; Bartels, J.; Christophers, E.; Schroder, J.M. Isolation and characterization of human beta -defensin-3, a novel human inducible peptide antibiotic. J. Biol. Chem. 2001, 276, 5707–5713. [Google Scholar] [CrossRef] [Green Version]
- Vandamme, D.; Landuyt, B.; Luyten, W.; Schoofs, L. A comprehensive summary of LL-37, the factoctum human cathelicidin peptide. Cell. Immunol. 2012, 280, 22–35. [Google Scholar] [CrossRef]
- Kang, J.; Dietz, M.J.; Li, B. Antimicrobial peptide LL-37 is bactericidal against Staphylococcus aureus biofilms. PLoS ONE 2019, 14, e0216676. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Hoffert, U.; Schwarz, T.; Schroder, J.M.; Glaser, R. Increased expression of human beta-defensin 3 in mollusca contagiosum. Clin. Exp. Dermatol. 2010, 35, 190–192. [Google Scholar] [CrossRef]
- Kirschner, N.; Poetzl, C.; Driesch, P.V.D.; Wladykowski, E.; Moll, I.; Behne, M.J.; Brandner, J.M. Alteration of tight junction proteins is an early event in psoriasis. Am. J. Pathol. 2009, 175, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Kubo, A.; Nagao, K.; Yokouchi, M.; Sasaki, H.; Amagai, M. External antigen uptake by Langerhans cells with reorganization of epidermal tight junction barriers. J. Exp. Med. 2009, 206, 2937–2946. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, N.; Houdek, P.; Fromm, M.; Moll, I.; Brandner, J.M. Tight junctions form a barrier in human epidermis. Eur. J. Cell Biol. 2010, 89, 839–842. [Google Scholar] [CrossRef]
- Ishida-Yamamoto, A.; Kishibe, M.; Murakami, M.; Honma, M.; Takahashi, H.; Iizuka, H. Lamellar granule secretion starts before the establishment of eight junction barrier for paracellular tracers in mammalian epidermis. PLoS ONE 2012, 7, e31641. [Google Scholar] [CrossRef]
- Sugawara, T.; Iwamoto, N.; Akashi, M.; Kojima, T.; Hisatsune, J.; Sugai, M.; Furuse, M. Tight junction dysfunction in the stratum granulosum leads to aberrant stratum corneum barrier function in claudin-1-deficient mice. J. Dermatol. Sci. 2013, 70, 12–18. [Google Scholar] [CrossRef]
- Yokouchi, M.; Kubo, A.; Kawasaki, H.; Yoshida, K.; Ishii, K.; Furuse, M.; Amagai, M. Epidermal tight junction barrier function is altered by skin inflammation, but not by filaggrin-deficient stratum corneum. J. Dermatol. Sci. 2015, 77, 28–36. [Google Scholar] [CrossRef]
- Chen, X.; Takai, T.; Xie, Y.; Niyonsaba, F.; Okumura, K.; Ogawa, H. Human antimicrobial peptide LL-37 modulates proinflammatory responses induced by cytokine milieus and double-stranded RNA in human keratinocytes. Biochem. Biophys. Res. Commun. 2013, 433, 532–537. [Google Scholar] [CrossRef]
- Sugarman, J.L.; Parish, L.C. Efficacy of a lipid-based barrier repair formulation in moderate-to-severe pediatric atopic dermatitis. J. Drugs Dermatol. 2009, 8, 1106–1111. [Google Scholar]
- Lowe, A.J.; Su, J.C.; Allen, K.J.; Abramson, M.J.; Cranswick, N.; Robertson, C.F.; Forster, D.; Varigos, G.; Hamilton, S.; Kennedy, R.; et al. A randomized trial of a barrier lipid replacement strategy for the prevention of atopic dermatitis and allergic sensitization: The PEBBLES pilot study. Br. J. Dermatol. 2018, 178, e19–e21. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, H.L.T.; Trujillo-Paez, J.V.; Umehara, Y.; Yue, H.; Peng, G.; Kiatsurayanon, C.; Chieosilapatham, P.; Song, P.; Okumura, K.; Ogawa, H.; et al. Role of Antimicrobial Peptides in Skin Barrier Repair in Individuals with Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 7607. https://doi.org/10.3390/ijms21207607
Nguyen HLT, Trujillo-Paez JV, Umehara Y, Yue H, Peng G, Kiatsurayanon C, Chieosilapatham P, Song P, Okumura K, Ogawa H, et al. Role of Antimicrobial Peptides in Skin Barrier Repair in Individuals with Atopic Dermatitis. International Journal of Molecular Sciences. 2020; 21(20):7607. https://doi.org/10.3390/ijms21207607
Chicago/Turabian StyleNguyen, Hai Le Thanh, Juan Valentin Trujillo-Paez, Yoshie Umehara, Hainan Yue, Ge Peng, Chanisa Kiatsurayanon, Panjit Chieosilapatham, Pu Song, Ko Okumura, Hideoki Ogawa, and et al. 2020. "Role of Antimicrobial Peptides in Skin Barrier Repair in Individuals with Atopic Dermatitis" International Journal of Molecular Sciences 21, no. 20: 7607. https://doi.org/10.3390/ijms21207607
APA StyleNguyen, H. L. T., Trujillo-Paez, J. V., Umehara, Y., Yue, H., Peng, G., Kiatsurayanon, C., Chieosilapatham, P., Song, P., Okumura, K., Ogawa, H., Ikeda, S., & Niyonsaba, F. (2020). Role of Antimicrobial Peptides in Skin Barrier Repair in Individuals with Atopic Dermatitis. International Journal of Molecular Sciences, 21(20), 7607. https://doi.org/10.3390/ijms21207607